

Abstract
The force balance between the extracellular microenvironment and the intracellular cytoskeleton controls the cell fate. We report a new (to our knowledge) mechanism of receptor force control in cell adhesion originating from friction between cell adhesion ligands and the supporting substrate. Adherent human endothelial cells have been studied experimentally on polymer substrates noncovalently coated with fluorescent-labeled fibronectin (FN). The cellular traction force correlated with the mobility of FN during cell-driven FN fibrillogenesis. The experimental findings have been explained within a mechanistic two-dimensional model of the load transfer at focal adhesion sites. Myosin motor activity in conjunction with sliding of FN ligands noncovalently coupled to the surface of the polymer substrates is shown to result in a controlled traction force of adherent cells. We conclude that the friction of adhesion ligands on the supporting substrate is important for mechanotransduction and cell development of adherent cells in vitro and in vivo.
Introduction
Mechanical cues of the extracellular microenvironment trigger the fate of living cells (1,2). This microenvironment—the so-called extracellular matrix (ECM)—consists of different proteins and glycosaminoglycans, which act as ligands and control cell adhesion and tissue integrity. The characteristics of the ECM, i.e., matrix stiffness, ligand density, ligand conformation, ligand anchorage, and lateral ligand distribution influence intracellular signaling pathways. Force regulation by myosin motors in the actin cytoskeleton is one of them to be directly affected in this context. These forces are transmitted from the cytoskeleton to the load-bearing ECM via a complex arrangement of different proteins in the adhesion site, including the transmembrane receptors, e.g., integrins (3). The ligand-receptor interaction, the characteristics of the intracellular components of the cytoskeleton, and the cell adhesion sites were extensively studied experimentally and by theoretical modeling (4,5). These studies included mainly static aspects of the interaction mechanisms. However, some dynamic aspects were already discussed as influencing factors, which also affect cell adhesion and mechanotransduction of extracellular signals to the cell interior. These approaches discussed viscoelastic behavior of the cell membrane as well as bond formation and rupture between adhesion receptors and ligands. Furthermore, bond strengthening by integrin receptors under applied force is a prominent example for the dynamics of adhesion-receptor binding to their ligands (6).
Another contribution to load transfer between the extracellular space and cells is related to the presence of ECM proteins. They are noncovalently anchored to the supporting matrix or material. Many of them are reorganized by the cells during development and repair of tissue (7,8). With the noncovalently anchorage via specific and nonspecific binding sites to the supporting matrix or material, they exhibit a certain degree of mobility. The molecular control of ligand attachment to the ECM might be a mechanism for regulating cell adhesion in vivo and in vitro. As it will be shown herein, we think that the noncovalent anchorage of adhesion ligands and its downstream impact on cellular force development and cell fate should be considered in cell adhesion-related phenomena.
In the following, we will discuss specific features of the regulation of cell traction forces, which may arise from noncovalently anchoring of ligands to the underlying materials surface (or the supporting ECM). We propose a model, where traction forces are caused by a myosin-driven movement of the ligands on the substrate surface. This movement results in friction forces in the range of pN per ligand.
Materials and Methods
Substrate preparation and characterization
Two types of substrates were used in the experiments: A), the in situ analysis of fibronectin (FN) reorganization was performed on stiff glass substrates coated with a thin film of maleic acid copolymers (MACP), and B), traction force measurements were performed on hydrogel layers coated with a similar thin film of MACP.
For substrate A, films of poly(octadecene-alt-maleic anhydride) (POMA; MW 40.000) (Polysciences, Warrington, PA), poly(propene-alt-maleic anhydride) (PPMA; MW 39.000), poly(styrene-alt-maleic anhydride) (PSMA; MW 20.000) (both special products of Leuna-Werke AG, Germany), and poly(ethylene-alt-maleic anhydride) (PEMA; MW 125.000) (Sigma Aldrich, Deisenhofen, Germany) were prepared on glass coverslips as described previously (9,10). Briefly, polymer layers were produced by spin-coating (RC5, Suess Microtec, Garching, Germany) of 0.08% (POMA), 0.08% (PSMA), 0.1% (PPMA), and 0.15% (PEMA) copolymer solutions in tetrahydrofuran (POMA and PSMA) (Fluka, Deisenhofen, Germany), ethylmethylketone (Merck, Darmstadt, Germany), and acetone/tetrahydrofuran (w/w 1/2, Acros Organics, Geel, Belgium), respectively, on glass coverslips after their surface modification with 3-aminopropyltriethoxysilane (ABCR, Karlsruhe, Germany).
For substrate B, the samples were prepared as described recently (11). Briefly, the preparation of the polyacrylamide (PAAm) hydrogel layer was performed similar to the standard procedure (12) by coating first freshly oxidized coverslips with (3-acryloxypropyl)trimethoxysilane (ABCR), and subsequent deposition of PAAm from stock solutions of 80% acrylamide (PlusOne Acrylamide PAGE, Amersham Biosciences, Piscataway, NJ), 1% bis-acrylamide solutions (Amersham Biosciences). Fluorescent microbeads (Fluoresbrite YG Microspheres 0.5 μm, Polysciences) were embedded in the PAAm layer. Following washing and drying at room temperature under vacuum for 30 min, monomolecular films of MACP on top of PAAm hydrogels were prepared by spin coating PSMA, PPMA, and PEMA solutions, see above. The MACP-PAAm coverslips were equilibrated for 24 h in phosphate buffer saline at pH 7.4 (PBS) (Biochrom, Berlin, Germany) to ensure: i), complete hydrolysis of the anhydride groups of the maleic anhydride copolymers to carboxylic acid groups, ii), removal of nonbound maleic acid polymer molecules, and iii), full reswelling of the PAAm gels under physiological buffer conditions. The final gel films were 70–100 μm thick. On the basis of our previous studies (13), the thickness of the maleic acid topcoat can be assumed as a few tens of nanometers. Hence, the single swollen MACP chains are comparable in length to the mesh size of the PAAm hydrogel. Because of this small thickness in comparison to the penetration depth of the mechanical deformation within the PAAm layer of ∼10 μm, we assume that the effect of the topcoat on the traction force measurement is negligible.
The stiffness of the hydrogel was determined by scanning force spectroscopy (Bioscope BS2-Z, Veeco, Santa Barbara, CA) using a pyramid-tipped SiNi cantilever and a Hertz cone model (Microlevers, Park Scientific Instruments, ThermoMicroscopes, Sunnyvale, CA, spring constant: ∼0.01 N/m) (11).
FN (purified from adult human plasma) was coated on the substrates by adsorption from a 50 μg/ml solution in PBS for 1 h at 37°C, resulting in high surface concentrations in the range from 200 to 300 ng/cm2 (11). For cellular reorganization experiments, FN was fluorescent labeled using 5-(and-6)-carboxytetramethylrhodamine (TAMRA, Invitrogen, Carlsbad, CA).
Cell culture
Human endothelial cells from the umbilical cord vein (HUVEC) were seeded in endothelial cell growth medium ECGM (Promocell, Heidelberg, Germany) containing 2% fetal calf serum (2 × 104 cells/cm2) on the FN-coated substrates. The cells were analyzed 30 or 60 min after seeding.
Traction force microscopy
Traction force microscopy of adherent cells was performed 60 min after seeding as described recently (11). Briefly, using an inverted microscope (Axiovert 200M, Carl Zeiss MicroImaging, Jena, Germany) with moveable stage and incubator, 3 to 5 cells per sample were imaged with the bead position before and after cell removal using trypsin-EDTA (Sigma-Aldrich). Hydrogel layers used had a Young's modulus of 2–7 kPa. The image set (with and without cell) was analyzed by the unconstrained Fourier-transform traction cytometry method introduced by Butler et al. (14) with improvements published elsewhere (15,16). Calculations were done using MATLAB (The MathWorks, Natick, MA) and the freely available software ImageJ (http://rsb.info.nih.gov/ij/). From the traction fields, the maximum traction stress per cell Tmax was determined.
In situ FN reorganization analysis
Adherent cells on substrates A coated with fluorescent-labeled FN were imaged 30 min after seeding in time intervals of 10 min for 30 s using a confocal laser scanning microscope (SP1, Leica Microsystems, Bensheim, Germany) with 40× oil immersion objective and incubator. Fluorescence intensities of image sequences were analyzed in the following way. The intensity increase of a growing fibril over time I(t) was measured in a specific area AFib defined by the final size of the fibril using the software ImageJ and the Time Series Analyzer Plugin. Intensity fluctuations from focus shifts were accounted for by subtracting the intensity I∞(t) measured on another position far away from the growing fibril. To exclude any impact of the cell membrane on I∞, the measurement was compared with the intensity I∞,out outside the region of the adherent cell. As expected, no significant differences were notified. The background intensity of the microscope setup was found to be negligible.
For each polymer surface, 10–20 fibrils were analyzed from 2 to 8 cells per substrate in four independent experiments.
FN displacement analysis for POMA and PSMA
For traction force microscopy, the POMA surface was substituted by PSMA. POMA polymer chains do not dissolve/swell in an aqueous medium (13). This would lead to a stiff top layer on the soft hydrogels. Hence, it is incompatible with the traction force measurements. In contrast, PSMA swells as short free single chains at physiological conditions (13). The FN anchorage was determined by displacement studies as described recently (17). Briefly, the fluorescence intensity of preadsorbed FN is followed over 48 h by confocal laser scanning microscopy in PBS buffer containing 50 μg/ml bovine serum albumin. The kinetics of the displacement of FN by albumin characterizes the anchorage strength of FN to the substrate surfaces (see also Fig. S1 in the Supporting Material). A similar kinetics was observed for POMA and PSMA, supporting their interchangeability for the traction force studies.
Results and Discussion
Control of traction force by physicochemical characteristics of substrate surface
In previous works (9,11), we suggested that adhesive forces of adherent cells can be affected by the anchorage of FN ligands to the underlying support. In (11) we already quantified traction forces of HUVECs on some MACP surfaces. We now extended these measurements to three MACP surfaces with gradually different physiochemical surface characteristics, which were also used in previous studies on stiff substrates (9,17,18). Traction stresses caused by HUVECs were determined 60 min after seeding on FN-coated polymer surfaces as exemplarily, shown in Fig. 1 a. As depicted in Fig. 1 b, we indeed found a monotonic dependence of the maximum cell traction stress Tmax on the type of polymer surface. A higher stress was observed on surfaces with lower polarity and higher hydrophobicity (10). The highest stress was measured on PSMA. The observed traction force dependency correlated with the anchorage strength of FN to the underlying MACP surface (17). The mean values of Tmax were in the range from 300 to 700 Pa. A comparison with a traction stress of 2 kPa reported in (19) for endothelial cells on covalently attached ligands suggests an intracellular upregulation of adhesion-related signals due to the stronger coupling of the ligand to the substrate as discussed previously (11). In comparison to earlier studies (9), POMA surfaces had to be substituted by PSMA, because POMA does not swell in aqueous conditions (13). Therefore, it does not allow a soft top coating on the hydrogel layer; see also the Materials and Methods. Fortunately, the PSMA coating provides the same FN anchorage strength as POMA (see Fig. S1).
Figure 1.
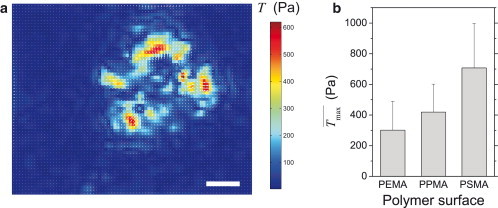
Traction stress of adherent HUVECs controlled by noncovalent anchorage of FN to polymer surfaces. (a) Traction stress map of an adherent cell on PEMA. Scale bar: 30 μm. (b) Mean maximum traction stress Tmax of HUVECs in dependence on polymer surface as determined from 3 to 6 independent experiments with each having 3–5 cells. Error bars indicate mean + SD.
Obviously, the change of FN anchorage in dependence on the physicochemical properties of the supporting polymer surface enabled a control of the traction stress. This finding is in line with our hypothesis that the noncovalent ligand anchorage to the supporting surface regulates cell adhesion forces (9). Note that the ligand density is no relevant parameter for control of traction stress in the present experiment (20) because the used FN ligand densities on the substrate surfaces were very high (in the saturation range of 200–300 ng/cm2).
A model of ligand friction on substrate surfaces
To explain our experimental findings, we propose a model where the interaction zone near a focal adhesion site (Fig. 2 a) is strongly simplified as schematically drawn in Fig. 2 b. The focal adhesions act therein as the primary sites for the occurrence of traction forces. Focal adhesions are known as clusters of integrin receptors (Fig. 2 a, right inset), which are bound extracellularly to the adhesion ligands (i.e., to FN). Intracellularly, these integrin clusters are bound to each other. They are further linked to the actin stress fibers. Many different proteins are known to be involved in this linking process, among them talin, vinculin, and α-actinin (3). In our model (Fig. 2 b), we ignore the details of this complex structure and assume a rigid link of the integrins to the force-generating actin stress fibers with their myosin motors. This is of course a strong simplification. However, it is supported by experimental data of disruption forces of the intracellular adhesion complex in the range of 100 pN (4). These values are much higher than a few pN as considered in our model for the adhesion ligands. Accordingly, the actin stress fiber, the integrins, and the other linking proteins are combined to a rigid intracellular force transmission, which is connected to the adhesion ligand via receptor-ligand interaction. The whole unit is driven by the myosin motor system with force acting against a sliding friction of the FN ligand. Thereby it moves along the surface of the supporting polymer substrate (cf. Fig. 2). In summary, our friction model includes the following assumptions: i), the cellular force apparatus causes the transport of the ligands, which are noncovalently anchored to the supporting polymer substrate; ii), the movement of ligands on the polymer surface is accompanied by a sliding friction force; and iii), the intracellular protein complex of the focal adhesion is assumed to be rigid.
Figure 2.
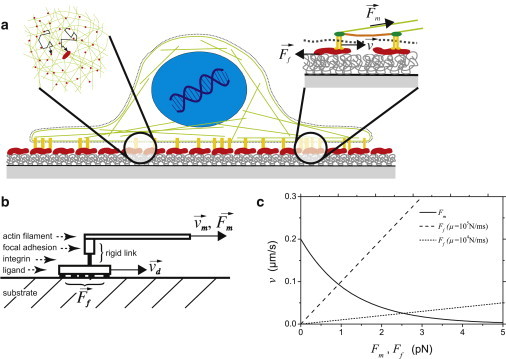
Model of frictional control of traction forces of adherent cells by noncovalent anchorage of adhesion ligands on surfaces. (a) Schemes of slow, random, and fast, directed motion of integrin-FN complexes within the interlayer between the cell membrane and the substrate surface. (Left inset) Random reorganization of FN ligands by a myosin-driven translocation along the actin cortex network. (Right inset) Stationary motion of integrin-FN complexes inside a focal adhesion with equilibrium between molecular friction at the ligand-polymer interface and myosin-motor forces transferred via a link of integrin-ligand complex, proteins insides the focal adhesion, and the cytoskeleton. (b) Scheme of the model for explaining the friction force at the ligand-substrate interface. (c) Plots of velocity v versus force for the myosin motor characteristic Fm (solid line) and for the linear friction model of FN sliding Ff (dashed lines). The intersections of the curves correspond to the force equilibrium (Eq. 3) and indicate force values in the pN range for ligand surface mobilities from 104 to 105 m/Ns.
When FN ligands are drawn along the polymer surface, their interaction with the polymer is characterized by many weak noncovalent bonds randomly distributed on the surface. Rupture of these bonds during FN movement corresponds to overcoming of small energy barriers by thermal activation. With molecular sliding steps δ of subnanometer size (typical repeating lengths of α-helix and β-sheet secondary structures of proteins are 5 to 7 Å) and relevant forces in the pN range, we get for the external work per activation event an upper estimate of for nm and pN. Because of this relation, the friction force can roughly be expressed as
| (1) |
where is the drift velocity of the ligand on the polymer surface and μ is the ligand mobility (see Supporting Material for a detailed explanation). This force-velocity relationship formally agrees with the linearized Tomlinson model (21).
During the time of the FN drift, the FN-integrin bond can be considered as stable because of the high binding affinity of the relevant integrin (α5β1) to FN (22,23). An estimate of the drift velocity can be derived from the motion of the myosin-driven cytoskeletal force apparatus. ATP-driven myosin II motors are acting via the link of the focal adhesion to the actin cytoskeleton. In a simplified manner, the myosin motor activity can be described by a velocity-force relation of biased motor proteins under external load Fm (24,25)
| (2) |
where am characterizes the myosin II step size along actin filaments of ∼7 nm (26), and v0 is the nonmuscle myosin II motor velocity without load of ∼0.2 μm/s (27). Here, we only use the described load dependence of motor step probability without looking at any detail of the myosin activity including internal drag (28). The latter should be negligible in comparison to the strong influence of external drag. This can be concluded from the difference between the FN mobility (see below) and the mobility governing the internal drag. The latter one is at least one order of magnitude larger (24). Note that the force-velocity relation (Eq. 2) cannot be linearized as in the case of the FN ligand friction. The work done by the myosin motors can be estimated by .
For the stationary motion of the ligand along the substrate surface, the force equilibrium has to be fulfilled
| (3) |
With the assumption of a rigid force transmission from the myosin-driven force apparatus to FN, we equalize Defining , one obtains from Eqs. 1–3
| (4) |
The intersections of the curves and in Fig. 2 represent solutions of Eq. 4; for two values of the FN mobility. One can see that a surface mobility μ of FN ligands in the range from 104 to 105 m/Ns results in reasonable receptor forces of a few pN.
Ligand reorganization at the cell-substrate interface described by diffusion
According to our proposed model, the friction force is determined by the velocity of FN and by the mobility of FN on the substrate surface (). The first one is governed by the myosin-motor system. As an essential result of the previous discussion, we have derived Eq. 4 as a relation between the friction force and the mobility. To validate this equation, force and mobility have to be determined independently. The friction force F can be derived from measurements of the traction stress Tmax and the knowledge of the receptor density.
To determine the mobility μ, we investigated the reorganization of randomly distributed individual FN ligands into larger fibrillar aggregates, known as FN fibrillogenesis (18,29–31). The whole process of FN fibrillogenesis, which involves integrin receptors, can roughly be divided into two parts. At first, FN ligands approach the focal adhesion by random motion. In the second step, FN molecules at the focal adhesion are stretched by tensile forces into an elongated conformation, enabling the exposure of cryptic binding sites for self-association into a fibrillar supramolecular structure. The exact mechanism and the degree of conformational changes are still a matter of debate.
We analyzed the first part of this process, the stochastic transport of FN to the focal adhesion. By that we could quantify FN mobility μ on the polymer surface. Because we were faced with a very low surface mobility of the large biopolymer molecules, we did not apply typical single molecule techniques like fluorescence correlation spectroscopy to investigate FN transport. Instead, we focused on the second part of FN fibrillogenesis, the fibril growth, which scales with the surface mobility of the FN ligands.
By using fluorescent labeled FN, we could in situ analyze FN fibril growth by means of a confocal laser scanning microscope with a temporal resolution of 30 s (cf. Fig. 3, a and b, and Movie S1). In our setup, only exogenous FN preadsorbed on the underlying polymer support was available for fibril formation. As stated previously, FN approaches a growing fibril by a two-dimensional (2D) random motion within the interlayer between the cell membrane and the polymer surface. For a quantitative analysis, we have described this process approximately by FN diffusion to a growing fibril with the fibril acting as perfect sink for FN. Actually, the size of the growing fibril changes. However, for an order of magnitude estimate of the corresponding FN diffusion coefficient D within the interlayer, we considered a constant size of the sink. The shape of the sink was approximated by a circle of radius R, which was correlated to the measurement area—the final size of the fibril. In our experiments, this radius R was ∼1 μm. At the fibril radius, a vanishing FN concentration was assumed. With these strongly simplifying assumptions, the aggregation rate of FN at the growing fibril in the early stage () is given by (32)
| (5) |
where is the FN area density at a large distance from the fibril, and the offset time accounts for a partially grown fibril.
Figure 3.
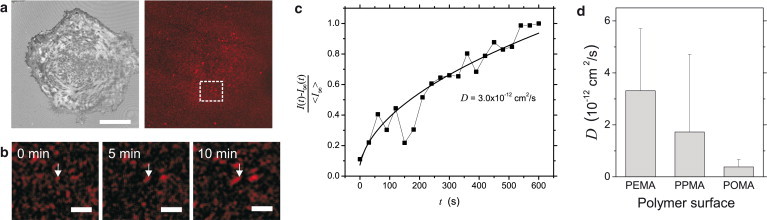
Diffusion coefficients of FN ligands at the cell-substrate interface measured by in situ analysis of FN fibrillogenesis. (a) Adherent HUVEC at 30 min after seeding visualized by reflection interference contrast microscopy and the corresponding fluorescence image of preadsorbed TAMRA-labeled FN. The area of snapshots in (b) and Movie S1 is indicated. Scale bar: 30 μm. (b) Snapshots of Movie S1of preadsorbed TAMRA-labeled FN after 0 min, 5 min, and 10 min. The arrow indicates a growing FN fibril. Scale bar: 5 μm. (c) Intensity of a growing FN fibril versus time on a PEMA surface. From the measured fibril intensity I(t) the background fluctuation I∞(t) has been subtracted and normalized to the mean background level . A fit of the intensity by using Eq. 5 in the Supporting Material yields a diffusion coefficient of D = 3.0 × 10−12 cm2/s. (d) Mean diffusion coefficients of FN on the different polymer surfaces. Error bars indicate mean + SD.
On the basis of the aggregation rate from Eq. 5, we expect a square root increase of the fluorescence intensity of the growing fibril over time (see Supporting Material for a more detailed explanation). As exemplarily shown in Fig. 3 c, such a dependency fits our measurements. The measured intensity in Fig. 3 c is shown with subtraction of background fluctuations and normalization by the mean background intensity. The fit allows determining the diffusion coefficient D of surface-anchored FN molecules. Fig. 3 d shows the corresponding values for the three different polymer surfaces. The diffusion coefficient increases with decreasing FN anchorage strength. The derived low diffusion coefficients of 0.3–3 × 10−12 cm2/s correlate with the comparatively high affinity of the big FN molecule to the polymer surfaces. Interestingly, they are in the range of literature values of diffusion of integrins on the ventral side of adherent cells in contact with FN ligands (33). Furthermore, they compare very well with diffusion-coefficient estimates within our recently established model of nanoscale FN fibrillogenesis (23).
From our estimates of the FN diffusion coefficient, we derived the FN mobility on the polymer surfaces by employing the Einstein relation μ = D/kT. The obtained values for the mobility are in the range from 104 to 105 m/Ns.
Interestingly, the measured diffusion coefficients support the applicability of the linear friction law Eq. 1 as shown in the following. It is assumed that the actin cortex at the cell membrane provides a framework for the random, nondirected motion of FN-integrins complexes toward the focal adhesions. Myosin motors inside the actin cortex are thought to act in a similar way as in actin stress fibers by pulling on actin filaments. However, the meshwork of the actin cortex resembles the trails for this type of transport. The 2D network of actin filaments in the actin cortex has a typical mesh size lmesh of ∼100 nm. In this case it can be assumed, that myosin motors only work with minimal load and therefore in a linear regime, see below. The mean velocity for the FN movement along segments of actin filaments within time t reads . On the other hand, the mean square displacement for a random walk in 2D is given by . Elimination of the time t yields the velocity . By means of the linear friction law Eq. 1, , and the Einstein relation , the friction force is estimated as , which yields a value of ∼0.2 pN for nm. Importantly, at such forces not only is much smaller than 1 and allows the application of Eq. 1. Now also Eq. 2 can be linearized, as . Hence, we can conclude that in the case of the slow, random motion of FN ligands along the actin cortex myosin motors has a different velocity-force relationship than discussed for the traction force control. Both processes are considered to be driven by myosin motors. The small motor forces during the slow, random motion of FN transport toward the focal adhesion lead to a linear velocity-force relation of myosin motors. In contrast, the fast, directed transport for the traction force control has to be described with a nonlinear velocity-force relationship of myosin motors as forces are not small.
Quantitative description of traction force control by ligand friction on the substrate surface
By inserting the Einstein relation into Eq. 4, one obtains the following relation between the measured diffusion coefficients and the resulting friction force F
| (6) |
Additionally, the friction force can be calculated from the measured traction stress by dividing the stress by the receptor area density inside the focal adhesions, which results in a force per receptor-ligand complex. The data of our measured diffusion coefficients and the function (Eq. 6) are displayed in Fig. 4. The plot shows that the data of our three experiments can be nicely fitted with an area density of integrin receptors of 270 μm−2. The good agreement between the experimental data and Eq. 6 supports our model of friction-controlled traction forces. Moreover, the calculated friction forces lie in a reasonable range of a few pN. The agreement of experiment and model does not exclude additional contributions to the traction force. Tensile interactions on stationary ligands can be in place, too. However, it seems that the frictional component dominates the maximum traction stress in our system.
Figure 4.
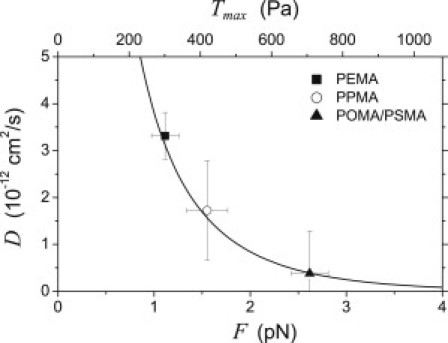
Diffusion coefficient D as a function of the traction force F. Comparison of experimental data with Eq. 6. The measured traction stress (Fig. 1.b) has been related to the traction force per FN-integrin complex by assuming 270 integrin receptors per μm2 inside a focal adhesion. Error bars indicate mean ± SE.
Sliding of focal adhesions
From our results, we can conclude that the motion of integrin-FN complexes inside the focal adhesions should lead to mobile focal adhesions. Indeed, we were able to observe sliding focal adhesions in our experiments by using in situ reflection interference contrast microscopy (see Fig. 5, Movie S2). The spots of dark contrast of focal adhesions in Fig. 5 shifted centripedally as one would expect from the traction stress caused by the actin stress fibers. This finding supports our model.
Figure 5.
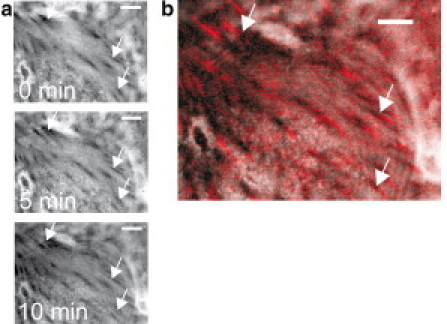
Sliding of focal adhesions. (a) Snapshots (at 0 min, 5 min, 10 min) of in situ reflection interference contrast microscopy of an adherent HUVEC (see also Movie S2) revealed a centripetally sliding of focal adhesions (characterized by the dark contrast). Stationary arrows are inserted as a guide to the eye. Scale bar: 10 μm. (b) Overlay of reflection interference contrast image and fluorescence image of TAMRA-labeled FN indicates the localization of the focal adhesion (black contrast) centripetally at the growing tip of FN fibril (red). Scale bar: 10 μm.
An overlay of focal adhesion pattern with FN fibrils revealed the position of the sliding focal adhesions at the growing tip of the FN fibrils (Fig. 5 b). This observation was supported by immunofluorescence staining of fixed samples for vinculin as typical marker protein in focal adhesions (Fig. S2).
Stationary and mobile focal adhesions were reported in the literature (29). Different mechanisms were discussed for mobile focal adhesions including disassembly and protein turnover. These mechanisms are mostly connected to rapidly migrating cells. In our experiments HUVECs migrate very slowly due to the high ligand density (18,34). Additionally, the orientation of the FN fibril relative to the focal adhesion into the direction of the cell border indicates a different growth mechanism of FN fibrils in comparison to previous reports (29). Therein FN fibrils were found to elongate centripedally from the focal adhesion. We think that the noncovalent anchorage of exogenously supplied FN and the frictional sliding as described previously are possible reasons for the different growth mechanism.
Finally, the velocity of focal adhesion translocations were analyzed and found to depend on the type of polymer surface, too. However, the corresponding velocity values of 0.002–0.003 μm/s (see Fig. S3) differed from the sliding velocity of FN ligands in our friction model (0.02 to 0.1 μm/s). We attribute this difference to the complexity and intrinsic dynamics of the focal adhesion and fibril assembly mechanism (3). A possible reason could be the permanent exchange of force-transmitting linkers due to protein turnover inside the focal adhesion. Although this process is assumed to be slow in comparison to the myosin movement, it could affect the slow overall translocation of the focal adhesion. Thereby, force-transmitting linkers can be exchanged and stretched molecules are able to relax. This process can lead to a stop-and-go behavior of the force-transmitting ligands, which could explain a slower overall translocation of the focal adhesion.
Conclusions
In summary, our study revealed traction forces and FN ligand reorganization to depend on the noncovalent anchorage of FN ligands to polymer substrates of different physicochemical surface characteristics. We have found a direct correlation of the ligand surface mobility and the traction force. We suggest that the interaction between FN ligands and the supporting material can be characterized by a thermally activated sliding of FN described by a linear friction law in connection with the myosin activity of the actin cytoskeleton. In this respect, the variation of ligand anchorage by means of substrate surface characteristics provides control of cell adhesion forces and downstream cell signaling. This finding could possibly affect current strategies in materials development for tissue engineering scaffolds.
Acknowledgments
We thank Juliane Drichel for technical assistance, Ina Uhlmann for stiffness measurement of polyacrylamide hydrogels, Hans-Joachim Schnittler for stimulating discussions, and Christina Müller for comments on the manuscript.
T.P. and M.K. were supported by the Deutsche Forschungsgemeinschaft (grant PO 713/2). C.W. was supported by the Federal Ministry of Science, Education and Technology of Germany (grant 0N4022, Center of Excellence in Biomaterials, Dresden).
The authors declare no conflict of interest.
Supporting Material
References
- 1.Hynes R.O. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Discher D.E., Mooney D.J., Zandstra P.W. Growth factors, matrices, and forces combine and control stem cells. Science. 2009;324:1673–1677. doi: 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geiger B., Spatz J.P., Bershadsky A.D. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 2009;10:21–33. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- 4.Evans E.A., Calderwood D.A. Forces and bond dynamics in cell adhesion. Science. 2007;316:1148–1153. doi: 10.1126/science.1137592. [DOI] [PubMed] [Google Scholar]
- 5.Colombelli J., Besser A., Stelzer E.H. Mechanosensing in actin stress fibers revealed by a close correlation between force and protein localization. J. Cell Sci. 2009;122:1665–1679. doi: 10.1242/jcs.042986. [DOI] [PubMed] [Google Scholar]
- 6.Friedland J.C., Lee M.H., Boettiger D. Mechanically activated integrin switch controls alpha5beta1 function. Science. 2009;323:642–644. doi: 10.1126/science.1168441. [DOI] [PubMed] [Google Scholar]
- 7.Daley W.P., Peters S.B., Larsen M. Extracellular matrix dynamics in development and regenerative medicine. J. Cell Sci. 2008;121:255–264. doi: 10.1242/jcs.006064. [DOI] [PubMed] [Google Scholar]
- 8.Ohashi T., Kiehart D.P., Erickson H.P. Dynamics and elasticity of the fibronectin matrix in living cell culture visualized by fibronectin-green fluorescent protein. Proc. Natl. Acad. Sci. USA. 1999;96:2153–2158. doi: 10.1073/pnas.96.5.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pompe T., Keller K., Werner C. Fibronectin fibril pattern displays the force balance of cell-matrix adhesion. Eur. Biophys. J. 2005;34:1049–1056. doi: 10.1007/s00249-005-0490-z. [DOI] [PubMed] [Google Scholar]
- 10.Pompe T., Zschoche S., Werner C. Maleic anhydride copolymers—a versatile platform for molecular biosurface engineering. Biomacromolecules. 2003;4:1072–1079. doi: 10.1021/bm034071c. [DOI] [PubMed] [Google Scholar]
- 11.Pompe T., Glorius S., Werner C. Dissecting the impact of matrix anchorage and elasticity in cell adhesion. Biophys. J. 2009;97:2154–2163. doi: 10.1016/j.bpj.2009.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pelham R.J., Jr., Wang Y.L. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA. 1997;94:13661–13665. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pompe T., Renner L., Werner C. Functional films of maleic anhydride copolymers under physiological conditions. Macromol. Biosci. 2005;5:890–895. doi: 10.1002/mabi.200500097. [DOI] [PubMed] [Google Scholar]
- 14.Butler J.P., Tolić-Nørrelykke I.M., Fredberg J.J. Traction fields, moments, and strain energy that cells exert on their surroundings. Am. J. Physiol. Cell Physiol. 2002;282:C595–C605. doi: 10.1152/ajpcell.00270.2001. [DOI] [PubMed] [Google Scholar]
- 15.Sabass B., Gardel M.L., Schwarz U.S. High resolution traction force microscopy based on experimental and computational advances. Biophys. J. 2008;94:207–220. doi: 10.1529/biophysj.107.113670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tolić-Nørrelykke I.M., Butler J.P., Wang N. Spatial and temporal traction response in human airway smooth muscle cells. Am. J. Physiol. Cell Physiol. 2002;283:C1254–C1266. doi: 10.1152/ajpcell.00169.2002. [DOI] [PubMed] [Google Scholar]
- 17.Renner L., Pompe T., Werner C. Dynamic alterations of fibronectin layers on copolymer substrates with graded physicochemical characteristics. Langmuir. 2004;20:2928–2933. doi: 10.1021/la0362627. [DOI] [PubMed] [Google Scholar]
- 18.Pompe T., Kobe F., Werner C. Fibronectin anchorage to polymer substrates controls the initial phase of endothelial cell adhesion. J. Biomed. Mater. Res. A. 2003;67:647–657. doi: 10.1002/jbm.a.10130. [DOI] [PubMed] [Google Scholar]
- 19.Reinhart-King C.A., Dembo M., Hammer D.A. Endothelial cell traction forces on RGD-derivatized polyacrylamide substrata. Langmuir. 2003;19:1573–1579. [Google Scholar]
- 20.Rajagopalan P., Marganski W.A., Wong J.Y. Direct comparison of the spread area, contractility, and migration of balb/c 3T3 fibroblasts adhered to fibronectin- and RGD-modified substrata. Biophys. J. 2004;87:2818–2827. doi: 10.1529/biophysj.103.037218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suda H. Origin of friction derived from rupture dynamics. Langmuir. 2001;17:6045–6047. [Google Scholar]
- 22.Li F., Redick S.D., Moy V.T. Force measurements of the α5β1 integrin-fibronectin interaction. Biophys. J. 2003;84:1252–1262. doi: 10.1016/S0006-3495(03)74940-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pompe T., Starruss J., Pompe W. Modeling of pattern development during fibronectin nanofibril formation. Biointerphases. 2006;1:93–97. doi: 10.1116/1.2345653. [DOI] [PubMed] [Google Scholar]
- 24.Bormuth V., Varga V., Schäffer E. Protein friction limits diffusive and directed movements of kinesin motors on microtubules. Science. 2009;325:870–873. doi: 10.1126/science.1174923. [DOI] [PubMed] [Google Scholar]
- 25.Stachowiak M.R., O'Shaughnessy B. Kinetics of stress fibers. N. J. Phys. 2008;10:025002. [Google Scholar]
- 26.Murphy C.T., Rock R.S., Spudich J.A. A myosin II mutation uncouples ATPase activity from motility and shortens step size. Nat. Cell Biol. 2001;3:311–315. doi: 10.1038/35060110. [DOI] [PubMed] [Google Scholar]
- 27.O'Connell C.B., Tyska M.J., Mooseker M.S. Myosin at work: motor adaptations for a variety of cellular functions. Biochim. Biophys. Acta. 2007;1773:615–630. doi: 10.1016/j.bbamcr.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 28.Howard J. Mechanical signaling in networks of motor and cytoskeletal proteins. Annu. Rev. Biophys. 2009;38:217–234. doi: 10.1146/annurev.biophys.050708.133732. [DOI] [PubMed] [Google Scholar]
- 29.Pankov R., Cukierman E., Yamada K.M. Integrin dynamics and matrix assembly: tensin-dependent translocation of alpha(5)beta(1) integrins promotes early fibronectin fibrillogenesis. J. Cell Biol. 2000;148:1075–1090. doi: 10.1083/jcb.148.5.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baneyx G., Baugh L., Vogel V. Fibronectin extension and unfolding within cell matrix fibrils controlled by cytoskeletal tension. Proc. Natl. Acad. Sci. USA. 2002;99:5139–5143. doi: 10.1073/pnas.072650799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mao Y., Schwarzbauer J.E. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005;24:389–399. doi: 10.1016/j.matbio.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 32.Crank J. Oxford University Press; London: 1970. The Mathematics of Diffusion. [Google Scholar]
- 33.Wiseman P.W., Brown C.M., Horwitz A.F. Spatial mapping of integrin interactions and dynamics during cell migration by image correlation microscopy. J. Cell Sci. 2004;117:5521–5534. doi: 10.1242/jcs.01416. [DOI] [PubMed] [Google Scholar]
- 34.DiMilla P.A., Barbee K., Lauffenburger D.A. Mathematical model for the effects of adhesion and mechanics on cell migration speed. Biophys. J. 1991;60:15–37. doi: 10.1016/S0006-3495(91)82027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.