

Abstract
Objective
Sjögren’s syndrome is a chronic autoimmune disorder characterized by progressive lymphocytic infiltration within the salivary and lacrimal glands. The current study was undertaken to investigate the effects of innate immunity activation on sialoadenitis in a mouse strain genetically susceptible for development of Sjögren’s syndrome-like disease.
Methods
Female New Zealand Black × New Zealand White F1 mice were repeatedly treated with toll-like 3 receptor agonist poly(I:C). Submandibular glands were investigated at different time points for sialoadenitis by immunohistochemistry and for gene expression of different chemokines by quantitative PCR. Submandibular gland infiltrating cells were characterized by flow cytometry.
Results
Poly(I:C) treatment significantly upregulated the expression of multiple chemokines within the submandibular glands. The severity and incidence of sialoadenitis was considerably higher in poly(I:C) treated mice. There was a preponderance of dendritic cells and NK cells in the initial inflammatory cell infiltrates and these were followed by CD4+ T cells.
Conclusions
Our data clearly demonstrates that systemic activation of innate immunity accelerates sialoadenitis in a mouse model for Sjögren’s syndrome-like disease. These findings suggest that chronic activation of innate immunity can influence certain features of Sjögren’s syndrome.
Keywords: Sjögren’s Syndrome, Innate Immunity, Sialoadenitis, mouse
Introduction
Sjögren’s syndrome (SS) is a chronic autoimmune disorder affecting exocrine glands. A progressive lymphocytic infiltration within salivary and lacrimal glands is a characteristic feature of the disorder (Gottenberg, 2009). While primary SS is generally restricted to the involvement of exocrine glands, patients with secondary SS have additional autoimmune disorders such as arthritis and lupus. Clinically, the major feature of SS is the presence of dry mouth and dry eye symptoms (Mariette and Gottenberg, 2010). Destruction of glandular architecture by inflammatory cell infiltrates is considered an important mechanism responsible for reducing salivary and lacrimal gland secretions. Action of inflammatory cytokines on acinar cells and deposition of anti-muscarinic receptor 3 autoantibodies within the tissue have also been proposed to contribute towards glandular dysfunction (Li et al., 2004; Dawson et al., 2006; Roescher et al., 2009). Clearly, a localized immune response within the salivary and lacrimal glands is critical for the pathogenesis of SS. A pivotal event in this pathway is the initiation of inflammatory cell infiltration within the exocrine glands.
The triggers responsible for initiating sialoadenitis and dacryoadenitis in SS patients are not known. Like several other autoimmune disorders, SS has a very strong genetic and gender bias, and interaction with environmental factors is considered critical for the development of disease (Voulgarelis and Tzioufas, 2010). Amongst the pro-inflammatory environmental factors, microbial infections, particularly those by viruses have been hypothesized to constitute an important trigger for SS. However, lack of strong evidence linking a specific viral infection with SS suggests that activation of innate immunity by diverse viruses might be the pathway triggering lymphocytic infiltration within the exocrine glands. In this study, innate immunity in New Zealand Black × New Zealand White F1 (NZB/W F1) mice, a strain susceptible for SS-like disease development, was activated by repeated treatment with polyinosinic:polycytidylic acid (poly(I:C)) and effects on salivary gland disease investigated. Poly(I:C) is synthetic double stranded RNA that activates innate immunity through the engagement of toll-like receptor 3 (TLR3), Retinoic Acid Inducible Gene 1 (RIG-1) and Melanoma Differentiation Antigen 5 (MDA5) (Alexopoulou et al., 2001; Yoneyama et al., 2004; Kato et al., 2006). Major consequences of poly(I:C) treatment are production of type I interferons, particularly IFN-β and pro-inflammatory cytokines such as IL-6 and TNF-α (Kumar et al., 2009).
Our data show that systemic treatment of NZB/W F1 mice with poly(I:C) accelerates the development of salivary gland disease. The findings from this study provide support to the thesis that in genetically susceptible individuals, excessive activation of innate immunity, such as that through repeated or chronic infections can be a predisposing factor for the development of SS.
Materials and Methods
Mice
All mouse experiments were approved by the Institutional Animal Care and Use Committee and were in accordance to National Institutes of Health guidelines. Female NZB/W F1 mice (6–8 week old) were obtained from Jackson laboratory (Bar Harbor, ME, USA). Mice were injected each time with 50μg of poly(I:C) (Invivogen Corp, San Diego, CA, USA) by intraperitoneal route on days 0, 2, 4, 6, 8, 10, and 12. Mice kept for long-term study (>3 weeks) were also injected on day 14. Control mice were injected similarly with PBS. Pilocarpine (Sigma-Aldrich Corp, St. Louis, MO, USA) induced saliva was measured as described previously (Deshmukh et al., 2009). Mice were euthanized at different time points and submandibular glands processed for histopathology, immunostaining, RNA isolation and flow cytometry.
Histopathology and Immunohistochemistry
Submandibular glands were evaluated for histopathology as described earlier with few modifications (Ohyama et al., 2006). Briefly, formalin fixed submandibular gland sections were stained with hematoxylin and eosin. The slides were scored by an individual (HB) blinded to experimental details. An aggregation of >50 inflammatory cells was considered a focus. The severity was graded on a scale of 0–5 as follows, grade 0: no disease; grade 0.5 to 1: 1–5 foci; grade 1.5 to 2.5: >5 foci without parenchymal destruction; grade 3 to 3.5: moderate parenchymal destruction and grade 4 to 5: extensive parenchymal destruction.
Flow Cytometry
Fluorochrome conjugated antibodies to CD45, CD4, CD8, CD11c, CD11b, CD3, NK1.1, and B220 were obtained from eBioscience (San Diego, CA USA) and used for staining and flow cytometry. Supplementary file 1 provides information on the clones, fluorochromes and isotypes of the antibodies used. Concentrations of each antibody used for staining were determined using splenocytes and corresponding isotype controls. Submandibular glands were collected in cold DMEM medium, minced and treated with type I Collagenase (Worthington Biochemical Corp, Lakewood, NJ, USA) for 20 min at 37°C to obtain single cells suspensions. Cells were stained following standard protocols. Greater than 105 events were acquired per sample and data was analyzed using FlowJo software (Tree Star, Inc., Ashland, OR, USA).
Gene Expression Analysis
Real-time PCR was used to determine the gene expression levels in submandibular glands of poly(I:C) and PBS treated mice as described previously (Deshmukh et al., 2009). Briefly, rapidly frozen submandibular glands were homogenized and RNA extracted using RNAeasy kit (Qiagen Inc., Valencia, CA, USA), which was followed by cDNA synthesis employing Quantitect Rev. Transcription kit (Qiagen Inc., Valencia, CA, USA). As a primary screen, a cDNA sample from each group was used to determine the expression levels of multiple chemokines using quantitative PCR array (SABiosciences, Frederick, MD, USA). Expression levels of chemokines showing highest fold change (top 8) were determined by employing Taqman gene expression assays (Applied Biosystems, Foster City, CA, USA). The fold change in gene expression was calculated using the 2[−Delta Delta C(T)] method (Livak and Schmittgen 2001). Submandibular gland cDNA from a normal B6 mouse was used as reference calibrator and glyceraldehyde phosphate dehydrogenase (GAPDH) was used as house keeping gene to normalize expression.
Statistical Analysis
Unpaired t test was used to determine statistical significance. A p<0.05 at 95% confidence interval was considered significant. For samples not passing normality test, statistical significance was determined using the Mann-Whitney test. Correlation analysis was done using Pearson test. All analyses were done using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA) and SigmaStat software (Systat Software, Inc., San Jose, CA, USA).
Results
Increased chemokine production in submandibular glands of poly(I:C) treated mice
Our previous study demonstrated that systemic treatment of NZB/W F1 mice with poly(I:C) caused a significant increase in type I IFN gene expression in the submandibular glands (Deshmukh et al., 2009). Thus, it was hypothesized that this treatment will also cause upregulated chemokine expression and inflammatory cell infiltration within the submandibular glands. Expression of different chemokine genes was analyzed by real time PCR at 2 weeks time point. A primary screen was performed by employing a PCR array (SABiosciences, Frederick, MD, USA) comprising of all chemokine genes. The top 8 chemokines showing differential expression were analyzed by real time PCR employing Taqman gene expression assays. Figure 1 shows that in submandibular glands of poly(I:C) treated mice, a significant increase occurs in the expression of Ccl7 (17.52 fold), Cxcl13 (9.71 fold), Ccl2 (9.55 fold), Ccl12 (6.09 fold), Ccl4 (4.49 fold), Ccl3 (4.13 fold), Cxcl10 (4.07 fold), and Ccl11 (2.92 fold).
Figure 1. Expression levels of multiple chemokines were upregulated in the submandibular glands of poly(I:C) treated mice.

Submandibular glands (5 mice per group) were obtained at 2 week time point and expression levels of different chemokines were determined by real time PCR using Taqman gene expression assays. Data are represented as mean ± SEM of relative gene expression. The ratio of mean gene expression level in poly(I:C) treated mice to mean gene expression level in PBS treated mice is shown as fold (X) increase in the top right corner of each graph. Amongst the chemokines analyzed, Ccl7 showed the highest fold change (17.52X) in expression and Ccl11 showed the lowest fold change (2.92X).
Sialoadenitis is accelerated in poly(I:C) treated mice
To investigate the effects of innate immunity activation on formation of inflammatory cell foci within the submandibular glands, poly(I:C) and PBS treated mice were euthanized at different time points. Hematoxylin and eosin stained submandibular gland sections were examined for the evidence of sialoadenitis and severity scored on a scale of 0 to 5. Submandibular gland inflammation was significantly accelerated in mice treated with poly(I:C) compared to PBS treated controls (figure 2a). Data from individual mice is shown in figure 2b. As early as 3 weeks after initiation of treatment (or at 11 weeks of age), 80% of poly(I:C) treated mice showed evidence of mild sialoadenitis. The severity of inflammation progressively increased with age showing an incidence of 80–100%. In contrast, in the PBS treated group, only one out of five mice showed evidence for mild sialoadenitis at 4 and 9 weeks after treatment. At 20 weeks after treatment, the incidence and severity of sialoadenitis in PBS treated mice was still significantly lower than poly(I:C) treated mice. The spontaneous sialoadenitis in control mice increased in severity at 26 weeks after treatment (or 34 weeks of age) (figure 2b). Representative pictures depicting different severity of sialoadenitis are shown in figure 2c (score = 0), 2d (score = 0.5), 2e (score = 2) and 2f (score = 4.5). These data clearly demonstrate that poly(I:C) treatment accelerates the development of sialoadenitis in NZB/W F1 mice.
Figure 2. NZB/W F1 mice treated with poly(I:C) develop severe and accelerated sialoadenitis.

Poly(I:C) and PBS treated NZB/W F1 mice were euthanized, at different time points after initial treatment and submandibular glands were analyzed for sialoadenitis. Severity of sialoadenitis was scored on a scale of 0 to 5 by an observer blinded to experimental details. The right top panel (a) shows mean disease score ± SEM and the right bottom panel (b) shows data from individual mice. In the top panel, mice euthanized between 20–23 weeks are plotted at 22 weeks on X-axis. Non parametric t test was used to determine statistical significance between poly(I:C) and PBS treated groups at indicated time points and a p<0.05 was considered significant. Representative pictures of H&E staining are shown in the right panel. c: PBS treated mice at 2 weeks after treatment showing normal salivary gland (score = 0). d: A single small, periductal/perivascular focus seen at 3 weeks after treatment (score = 0.5). e: A moderate sized focus of inflammation with or without multiple foci, showing destruction of glandular structures at 9 weeks (score = 2). f: A portion of one of the large foci of inflammation replacing large portions of glandular tissue with lymphocytes seen at 20–23 weeks after treatment (score = 4.5). Scale bar = 20microns. Inset: – shows the entire focus at a lower magnification (Scale bar = 200 microns).
Dendritic cells and NK cells dominate the early inflammatory cell infiltrates
Our previous study demonstrated that 1 week after poly(I:C) treatment, lymphocytic foci were not detected within the submandibular glands by immunohistochemistry (Deshmukh et al., 2009). The data presented in this investigation shows that by 3 weeks after treatment, mild sialoadenitis (mean focus score ± SEM of 0.5 ± 0.15) was evident in poly(I:C) treated mice. Thus, to characterize early inflammatory cells, flow cytometry was used. The details on antibodies used for staining are provided in supplementary file 1. Submandibular glands were harvested and single cell suspensions stained with antibody to CD45 and for T cells (CD4, CD8), B cells (B220), macrophages (CD11b), dendritic cells (CD11c), NK cells (NK1.1+ and CD3 negative), and NKT cells (NK1.1 and CD3). At 2 week time point, a significantly increased frequency of CD11chi dendritic cells and NK cells was observed in the submandibular glands of poly(I:C) treated mice (figure 3). At this time point the frequency of CD4+T cell in the submandibular glands of poly(I:C) and PBS treated mice were comparable. However, by 3 weeks, CD4+ T cells were significantly increased in poly(I:C) treated mice compared with the PBS treated group (p=0.0033), as well as when compared with group of poly(I:C) treated mice at 2 week time point (p=0.011) (Figure 3d). The setting of gates for analysis of cell populations is shown in supplementary file 2. At both time points, differences in frequencies of B cells and macrophages between poly(I:C) and PBS treated groups of mice were statistically not significant (data not shown). However, at 20 week time point, lymphocytic foci within the submandibular glands from poly(I:C) treated mice show considerable B cell infiltration (Supplementary File 3). Characterization of different subsets of B cells infiltrating the salivary glands, their kinetics and association with localized chemokine production will be pursued in future studies. This will provide valuable information on how activation of innate immunity and early inflammatory cells influence B cell infiltration within the salivary glands.
Figure 3. Kinetics of early inflammatory cell infiltration within submandibular glands of poly(I:C) and PBS treated mice.

Submandibular glands from poly(I:C) and PBS treated mice were obtained at 2 week (a–c) and 3 week (d) after initial treatment. Single cell suspensions were prepared and infiltrating cells were characterized by flow cytometry. The gates set up for analysis are shown in supplementary file 2. At 2 week time point, in comparison with the PBS group, there were significantly more dendritic (a) and NK cells (b) in the submandibular glands of poly(I:C) group. At 3 week time point, frequency of CD4+ T cells was significantly higher in the poly(I:C) treated group (d). Also, frequency of CD4+ T cells in poly(I:C) mice was higher at 3 weeks compared to poly(I:C) mice at 2 weeks (p=0.011) after treatment. Frequency of CD4+ T cells in PBS treated groups at 2 week and 3 week time points were not significantly different (p=0.21).
Glandular dysfunction in poly(I:C) treated mice
Our previous studies have demonstrated that poly(I:C) induces an immediate loss (within 1 week after treatment) in glandular function (Deshmukh et al., 2009). However, the salivary gland function recovers within 2–3 weeks after poly(I:C) treatment is stopped. This temporary loss of function was independent of lymphocytic infiltration and autoantibody production, demonstrating that it was through the transient effects of interferons and proinflammatory cytokines induced by poly(I:C). To determine whether accelerated sialoadenitis affected glandular function, pilocarpine induced saliva was measured at different time points. Figure 4 shows data from 2 different cohorts of mice at 12 week and 20 week time points. At 12 weeks, the differences in mean saliva volume between PBS and poly(I:C) treated groups were not significantly different. However, by 20 weeks after treatment, several mice in the poly(I:C) treated group showed evidence for glandular hypofunction and this group had significantly lower mean saliva volume than the PBS treated group (p=0.0101). This trend and statistical significance stayed valid even when the 2 outlier mice with greatest loss in function were not included in the analysis (p=0.0431). These data suggest that a moderate loss in glandular function occurs despite severe lymphocytic infiltration. Moreover, no correlation was evident between the severity of sialoadenitis and the extent of loss of function.
Figure 4. Despite severe inflammation, only moderate loss of salivary gland function occurs in poly(I:C) treated mice.
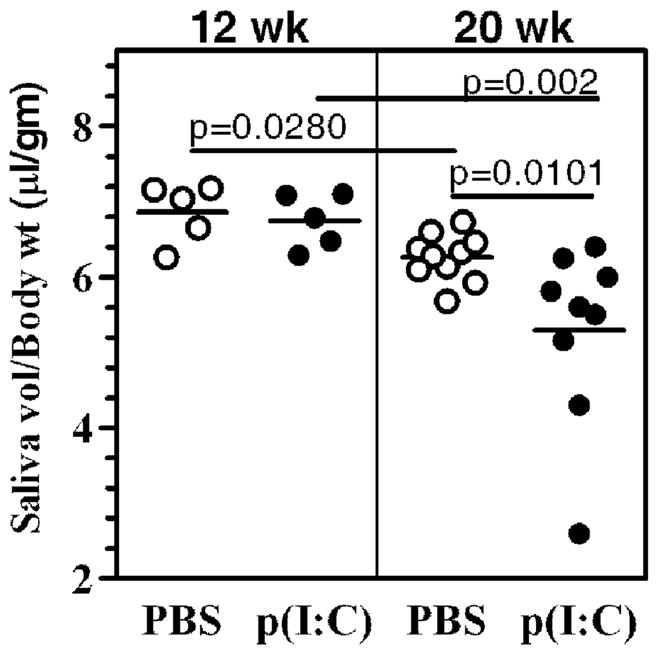
Pilocarpine induced saliva volumes were measured in 2 different cohorts of poly(I:C) (●) or PBS (○) treated mice, at 12 and 20 weeks after treatment. The data are represented as ratio of saliva volume (μl) to body weight (gm). At 12 weeks, the mean saliva volumes between poly(I:C) and PBS treated groups were not significantly different. At 20 weeks the mean saliva volume in poly(I:C) treated group was significantly lower (p=0.0101) than that in PBS treated group. This trend and statistical significance (p=0.04) stayed valid even when the 2 mice with the greatest drop in function were not included in the analysis.
Discussion
In SS, events initiating inflammatory cell infiltration within the salivary glands are not known. Although a role for viral or bacterial infections is suspected, it is difficult to prove this supposition as considerable time may have lapsed between the initiation of the disease and appearance of clinical symptoms. Also, no unique infectious agent has been identified to be convincingly associated with SS. Thus, we hypothesized that excessive activation of innate immunity by chronic or repeated exposures to microbial stimuli, facilitates lymphocytic infiltration within the salivary glands. In this study, we show that repeated treatment of young NZB/W F1 mice with TLR3 agonist poly(I:C), accelerates the development of sialoadenitis. The NZB/W F1 mouse strain has genetic susceptibility to develop autoimmunity and was amongst the first mouse model described for SS (Kessler 1968). Previous studies have demonstrated that female mice show presence of lymphocytic foci mainly within their submandibular glands, after 20–22 weeks of age (Kessler 1968, Jonsson et al., 1987). This observation was reproduced in our cohort mice (figure 2). PBS treated mice developed sialoadenitis between 20–28 weeks of age. However, the striking finding was that in poly(I:C) treated mice, lymphocytic foci were detected around 11 weeks of age (3 weeks after first treatment) and they increased in severity with time.
Systemic treatment of mice with poly(I:C) is a well established model to mimic activation of innate immunity by viral infections. It has been used to modulate autoimmune responses in mouse models of lupus (Steinberg et al., 1969, Patole et al., 2005, Jorgensen et al., 2006), pancreatitis (Nishio et al., 2011), type I diabetes (Devendra and Eisenbarth 2004, Moriyama et al., 2002, Sobel et al., 1992), arthritis (Yarilina et al., 2007) and experimental autoimmune encephalomyelitis (Touil et al., 2006). These studies demonstrate that systemic treatment with poly(I:C) causes a generalized inflammation in the whole mouse. Our previous study had shown that this also causes activation of innate immune responses within the submandibular glands and rapidly induces a transient salivary gland hypofunction in mice (Deshmukh et al., 2009). The glandular hypofunction was dependent on innate immune responses as withdrawal of poly(I:C) treatment completely restored salivary gland function. In the present study, effects of poly(I:C) treatment on chemokine production and inflammatory cell infiltration within the submandibular glands were investigated. Poly(I:C) treatment caused significant upregulation in the expression of different chemokine genes that influence the inflammatory cell infiltration within the submandibular glands. Expression level of Ccl7 was the highest. CCL7 binds chemokine receptors CCR1, CCR2, and CCR3 (Kim, 2004). These receptors are known to be expressed on activated NK cells and immature dendritic cells (Kim, 2004). In addition, CCR2 also binds CCL2 and CCL12, and expression levels of these chemokines were also upregulated in poly(I:C) treated mice. Similarly, gene expression levels of Ccl3 (binds CCR1 and CCR5), and Ccl11 (binds CCR3) were also upregulated. Thus, it is possible that cumulative action of these chemokines is responsible for initiating inflammatory cell infiltration within the submandibular glands. The earliest infiltrates were predominated by dendritic cells and NK cells in the submandibular glands of poly(I:C) treated mice. At later time point lymphocytic foci within the submandibular glands showed significant presence of B cells (Supplementary file 3). Although precise kinetics of B cell infiltration in this model is not known, the data suggests that activated NK cells and dendritic cells infiltrate the submandibular glands and influence the adaptive immune response that occurs at later stage. Both of these cell types can contribute towards chemokine production and thereby influence the infiltration by other cell types. In addition, activated dendritic cells can present antigens and activate CD4+ T cells, which then can interact with salivary gland infiltrating B cells resulting in a localized adaptive immune response. Similar to our findings, in the NOD mouse model for SS, it has been demonstrated that dendritic cells predominate the early stage of inflammatory cell infiltrates within the submandibular glands (van Blokland et al., 2000). However, this was not the case in MRL lpr/lpr mouse model, suggesting that each mouse model for SS might be representative of the variations seen in patient populations (van Blokland et al., 2000).
Modulation of chemokine production in the submandibular glands following stimulation with TLR agonist and its consequences for T cell infiltration have been demonstrated in Scurfy mice (Sharma et al., 2009). Scurfy mice, deficient in the transcription factor Foxp3, are defective in regulatory T cells and develop multi organ inflammation. Surprisingly, inflammation was not observed in the submandibular glands and only occurred following oral treatment with LPS (Sharma et al., 2006). In this model it was demonstrated that following oral LPS treatment, significant changes occurred in the expression of multiple chemokines within the submandibular glands. Since concurrent changes in the expression of respective chemokine receptors were not seen in the CD4+ T cells, chemokine expression in submandibular glands was considered a major step for initiating lymphocytic infiltration. Our studies employing poly(I:C) suggest the possibility that following viral infections, upregulated chemokine production within the salivary glands can be responsible for initiating inflammatory cell infiltration. In genetically susceptible hosts, over a period of time, this can lead to the development of SS.
Glandular hypofunction leading to reduced saliva production is a major reason for dry mouth in SS. At 20 week time point, the mean saliva volume in the poly(I:C) treated mice was significantly lower than that in the PBS treated mice (figure 4). However, it should be noted that despite severe lymphocytic infiltration in poly(I:C) treated mice at this time point (figure 2), the loss of salivary gland function was modest (figure 4). Moreover, we did not see any correlation between the disease severity score and the amount of stimulated saliva produced. This dissociation between severity of inflammation and loss of function is very similar to that observed in several SS patients (Nikolov et al., 2009). Indeed, we have previously demonstrated that in NZB/W F1 mice, in absence of any inflammatory foci, activation of innate immunity by poly(I:C) is sufficient to cause a rapid but transient salivary gland hypofunction (Deshmukh et al., 2009). We have also observed this dissociation in NZB/W F1 mice treated with Freund’s incomplete adjuvant (IFA) (Deshmukh et al., 2008). By 7–8 weeks after treatment despite very mild sialoadenitis, IFA treated mice developed significant glandular hypofunction in comparison with PBS treated mice. Considered together, these data further emphasize that mechanisms other than acinar cell destruction are involved in inducing glandular dysfunction in SS. Clearly, the NZB/W F1 mouse model will prove to be a valuable tool for investigating these mechanisms.
Recently it was shown that repeated treatment of mice with heat killed E. coli induced autoimmune pancreatitis (Haruta et al., 2010). In these mice at 6–12 months after treatment, inflammatory cell infiltrates were also observed in the salivary glands. Also, as discussed above, scurfy mice fed LPS developed sialoadenitis (Sharma et al., 2006). While these 2 investigations highlight innate immunity activated through bacterial products, our model mimics innate immunity activated by viral infection. Collectively, these studies suggest that in genetically susceptible individuals, chronic activation of innate immunity by microbial stimuli can contribute towards the development and exacerbation of SS. Thus, clinically, dampening excessive activation of innate immunity as well as aggressive treatment of chronic infections should be undertaken to avoid possible development of autoimmunity at a later time.
Supplementary Material
A standard live cell gate was set based on forward and side scatter and subsets are represented as a frequency within the live gate. Contour plots show representative staining profiles of submandibular gland cells from each group. Gates set up for dendritic cells (CD11c high) and NK cells (NK1.1+ CD3 negative) at 2 wks and CD4+ T cells and CD19+ B cells at 3 wks are shown. Spleen cells were used as a positive control for all analyses and are shown in the right panel.
{kind=link}
Submandibular glands were obtained at 20 weeks after initial treatment, fixed in 4% paraformaldehyde and 5μm sections were stained for B cells (anti-B220, green) and T cells (anti-CD4, red). The nuclei (blue) were stained with DAPI. Panel A shows representative staining from a PBS treated mouse with very low sialoadenitis score. Panel B shows a lymphocytic focus in a poly(I:C) treated mouse with a cluster of B cells that is surrounded by CD4+ T cells.
{kind=link}
Acknowledgments
This study was supported by grants from the National Institutes of Health, USA, R21DE019883 (USD), R01AI079621 (USD), R01DK069769 (HB) and an Innovative Research Grant from the Sjögren’s Syndrome Foundation USA (USD).
References
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Dawson LJ, Stanbury J, Venn N, Hasdimir B, Rogers SN, Smith PM. Antimuscarinic antibodies in primary sjogren’s syndrome reversibly inhibit the mechanism of fluid secretion by human submandibular salivary acinar cells. Arthritis Rheum. 2006;54:1165–1173. doi: 10.1002/art.21764. [DOI] [PubMed] [Google Scholar]
- Deshmukh US, Nandula SR, Thimmalapura PR, Scindia YM, Bagavant H. Activation of innate immune responses through toll-like receptor 3 causes a rapid loss of salivary gland function. J Oral Pathol Med. 2009;38:42–47. doi: 10.1111/j.1600-0714.2008.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh US, Ohyama Y, Bagavant H, Guo X, Gaskin F, Fu SM. Inflammatory stimuli accelerate sjogren’s syndrome-like disease in (NZB × NZW)F1 mice. Arthritis Rheum. 2008;58:1318–1323. doi: 10.1002/art.23368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devendra D, Eisenbarth GS. Interferon alpha--a potential link in the pathogenesis of viral-induced type 1 diabetes and autoimmunity. Clin Immunol. 2004;111:225–233. doi: 10.1016/j.clim.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Gottenberg JE. Primary sjogren’s syndrome: Pathophysiological, clinical and therapeutic advances. Joint Bone Spine. 2009;76:591–594. doi: 10.1016/j.jbspin.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Haruta I, Yanagisawa N, Kawamura S, Furukawa T, Shimizu K, Kato H, Kobayashi M, Shiratori K, Yagi J. A mouse model of autoimmune pancreatitis with salivary gland involvement triggered by innate immunity via persistent exposure to avirulent bacteria. Lab Invest. 2010;90:1757–1769. doi: 10.1038/labinvest.2010.153. [DOI] [PubMed] [Google Scholar]
- Jonsson R, Tarkowski A, Backman K, Klareskog L. Immunohistochemical characterization of sialadenitis in NZB X NZW F1 mice. Clin Immunol Immunopathol. 1987;42:93–101. doi: 10.1016/0090-1229(87)90176-0. [DOI] [PubMed] [Google Scholar]
- Jorgensen TN, Thurman J, Izui S, Falta MT, Metzger TE, Flannery SA, Kappler J, Marrack P, Kotzin BL. Genetic susceptibility to polyI:C-induced IFNalpha/beta-dependent accelerated disease in lupus-prone mice. Genes Immun. 2006;7:555–567. doi: 10.1038/sj.gene.6364329. [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Kessler HS. A laboratory model for sjogren’s syndrome. Am J Pathol. 1968;52:671–685. [PMC free article] [PubMed] [Google Scholar]
- Kim CH. Chemokine-chemokine receptor network in immune cell trafficking. Curr Drug Targets Immune Endocr Metabol Disord. 2004;4:343–361. doi: 10.2174/1568008043339712. [DOI] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- Li J, Ha YM, Ku NY, Choi SY, Lee SJ, Oh SB, Kim JS, Lee JH, Lee EB, Song YW, Park K. Inhibitory effects of autoantibodies on the muscarinic receptors in sjogren’s syndrome. Lab Invest. 2004;84:1430–1438. doi: 10.1038/labinvest.3700173. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mariette X, Gottenberg JE. Pathogenesis of sjogren’s syndrome and therapeutic consequences. Curr Opin Rheumatol. 2010;22:471–477. doi: 10.1097/BOR.0b013e32833c36c5. [DOI] [PubMed] [Google Scholar]
- Moriyama H, Wen L, Abiru N, Liu E, Yu L, Miao D, Gianani R, Wong FS, Eisenbarth GS. Induction and acceleration of insulitis/diabetes in mice with a viral mimic (polyinosinic-polycytidylic acid) and an insulin self-peptide. Proc Natl Acad Sci USA. 2002;99:5539–5544. doi: 10.1073/pnas.082120099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolov NP, Alevizos I, Grisius M, Atkinson J, Cotrim A, Baum BJ, Brahim J, Kleiner D, Bebris L, Illei GG. Arthritis Rheum. Vol. 60. Nikolov NP; 2009. Uncoupling Salivary Gland Dysfunction From Inflammation in a Large Cohort of Primary Sjgrens Syndrome Patients; p. 480. [DOI] [Google Scholar]
- Nishio A, Asada M, Uchida K, Fukui T, Chiba T, Okazaki K. The role of innate immunity in the pathogenesis of experimental autoimmune pancreatitis in mice. Pancreas. 2011;40:95–102. doi: 10.1097/MPA.0b013e3181f3a5d4. [DOI] [PubMed] [Google Scholar]
- Ohyama Y, Carroll VA, Deshmukh U, Gaskin F, Brown MG, Fu SM. Severe focal sialadenitis and dacryoadenitis in NZM2328 mice induced by MCMV: A novel model for human sjogren’s syndrome. J Immunol. 2006;177:7391–7397. doi: 10.4049/jimmunol.177.10.7391. [DOI] [PubMed] [Google Scholar]
- Patole PS, Grone HJ, Segerer S, Ciubar R, Belemezova E, Henger A, Kretzler M, Schlondorff D, Anders HJ. Viral double-stranded RNA aggravates lupus nephritis through toll-like receptor 3 on glomerular mesangial cells and antigen-presenting cells. J Am Soc Nephrol. 2005;16:1326–1338. doi: 10.1681/ASN.2004100820. [DOI] [PubMed] [Google Scholar]
- Roescher N, Tak PP, Illei GG. Cytokines in sjogren’s syndrome. Oral Dis. 2009;15:519–526. doi: 10.1111/j.1601-0825.2009.01582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Deshmukh US, Zheng L, Fu SM, Ju ST. X-linked Foxp3 (scurfy) mutation dominantly inhibits submandibular gland development and inflammation respectively through adaptive and innate immune mechanisms. J Immunol. 2009;183:3212–3218. doi: 10.4049/jimmunol.0804355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Zheng L, Guo X, Fu SM, Ju ST, Jarjour WN. Novel animal models for sjogren’s syndrome: Expression and transfer of salivary gland dysfunction from regulatory T cell-deficient mice. J Autoimmun. 2006;27:289–296. doi: 10.1016/j.jaut.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel DO, Newsome J, Ewel CH, Bellanti JA, Abbassi V, Creswell K, Blair O. Poly I:C induces development of diabetes mellitus in BB rat. Diabetes. 1992;41:515–520. doi: 10.2337/diab.41.4.515. [DOI] [PubMed] [Google Scholar]
- Steinberg AD, Baron S, Talal N. The pathogenesis of autoimmunity in new zealand mice, I. induction of antinucleic acid antibodies by polyinosinic-polycytidylic acid. Proc Natl Acad Sci USA. 1969;63:1102–1107. doi: 10.1073/pnas.63.4.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touil T, Fitzgerald D, Zhang GX, Rostami A, Gran B. Cutting edge: TLR3 stimulation suppresses experimental autoimmune encephalomyelitis by inducing endogenous IFN-beta. J Immunol. 2006;177:7505–7509. doi: 10.4049/jimmunol.177.11.7505. [DOI] [PubMed] [Google Scholar]
- van Blokland SC, van Helden-Meeuwsen CG, Wierenga-Wolf AF, Drexhage HA, Hooijkaas H, van de Merwe JP, Versnel MA. Two different types of sialoadenitis in the NOD- and MRL/lpr mouse models for sjogren’s syndrome: A differential role for dendritic cells in the initiation of sialoadenitis? Lab Invest. 2000;80:575–585. doi: 10.1038/labinvest.3780062. [DOI] [PubMed] [Google Scholar]
- Voulgarelis M, Tzioufas AG. Pathogenetic mechanisms in the initiation and perpetuation of sjogren’s syndrome. Nat Rev Rheumatol. 2010;6:529–537. doi: 10.1038/nrrheum.2010.118. [DOI] [PubMed] [Google Scholar]
- Yarilina A, DiCarlo E, Ivashkiv LB. Suppression of the effector phase of inflammatory arthritis by double-stranded RNA is mediated by type I IFNs. J Immunol. 2007;178:2204–2211. doi: 10.4049/jimmunol.178.4.2204. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A standard live cell gate was set based on forward and side scatter and subsets are represented as a frequency within the live gate. Contour plots show representative staining profiles of submandibular gland cells from each group. Gates set up for dendritic cells (CD11c high) and NK cells (NK1.1+ CD3 negative) at 2 wks and CD4+ T cells and CD19+ B cells at 3 wks are shown. Spleen cells were used as a positive control for all analyses and are shown in the right panel.
Submandibular glands were obtained at 20 weeks after initial treatment, fixed in 4% paraformaldehyde and 5μm sections were stained for B cells (anti-B220, green) and T cells (anti-CD4, red). The nuclei (blue) were stained with DAPI. Panel A shows representative staining from a PBS treated mouse with very low sialoadenitis score. Panel B shows a lymphocytic focus in a poly(I:C) treated mouse with a cluster of B cells that is surrounded by CD4+ T cells.