

Abstract
Adeno-associated virus (AAV) provides a promising platform for clinical treatment of neurological disorders owing to its established efficacy and lack of apparent pathogenicity. To use viral vectors in treating neurological disease, however, transduction must occur under neuropathological conditions. Previous studies in rodents have shown that AAV5 more efficiently transduces cells in the hippocampus and piriform cortex than AAV2. Using the kainic acid (KA) model of temporal lobe epilepsy and AAV2 and 5 carrying a hybrid chicken β-actin promoter driving green fluorescent protein (GFP), we found that limbic seizure activity caused substantial neuropathology and resulted in a significant reduction in subsequent AAV5 transduction. Nonetheless, this reduced transduction still was greater than AAV2 transduction in control rats. Although KA seizures compromise blood–brain barrier function, potentially increasing exposure of target tissue to circulating neutralizing antibodies, we observed no interaction between KA seizure-induced damage and immunization status on AAV transduction. Finally, while we confirmed the near total neuronal-specific transgene expression for both serotypes in control rats, AAV5–GFP expression was increasingly localized to astrocytes in seizure-damaged areas. Thus, the pathological milieu of the injured brain can reduce transduction efficacy and alter viral tropism- both relevant concerns when considering viral vector gene therapy for neurological disorders.
Keywords: viral vector gene transfer, neuronal cell death, neurodegeneration, neutralizing antibodies, kainic acid, glia
INTRODUCTION
Viral vector gene therapy has shown substantial promise for the treatment of a wide range of neurological disorders, from epilepsy and lysosomal storage disorders1–5 to Parkinson’s and Alzheimer’s disease.6–8 When working with viral vector-based therapies for neurological diseases, an important consideration is that the product must often be delivered to areas experiencing extensive neurodegeneration and neuroinflammation. Such is the case in considering treatment of temporal lobe epilepsy with hippocampal sclerosis, in which neuronal loss and reactive gliosis can be extensive.9 In such disease states, the accompanying blood–brain barrier (BBB) compromise can also provide access of normally restricted serum factors to the central nervous system (CNS).10 Furthermore, degenerative conditions or disease-specific factors can alter cell-surface receptor densities and introduce immune factors that can change the overall milieu of the target area.11 Though generally overlooked, a neuropathological brain environment may significantly impact the success of gene transfer.
To date, the adeno-associated virus 2 (AAV2) serotype has dominated AAV-based clinical trials because of its long history of study and recent clinical success.12 However, studies have shown in both rodent and non-human primates that other AAV serotypes are capable of CNS transduction efficiencies superior to AAV2 in the context of direct parenchymal injection.13 Moreover, neutralizing antibodies (nAbs) against AAV2 are found circulating in the majority of the human population, presenting additional constraints on this serotype for use in human CNS application.14 These issues have provided the impetus to seek alternative serotypes for therapeutic use.
It is currently unknown whether AAV differentially transduces target tissue in a neurodegenerative milieu, and whether such differences are common or differentially observed across serotypes. The rat kainic acid (KA) model of temporal lobe epilepsy not only results in a chronic spontaneous seizure phenotype, but also induces a neurodegenerative and neuroinflammatory phenotype15–17 comparable with that observed in some patients with temporal lobe epilepsy. Neuronal cell loss may restrict or redirect viral vector transduction, and BBB compromise could introduce vascular factors such as vector-nAbs that could hinder successful gene transfer. Thus, the present studies quantitatively and qualitatively examine the effects of KA-induced seizures on subsequent AAV2 and AAV5 transduction of the rat dorsal hippocampus (HC) and piriform/entorhinal cortex to determine the influence of neuropathological target tissue on viral vector-based gene expression.
RESULTS
KA seizure-induced neuronal cell death leads to loss of AAV transduction
The peripheral administration of KA (10 mg kg−1, intraperitoneal (i.p.)) produced the expected limbic seizure activity, and 2 weeks later, AAV2–chicken β-actin promoter (CBA)–green fluorescent protein (GFP; 2 μl; 1.8×109 vector genomes (vgs); left hemisphere) and AAV5–CBA–GFP (2 μl; 1.8×109 vgs; right hemisphere) were infused into the dorsal HC or piriform cortex (PC). A further 2 weeks later, immunohistochemistry for the neuronal marker NeuN confirmed the expected neuronal loss in both the dorsal HC (hilus, CA3 and CA1) and PC of KA-treated rats (Figure 1). Other damaged loci included the amygdala and dorsal thalamic nuclei (not shown), consistent with previous literature.18 Evaluation of GFP transduction volume (defined as the volume of brain tissue containing immunohistochemical evidence of GFP protein) of AAV2 and AAV5 in control and KA-seized animals found that the volume of AAV5 transduction far surpassed that of AAV2 in both the dorsal HC (serotype main effect: F1,8=55.02, P<0.0001) and PC (serotype main effect: F1,8=21.87, P=0.002; Figure 2). Comparisons between the KA-treated groups and -untreated controls showed a significant loss of transduction volume in KA-responsive rats compared with controls in the dorsal HC (seizure main effect: F1,8=16.25, P=0.004). Although a similar pattern of diminished transduction volume was found in PC, the difference in loss of volume compared with controls was not statistically significant (seizure main effect: F1,8=1.94, P=0.201). Finally, there was an interaction of serotype and seizure-induced damage on transduction efficiency in the HC (serotype×damage interaction: F1,8=9.01, P=0.017). Specifically, the deleterious effect of seizures on AAV transduction volume was prominent in the AAV5-treated hemisphere, but there was no effect of seizures on AAV2 transduction.
Figure 1.

KA-induced neuronal cell loss in the dorsal hippocampus and piriform cortex. Panels show NeuN immunohistochemistry of the dorsal hippocampus (a, b) and piriform cortex (d, e) of control (a, d) and KA-treated (b, e) rats. Although this image is representative of the pattern of neuronal cell loss in many animals, others showed less extensive CA1 damage and/or greater hilar or CA3 damage. Panels c, f show the level of the dorsal hippocampus and piriform cortex, respectively, adapted from the atlas of Paxinos and Watson.43 Outlines in gray (c) illustrate the boundaries used for stereological analysis of NeuN-positive and GFP-positive cells.
Figure 2.

AAV2 and AAV5 transduction volumes in control and KA-damaged dorsal hippocampus and piriform cortex. The upper portion of panels a, b show representative GFP-immunostained sections, revealing the prominent difference between AAV2 and AAV5 transduction of the dorsal hippocampus and piriform cortex (respectively). The lower representative images in panels a, b demonstrate that rats having experienced KA seizure damage overall showed loss of transduction volume compared with control-treated animals. Panels c, d graph the transduction volumes of AAV2 and AAV5 in the hippocampus (c) and piriform cortex (d) of control and seizure damaged animals. Transduction volume is defined as the entire GFP-immunostained area, including cellular process and cell bodies. In both panels c, d, groups with the same letter are not significantly different from one another, whereas groups with different letters are significantly different from one another (P<0.05). Error bars represent the s.e.m.
Given the transduction volume data, we sought to determine whether the loss of AAV5 transduction volume was because of the decreases in the number of transduced cells. To make this determination, numbers of GFP-positive cells were stereologically counted in subsets of dorsal HC sections in tissue derived from control and KA-treated rats (Figure 3). The total number of GFP-positive cells in CA1 dropped significantly in KA-treated rats compared with control rats (t13=10.26, P=0.012), with a similar, but nonsignificant pattern of loss in the dentate hilus. When the total number of neurons was quantified, a significant loss of NeuN-stained cells was found in both CA1 (CA1: t18=6.31, P<0.0001) and the dentate hilus (hilus: t (19)=3.044, P=0.007). Thus, as seen in Figures 3a and b, the neuronal viability pattern matched the pattern of GFP-positive cells. Moreover, in CA1, a significant, positive correlation was found between the number of neurons and GFP-positive cells (r=0.734, P=0.002). A positive, but nonsignificant correlation also was found in the dentate hilus (r=0.343, P=0.177), wherein a lesser extent of cell loss was observed. Together, these results show that hippocampal neuronal cell loss results in fewer transgene-expressing neurons.
Figure 3.
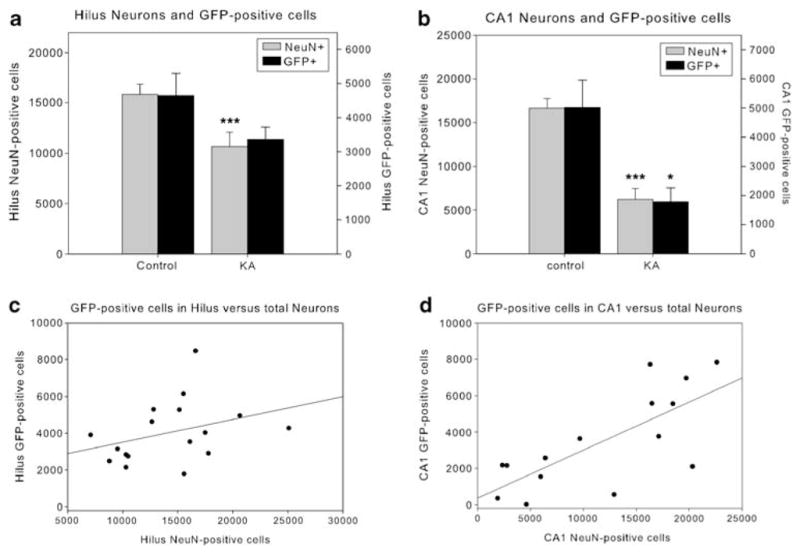
Quantification of AAV5-transduced cells in the dorsal hippocampus in relationship to neuronal viability. The overall number of NeuN- and GFP-positive cells decreased in the hippocampus of KA seizure-damaged animals compared with controls, and in CA1, there is a strong correlation between the number of NeuN- and GFP-positive cells. Panels a, b graph the number of neurons (NeuN, left y axis) and GFP-positive cells (right y axis) in the hilus (a) and CA1 (b) of control and KA seizure-damaged animals. Panels c, d show a regression between NeuN- and GFP-positive cells in the hilus (c) and CA1 (d). ***, *, significantly different from untreated control group (P<0.01, <0.05, respectively). Error bars represent the s.e.m.
KA seizures influence localization of AAV5-based GFP expression in the HC
Previous reports have shown that with a CBA promoter intracranially administered AAV5 results predominantly in neuronal transgene expression in rats.13 We labeled brain sections for astrocyte (glial fibrillary acidic protein (GFAP)), neuron (NeuN) and microglia (OX42) markers to localize AAV5–GFP-positive cells. In both the dorsal HC and PC, GFP-positive cells were predominantly neuronal (not shown). There was much more OX42 fluorescent label in kainate-treated versus control rats, and the OX42 levels were highest in hippocampal, piriform/entorhinal and dorsal thalamic areas, suggestive of activated microglia. However, we observed no GFP–OX42 co-localization (not shown). Hippocampal astrocytes of kainate-treated rats had larger cell bodies and thicker processes than those of control rats (Figure 4). Strikingly, GFP–GFAP co-localization was abundant in both the HC and PC of kainate-treated rats; no such co-localization was observed in control animals (Figure 4). GFP-expressing astrocytes were frequently observed in the hilar region of the hippocampal dentate gyrus, and in the hippocampal fissure. We observed several, but fewer GFP-positive astrocytes in the PC of these same tissue samples.
Figure 4.
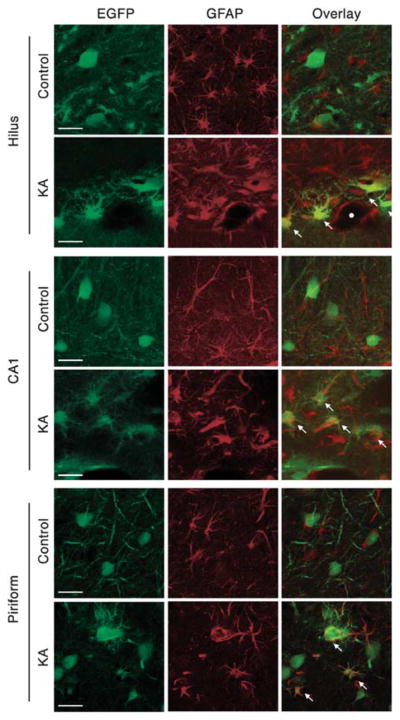
AAV5–GFP localizes to astrocytes in the KA seizure-damaged hippocampus and piriform cortex. In the dentate hilus, CA1, and piriform cortex of control animals, GFP-positive cells co-localize to neurons (NeuN, not shown), but not to astrocytes (GFAP). However, GFP does co-localize to astrocytes in the KA-damaged hippocampus and piriform cortex. Kainate-treated rats show astrocyte hypertrophy and fewer GFP-labeled cellular processes (presumably neuronal). White arrows point to astrocyte-localized GFP-positive cells. The white circle depicts a needle track in the hilus. Scale bars represent 25 μm.
Effect of AAV2-immunization and KA-induced seizures on AAV2 transduction of the HC and PC
KA-induced seizures result in BBB compromise, leading to extravasation of serum factors into the parenchyma19,20 and potentially greater penetration of circulating AAV2-nAbs. AAV2 immunization was accomplished by i.p. injection of 1×1011 vgs of AAV2–CBA–luciferase at 1 month and 2 weeks before the KA treatment. This procedure resulted in the presence of serum AAV2-nAbs at the time of intracranial vector injection (Figures 5a and b). As expected, no detectable levels of AAV2-nAbs were found in non-immunized rats (Figure 5b). Also, no rats had any detectable levels of serum-nAbs against AAV2 at the time of the first i.p. immunization. However, on the basis of GFP-immunohistochemistry, a significant effect of pre-immunization was found for AAV2-based GFP expression (transduction volume) in both the HC (F1,15=5.29, P=0.04) and PC (F1,15=7.73, P=0.014). In contrast, there was no effect of AAV2 pre-immunization on AAV5 transduction (data not shown, but illustrated in Figure 5d). In addition, there was no effect of seizures or interaction of seizure and immunization status on transduction volume in either brain area, although there was a trend for decreased transduction volume in the seizure-damaged PC (P=0.054). As seizure status did not interact with immunization status to alter vector transduction, immunoglobulin G (IgG) extravasation was examined to verify that KA seizures resulted in permeability of the BBB. We observed minimal IgG in the HC or PC of non-immunized, control-treated rats, whereas some IgG extravasation was found in the PC of non-immunized, seized rats (data not shown). Surprisingly, a striking presence of rat IgG was found diffusely spread in both control and seizure-exposed rats throughout the AAV2-injected dorsal HC and PC (Figure 5d). In contrast, there was little to no IgG reactivity in the AAV5-injected hemisphere (Figure 5d). Furthermore, with regard to AAV2, we found that GFP localization remained limited to neurons in both pre-immunized and pre-immunized, seizure damaged animals (not shown).
Figure 5.
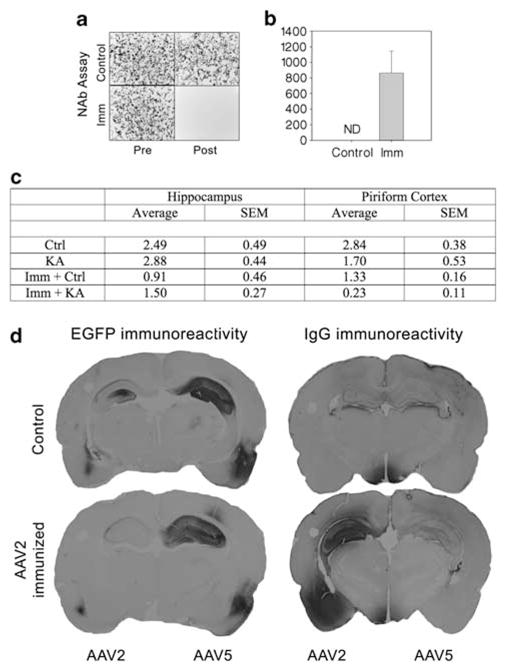
Effects of AAV2 immunization on parenchymal IgG and AAV transduction. AAV2 transduction in the dorsal hippocampus and piriform cortex of immunized (Imm) and KA-pretreated rats. Panel a shows representative images of GFP-fluorescent HEK 293 cells from the neutralizing antibody (nAb) titering assay. Upper panels show cells at 24 h post treatment with AAV2 pre-incubated serum from non-immunized animals before the first treatment (left) and at the time of vector injection at 6 weeks later (right). Lower panels show cells at 24 h post treatment with AAV2 pre-incubated serum from immunized animals before the first treatment (left) and at the time of vector injection at 6 weeks later (right). Panel b shows the average nAb titers found in non-immunized and immunized rats at the time of intracranial AAV injection (ND=none detected). Panel c shows that AAV2-immunized rats had far less AAV2 transduction volume in both the hippocampus and piriform cortex than non-immunized rats. However, there was no interaction between pre-immunization and seizure damage on AAV2 transduction in the hippocampus or piriform cortex, suggesting that seizure-associated BBB damage is not sufficient to further impact kainic AAV2 transduction in the brain of the AAV2-pre-immunized animal. Panel d shows representative GFP- and IgG-immunostained coronal sections of the rat brain at the level of hippocampus and piriform cortex. Non-immunized rats showed greater AAV5-based GFP expression than AAV2 in the hippocampus and piriform (panel d, upper left section). In addition, non-immunized rats showed little IgG extravasation in either the hippocampus or piriform cortex (panel d, upper right section). In contrast, rats with circulating AAV2-nAbs showed very low relative AAV2 transduction in these areas (panel d, lower left section), and had a striking IgG antibody presence in AAV2-injected parenchyma areas of the dorsal hippocampus and piriform cortex (panel d, lower right section). IgG antibody presence in the AAV5-injected side of the brain was absent, and there was no effect of AAV2-nAb presence on AAV5 transduction. Ctrl, control; EGFP, enhanced GFP.
DISCUSSION
Previous studies in rats have demonstrated the increased transduction efficacy of AAV5 when compared with that of AAV2 (refs 13, 21) throughout a variety of brain areas, and indeed the present results confirm these findings in the HC and PC. Because AAV-mediated delivery of therapeutic genes to both the HC and PC significantly attenuates limbic seizure activity,22,23 the transduction efficacy of AAV2 and AAV5 needed to be determined under conditions of prior seizure-induced neuropathology. The present results show that even though the transduction volumes for both AAV2 and AAV5 in the PC decrease substantially, AAV5 transduction volume within this seizure damaged tissue was still greater than AAV2 transduction volume in healthy tissue. Thus, in the context of temporal lobe epilepsy animal models, AAV5 results in a greater transgene-expressing volume in both control and seizure-damaged HC and PC than AAV2.
Certainly, the present findings demonstrate that seizure-induced neuronal cell death results in fewer AAV5-transduced cells. However, another component to loss of transduced cells could involve changes to the cellular milieu. In the damaged brain, and specifically in the presence of dying or injured cells, glia assume unique phenotypes,24 which may passively or actively limit target cell uptake of a viral vector.25 In the presence of dead or dying cells, microglia become activated and take on a phagocytic phenotype,26 which may result in active vector clearance. An early study tracking fluorescently labeled AAV2 capsids found that in untreated control rats, microglial uptake of viral capsids occurs within 24 h of injection.27 Thus, it is possible that activated microglia could increase virus clearance. In addition, neuropathology can alter the cellular and extracellular microenvironments, which could significantly influence viral vector tropism. For example, nerve injury induces an upregulation of integrin α-subunits in microglia,28 whereas pilocarpine-induced seizure damage to the HC increases the expression of α-5 integrin subunits on astrocytes.29 Because αVβ5 integrin serves as the AAV2 co-receptor,30 pathology-induced shifts in cellular integrin expression could significantly influence the pattern of viral vector transduction. Similarly, a pathological environment can influence promoter function. Through the use of an astrocyte-specific promoter, gene expression can be achieved in astrocytes even in AAV serotypes that normally drive neuronal-specific expression under a constitutive promoter (such as the CBA promoter).31,32 In the present findings with AAV5, seizure-induced pathology in the HC resulted in some astrocytic GFP even though a constitutive promoter drove gene expression. Thus, for any gene therapy that requires a specific cellular transduction pattern, the required gene expression pattern should be validated in the presence of the appropriate neuropathological milieu.
Many neurological disorders including epilepsy33 cause a compromise in the BBB sufficient to allow the extravasation of peripheral immune cells and serum factors into the CNS. Because a substantial portion of the human population have circulating antibodies to AAV2, it is possible that in cases of epilepsy-neutralizing AAV antibodies would have greater access to the CNS. Of the currently recognized AAV serotypes, immunoglobulins against AAV2 are the most prevalent in the human population (72%), and those against AAV5 the least prevalent (3.2%).14 Previous studies using adjuvant-based immunization strategy (median nAb titers of 51 200) have shown that AAV2 transduction via intracranial injection into the striatum is completely blocked,34 whereas studies adopting less rigorous immunization strategies found that pre-existing immunity to AAV decreased transgene expression in the striatum only in animals with pre-existing nAb titers above 1208.35 The present immunization strategy produced nAb titers of ~850, and this level of nAb proved sufficient to significantly reduce transduction volume of AAV2 in both the HC and PC. These nAb titers are similar with those found in the human population, in which >75% of healthy subjects in one study had nAb titers >400.14 However, the present results show that the documented BBB compromise caused by KA-induced seizures did not significantly facilitate immune clearance of AAV2 beyond that observed in immunized, non-KA-treated controls.
As IgG class antibodies comprise a major source of the human immune response to AAV2, it was possible that IgG could be associated with the observed loss of AAV2 transduction. Immunohistochemistry against rat IgG showed that in AAV2-immunized rats IgG was present in areas of the AAV2 injections. However, IgG was absent in non-immunized animals, as well as in the area of AAV5 injection for both immunized and non-immunized animals. Thus, this IgG extravasation into the CNS was because of presence of AAV2 but not because of the injection procedure alone. Because the immunizing stimulus used the same capsid but a different packaged gene sequence, the IgG likely is selective to the AAV2 capsid and not the transgene product. Clearly, prior immunization to AAV2 provokes a significant IgG response on subsequent intracranial AAV2 injection.
In conclusion, the present studies establish that AAV5 vectors exhibit a number of advantages over AAV2 vectors particularly in brain areas exhibiting seizure-induced damage. Thus, neuropathological states that result from specific CNS disorders must be considered in the application of any proposed viral vector gene therapy for neurological disorders.
MATERIALS AND METHODS
Recombinant virus production and quantification
Virus was produced in HEK-293 cells as previously described.36–38 Briefly, polyethylenimine (linear molecular weight, ~25 000) was used to for triple transfection of the pXR2/pXR5 rep and cap plasmid, the pXX6-80 helper plasmid, and GFP or luciferase (with pXR2 only for immunization) reporter plasmids containing either enhanced GFP or firefly luciferase transgenes under the hybrid CBA promoter and flanked by inverted terminal repeats. At 72 h post-transfection, cells were harvested and virus was purified by cesium chloride gradient density centrifugation. After identifying peak fractions by dot blot hybridization, virus was dialyzed into 1×phosphate-buffered saline (PBS) and 5% sorbitol. Titers were calculated by quantitative PCR according to established procedures38 using a LightCycler 480 using TaqMan PCR reaction mix (Applied Biosystems, Carlsbad, CA, USA) and primers designed against the SV40 polyA sequence: (forward) 5′-AGCAATAGCATCACAAATTTCACAA-3′, and (reverse) 5′-CCAGACATGATAAGATACATTGATGAGTT-3′. Adenovirus was generously provided by Pat Hearing and produced and purified as previously described.39,40
Immunization, KA administration, stereotactic injection, perfusion, fixation
A total of 20 male Sprague–Dawley rats (Charles River, Morrisville, NC, USA) with a starting weight of ~250 g (at time of immunization) were maintained in a 12-h light–dark cycle and had free access to water and food. All care and procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and all procedures received approval by the University of North Carolina Institutional Animal Care and Use Committee. To immunize rats against AAV2, 1×1011 vgs of AAV2 (suspended in 200 μl PBS) containing a firefly luciferase transgene were delivered through i.p. injection (week 0) followed by an equivalent booster injection at 2 weeks later. Control rats received PBS injection only. At week 4, rats received an i.p. injection of KA (10 mg kg−1, n=18) or PBS (control, n=4). At 1 h after the first class, IV seizure (based on the Racine Motor Seizure Grading Scale41) seizing rats were given 25 mg kg−1 pentobarbital i.p. Rats treated with KA but not showing seizure behaviors (n=7) were included with control animals. The duration and quantity of convulsive seizures varied somewhat between animals, with most rats having between 1 and 3 min of total class IV/V seizure duration. At 2 weeks after KA treatment (week 6), rats received intracranial viral vector infusions. For intracranial infusions, rats were anesthetized with isofluorane and placed into a stereotaxic frame. Using a 32-gauge stainless steel injector and a Fisher scientific infusion pump, rats received 2 μl (1.8×109 vgs) per hemisphere AAV2–CBA–GFP (left hemisphere) and AA5–CBA–GFP (right hemisphere) into the HC (bregma −4.2 mm, lateral+/−3.0 mm, skull −3.7 mm) followed by an equivalent bilateral injection in the PC (bregma −3.2 mm, lateral +/−6.0 mm, skull −7.8 mm).37 The infusion rate was 0.2 μl min−1, and the injector was left in place for 3 min after each infusion to allow diffusion from the injector. At 2 weeks after intracranial infusion, animals were killed using an overdose of pentobarbital (100 mg kg−1 pentobarbital, i.p.) and were perfused transcardially with ice-cold 100 mM sodium PBS (pH 7.4), followed by 4% paraformaldehyde in PBS (pH 7.4) as a fixative.
Immunohistochemistry
After brains were post fixed for 48 h at 4 °C, 40 μm coronal sections were cut using a vibrating blade microtome and subjected to immunohistochemical processing for GFP. Separate colorimetric GFP and neuronal detection was performed by incubating free-floating sections with a rabbit polyclonal antibody to GFP (1:1000, Millipore, Bedford, MA, USA), a mouse monoclonal antibody to NeuN (1:1000, Chemicon, Temecula, CA, USA), or a biotinylated antibody to rat IgG (1:250, Vector Laboratories, Burlingame, CA, USA). Sections were blocked in 10% normal goat or horse serum (Vector Laboratories), respectively, and 0.1% Triton X-100 in PBS for 1 h. The sections were then incubated with primary antibody (prepared in 3% normal serum, 0.2% Triton X-100) for 48–72 h at 4 °C with gentle agitation. Immunoperoxidase staining was then performed using the anti-rabbit Vectastain Elite ABC kit (Vector Laboratories) as described in the manufacturer’s instructions, with 3,3′-diaminobenzidine substrate and nickel–cobalt intensification of the reaction product. A rat-adsorbed biotinylated anti-mouse secondary (Vector Laboratories) was substituted for the Vectastain secondary for use with the NeuN antibody to avoid crossreactivity of the secondary antibody with possible extravasated rat IgG. No additional secondary antibodies were required for the anti-rat IgG reaction, however the Vectastain Elite ABC kit was used for streptavidin/biotin conjugation. Brain sections were mounted on slides, dehydrated and coverslipped. Sections used for representative illustrations were digitized using either a Leica MX16FA Stereo Microscope/Macroscope with a ×0.63 objective (Leica, Mannheim, Germany) and Cool Snap HQ2 black and white camera, and or Nikon Microphot SA with a ×2 objective, Nikon DXM 1200 color camera (Nikon, Melville, NY, USA), and ACT-1 software (Michael Hooker Microscopy Core Facility, University of North Carolina at Chapel Hill, NC, USA).
GFP fluorescence co-localization to specific brain cell subsets was performed by incubating sections adjacent to the 3,3′-diaminobenzidine-stained sections simultaneously with antibodies targeting the neuronal marker NeuN (1:1000; mouse monoclonal; Chemicon), with antibodies targeting the astrocyte marker GFAP (anti-GFAP, 1:4000; rabbit polyclonal; Dako, Glostrup, Denmark). Sections were first blocked as described above, and then incubated at 4 °C for 24 h in appropriate primary antibodies. After PBS rinsing and a second 1-h block, sections were then co-incubated for 1 h at 4 °C with Alexafluor 350-conjugated goat-anti-rabbit IgG (secondary to GFAP antibody; 1:500; Invitrogen, Carlsbad, CA, USA) and/or Alexafluor 594-conjugated goat anti-mouse IgG (secondary to NeuN; 1:500; Invitrogen) in PBS with 3% goat serum. Rinsed sections were mounted and fluorescence was visualized by confocal microscopy. Confocal imaging was performed at the Michael Hooker Microscopy Center at UNC-Chapel Hill using a Leica SP2 laser scanning confocal microscope (Leica). Images were processed using the LAS-AF (Leica) and Adobe Photoshop software. Native GFP and dual-label fluorescence images were reconstructed from single scan (line averaging=4) sections using a ×16 oil objective at an ×8 zoom.
Analysis of nAb titers
To ensure consistency, AAV2 prepared from a single batch preparation was used for both intracranial injections and also the in vitro neutralization assay. Sera were obtained from rats through the tail vein both immediately prior to the first i.p. AAV2 injection (for immunization) and during intracranial injection. AAV-nAb titers were obtained using methods similar to Moskalenko et al.42 and McPhee et al.39 Briefly, sera were heated to 56 °C for 30 min to inactivate complement, serially diluted in PBS (1:3 and 1:30 through 1:30 720 in 2×dilution steps) and mixed with equivalent volumes of AAV2 (multiplicity of infection=300) containing the enhanced GFP complementary DNA. The mixture was incubated for 2 h at 4 °C and added to HEK293 cells along with adenovirus (Ad5 dl309, multiplicity of infection=30). nAB titer was defined as the lowest dilution of sera at which an ~50% decline of immunofluorescence was detected, when compared with control (no sera) samples.
Anatomical demarcation and stereological measurement of transduction volume and cell counts
AAV transduction volumes were estimated stereologically by the Cavalieri method using Stereologer software (Stereology Resource Center, Inc., Chester, MD, USA). The entire area of 3,3′-diaminobenzidine-stained GFP immunoreactivity, including cell bodies and neuronal processes was encircled on every sixth serial section within the region of interest under ×4 magnification. The circumscribed area containing GFP-positive neuronal processes was greater than the area of transduced cell bodies alone. Generally HC-targeted AAV was contained within the HC, with very little spread of 3,3′-diaminobenzidine label into surrounding thalamic and cortical areas. Extra-hippocampal areas were not included in transduction volume calculations. Transduced areas around the piriform-targeted areas often included entorhinal cortex and amygdala areas as well as the PC. The total area of contiguous transduction in these general brain areas was counted.
AAV transduction volume in the dorsal HC and PC was determined separately from cell counting data collection. Both NeuN- and GFP-positive cells were counted in the same manner. Anatomical borders were determined using a rat brain atlas.7,43 Dorsal CA1 NeuN-positive and GFP-positive cells were counted within the pyramidal cell layer, in sections encompassing approximately bregma −3.14 through bregma −4.3, rostrocaudal. Boundaries for CA1 and CA2 were estimated, and thus a small fraction of the cell counts may be based on dorsal CA2. Rostrocaudal selection of the starting brain section was consistent across all samples, and subsequently measured sections were predetermined by the stereological software. Dentate hilus anatomical boundaries were determined by drawing a line from the inner to the outer blades of the dentate gyrus granule cell layers and capturing the entire area within (but not including) the granule cell layers (see Figure 1 for illustrations on the outlining of CA1 and the dentate hilus).
Stereology was performed similarly to Zhao et al.44 The computer-based system included Stereologer software, and the equipment consisted of an Olympus IX70 microscope with an Applied Scientific Instrumentation (Eugene, OR, USA) motorized stage and color CCD camera (Sony Corp., Teaneck, NJ, USA). The stereological software controlled operation of the system. We used the optical fractionator method45 for making unbiased estimates of particle counts.43,46 A series of pilots on these tissue sections generated optimal dissector dimensions and sampling frequency to incur a coefficient of error below 0.10 (10% method error) for both volume and cell number estimates a standard consistent with the software handbook guidelines, as well as previous work using similar tissue and software.44 The sampling parameters were as follows: sampling frequency=0.17 (6 sample sections out of 36 total sections) for GFP-positive cell quantification and 0.08 (3 sample sections out of 36 total sections) for NeuN-positive cells. The height of each dissector was 20 μm, the guard height was 1 μm, the dissector frame area was set at 50% (screen height),2 and dissectors spaced 300 μm apart were randomly generated by the Stereologer program. A ×5 objective was used to outline the region of interest. A ×10 objective was used for GFP-positive cell counting, and a ×20 objective was used for NeuN-positive cell counting.
Statistical analysis
Two-way analysis of variance (AAV serotype (within subjects) ×treatment (between subjects); Pillai’s trace) were performed in comparing volume transduction between AAV2 and AAV5 in the dorsal HC and PC. A student t-test was used to analyze NeuN-positive and GFP-positive cells in the hilus and CA1 of the dorsal HC, and a Pearson’s correlation analysis was performed when comparing NeuN-positive to GFP-positive cells. Finally, a two-way factorial analysis of variance (treatment×immunization status) was used to analyze AAV2 volume transduction in the dorsal HC and PC. Post hoc analyses of transduction volumes were performed using Fisher’s least significant differences tests.
Acknowledgments
We thank Dr Chengwen Li and Dr Matthew Hirsch of the UNC Chapel Hill Gene Therapy Center, and Michael Chua and Neal Kramarcy of the UNC Chapel Hill Michael Hooker Microscopy Core for technical advice, Swati Yadav for calculation of viral titers by qPCR, and Aadra Bhatt for manuscript review. This research was funded NINDS Grant NS35633 (TJM).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Noe F, Pool AH, Nissinen J, Gobbi M, Bland R, Rizzi M, et al. Neuropeptide Y gene therapy decreases chronic spontaneous seizures in a rat model of temporal lobe epilepsy. Brain. 2008;131 (part 6):1506–1515. doi: 10.1093/brain/awn079. [DOI] [PubMed] [Google Scholar]
- 2.McCown TJ. Adeno-associated virus-mediated expression and constitutive secretion of galanin suppresses limbic seizure activity in vivo. Mol Ther. 2006;14:63–68. doi: 10.1016/j.ymthe.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 3.McCown TJ. Adeno-associated virus vector-mediated expression and constitutive secretion of galanin suppresses limbic seizure activity. Neurotherapeutics. 2009;6:307–311. doi: 10.1016/j.nurt.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimpo M, Ikeda U, Maeda Y, Takahashi M, Miyashita H, Mizukami H, et al. AAV-mediated VEGF gene transfer into skeletal muscle stimulates angiogenesis and improves blood flow in a rat hindlimb ischemia model. Cardiovasc Res. 2002;53:993–1001. doi: 10.1016/s0008-6363(01)00546-6. [DOI] [PubMed] [Google Scholar]
- 5.McPhee SW, Janson CG, Li C, Samulski RJ, Camp AS, Francis J, et al. Immune responses to AAV in a phase I study for Canavan disease. J Gene Med. 2006;8:577–588. doi: 10.1002/jgm.885. [DOI] [PubMed] [Google Scholar]
- 6.Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet. 2007;369:2097–2105. doi: 10.1016/S0140-6736(07)60982-9. [DOI] [PubMed] [Google Scholar]
- 7.Lawlor PA, Bland RJ, Das P, Price RW, Holloway V, Smithson L, et al. Novel rat Alzheimer’s disease models based on AAV-mediated gene transfer to selectively increase hippocampal Abeta levels. Mol Neurodegener. 2007;2:11. doi: 10.1186/1750-1326-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryan DA, Mastrangelo MA, Narrow WC, Sullivan MA, Federoff HJ, Bowers WJ. Abeta-directed single-chain antibody delivery via a serotype-1 AAV vector improves learning behavior and pathology in Alzheimer’s disease mice. Mol Ther. 2010;18:1471–1481. doi: 10.1038/mt.2010.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crespel A, Coubes P, Rousset MC, Brana C, Rougier A, Rondouin G, et al. Inflammatory reactions in human medial temporal lobe epilepsy with hippocampal sclerosis. Brain Res. 2002;952:159–169. doi: 10.1016/s0006-8993(02)03050-0. [DOI] [PubMed] [Google Scholar]
- 10.van Vliet EA, da Costa Araujo S, Redeker S, van Schaik R, Aronica E, Gorter JA. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain. 2007;130 (part 2):521–534. doi: 10.1093/brain/awl318. [DOI] [PubMed] [Google Scholar]
- 11.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease–a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 12.Mitchell AM, Nicolson SC, Warischalk JK, Samulski RJ. AAV’s Anatomy: roadmap for optimizing vectors for translational success. Curr Gene Ther. 2010;10:319–340. doi: 10.2174/156652310793180706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, et al. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther. 2004;10:302–317. doi: 10.1016/j.ymthe.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 14.Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 15.Oprica M, Eriksson C, Schultzberg M. Inflammatory mechanisms associated with brain damage induced by kainic acid with special reference to the interleukin-1 system. J Cell Mol Med. 2003;7:127–140. doi: 10.1111/j.1582-4934.2003.tb00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minami M, Kuraishi Y, Satoh M. Effects of kainic acid on messenger RNA levels of IL-1 beta, IL-6, TNF alpha and LIF in the rat brain. Biochem Biophys Res Commun. 1991;176:593–598. doi: 10.1016/s0006-291x(05)80225-6. [DOI] [PubMed] [Google Scholar]
- 17.Sperk G, Lassmann H, Baran H, Seitelberger F, Hornykiewicz O. Kainic acid-induced seizures: dose-relationship of behavioural, neurochemical and histopathological changes. Brain Res. 1985;338:289–295. doi: 10.1016/0006-8993(85)90159-3. [DOI] [PubMed] [Google Scholar]
- 18.Nadler JV. Minireview. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci. 1981;29:2031–2042. doi: 10.1016/0024-3205(81)90659-7. [DOI] [PubMed] [Google Scholar]
- 19.Nitsch C, Klatzo I. Regional patterns of blood-brain barrier breakdown during epilepti-form seizures induced by various convulsive agents. J Neurol Sci. 1983;59:305–322. doi: 10.1016/0022-510x(83)90016-3. [DOI] [PubMed] [Google Scholar]
- 20.Ruth RE. Increased cerebrovascular permeability to protein during systemic kainic acid seizures. Epilepsia. 1984;25:259–268. doi: 10.1111/j.1528-1157.1984.tb04185.x. [DOI] [PubMed] [Google Scholar]
- 21.Klein RL, Dayton RD, Leidenheimer NJ, Jansen K, Golde TE, Zweig RM. Efficient neuronal gene transfer with AAV8 leads to neurotoxic levels of tau or green fluorescent proteins. Mol Ther. 2006;13:517–527. doi: 10.1016/j.ymthe.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haberman RP, Samulski RJ, McCown TJ. Attenuation of seizures and neuronal death by adeno-associated virus vector galanin expression and secretion. Nat Med. 2003;9:1076–1080. doi: 10.1038/nm901. [DOI] [PubMed] [Google Scholar]
- 23.Richichi C, Lin EJ, Stefanin D, Colella D, Ravizza T, Grignaschi G, et al. Anticonvulsant and antiepileptogenic effects mediated by adeno-associated virus vector neuropeptide Y expression in the rat hippocampus. J Neurosci. 2004;24:3051–3059. doi: 10.1523/JNEUROSCI.4056-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 25.Hendriks WT, Eggers R, Verhaagen J, Boer GJ. Gene transfer to the spinal cord neural scar with lentiviral vectors: predominant transgene expression in astrocytes but not in meningeal cells. J Neurosci Res. 2007;85:3041–3052. doi: 10.1002/jnr.21432. [DOI] [PubMed] [Google Scholar]
- 26.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 27.Bartlett JS, Samulski RJ, McCown TJ. Selective and rapid uptake of adeno-associated virus type 2 in brain. Hum Gene Ther. 1998;9:1181–1186. doi: 10.1089/hum.1998.9.8-1181. [DOI] [PubMed] [Google Scholar]
- 28.Kloss CU, Werner A, Klein MA, Shen J, Menuz K, Probst JC, et al. Integrin family of cell adhesion molecules in the injured brain: regulation and cellular localization in the normal and regenerating mouse facial motor nucleus. J Comp Neurol. 1999;411:162–178. doi: 10.1002/(sici)1096-9861(19990816)411:1<162::aid-cne12>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 29.Fasen K, Elger CE, Lie AA. Distribution of alpha and beta integrin subunits in the adult rat hippocampus after pilocarpine-induced neuronal cell loss, axonal reorganization and reactive astrogliosis. Acta Neuropathol. 2003;106:319–322. doi: 10.1007/s00401-003-0733-y. [DOI] [PubMed] [Google Scholar]
- 30.Summerford C, Bartlett JS, Samulski RJ. AlphaVbeta5 integrin: a co-receptor for adeno-associated virus type 2 infection. Nat Med. 1999;5:78–82. doi: 10.1038/4768. [DOI] [PubMed] [Google Scholar]
- 31.Weller ML, Stone IM, Goss A, Rau T, Rova C, Poulsen DJ. Selective overexpression of excitatory amino acid transporter 2 (EAAT2) in astrocytes enhances neuroprotection from moderate but not severe hypoxia-ischemia. Neuroscience. 2008;155:1204–1211. doi: 10.1016/j.neuroscience.2008.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lawlor PA, Bland RJ, Mouravlev A, Young D, During MJ. Efficient gene delivery and selective transduction of glial cells in the mammalian brain by AAV serotypes isolated from nonhuman primates. Mol Ther. 2009;17:1692–1702. doi: 10.1038/mt.2009.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mihaly A, Bozoky B. Immunohistochemical localization of extravasated serum albumin in the hippocampus of human subjects with partial and generalized epilepsies and epileptiform convulsions. Acta Neuropathol. 1984;65:25–34. doi: 10.1007/BF00689824. [DOI] [PubMed] [Google Scholar]
- 34.Peden CS, Burger C, Muzyczka N, Mandel RJ. Circulating anti-wild-type adeno-associated virus type 2 (AAV2) antibodies inhibit recombinant AAV2 (rAAV2)-mediated, but not rAAV5-mediated, gene transfer in the brain. J Virol. 2004;78:6344–6359. doi: 10.1128/JVI.78.12.6344-6359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanftner LM, Suzuki BM, Doroudchi MM, Feng L, McClelland A, Forsayeth JR, et al. Striatal delivery of rAAV-hAADC to rats with preexisting immunity to AAV. Mol Ther. 2004;9:403–409. doi: 10.1016/j.ymthe.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 36.Grieger JC, Choi VW, Samulski RJ. Production and characterization of adeno-associated viral vectors. Nat Protoc. 2006;1:1412–1428. doi: 10.1038/nprot.2006.207. [DOI] [PubMed] [Google Scholar]
- 37.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson JS, Li C, DiPrimio N, Weinberg MS, McCown TJ, Samulski RJ. Mutagenesis of adeno-associated virus type 2 capsid protein VP1 uncovers new roles for basic amino acids in trafficking and cell-specific transduction. J Virol. 2010;84:8888–8902. doi: 10.1128/JVI.00687-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrari FK, Samulski T, Shenk T, Samulski RJ. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J Virol. 1996;70:3227–3234. doi: 10.1128/jvi.70.5.3227-3234.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang MM, Hearing P. Adenovirus early region 4 encodes two gene products with redundant effects in lytic infection. J Virol. 1989;63:2605–2615. doi: 10.1128/jvi.63.6.2605-2615.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 42.Moskalenko M, Chen L, van Roey M, Donahue BA, Snyder RO, McArthur JG, et al. Epitope mapping of human anti-adeno-associated virus type 2 neutralizing antibodies: implications for gene therapy and virus structure. J Virol. 2000;74:1761–1766. doi: 10.1128/jvi.74.4.1761-1766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paxinos GWC. The Rat Brain in Stereotaxic Coordinates. 4. Academic Press; San Diego: 1998. [Google Scholar]
- 44.Zhao X, Ahram A, Berman RF, Muizelaar JP, Lyeth BG. Early loss of astrocytes after experimental traumatic brain injury. Glia. 2003;44:140–152. doi: 10.1002/glia.10283. [DOI] [PubMed] [Google Scholar]
- 45.West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 46.Sterio DC. The unbiased estimation of number and sizes of arbitrary particles using the disector. J Microsc. 1984;134 (part 2):127–136. doi: 10.1111/j.1365-2818.1984.tb02501.x. [DOI] [PubMed] [Google Scholar]