

Abstract
The primary electron donor P700 in photosystem I is composed of two chlorophylls, PA and PB. P700 forms the cationic [PA/PB]•+ state as a result of light-induced electron transfer. We obtained a PA•+/PB•+ ratio of 28:72 and a spin distribution of 22:78 for the entire PSI protein-pigment complex. By considering the influence of the protein components on the redox potential for one-electron oxidation of PA/PB monomers, we found that the following three factors significantly contributed to a large PB•+ population relative to PA•+: 1), Thr-A743 forming a H-bond with PA; 2), PA as a chlorophyll a epimer; and 3), a conserved PsaA/PsaB pair, the Arg-A750/Ser-B734 residue. In addition, 4), the methyl-ester groups of the accessory chlorophylls A−1A/A−1B significantly stabilized the cationic [PA/PB]•+ state and 5), the methyl-ester group orientations were completely different in A−1A and A−1B as seen in the crystal structure. When the methyl-ester group was rotated, the spin-density distribution over PA/PB ranged from 22:78 to 15:85.
Introduction
In oxygenic photosynthesis, photosystem I (PSI) participates in the conversion of light to chemical energy with photosystem II (PSII). Light-induced electron transfer occurs along a series of cofactors bound to the PsaA and PsaB subunits of PSI. On the lumen side, there exists a chlorophyll dimer P700 composed of two chlorophylls PA and PB. PB is chlorophyll a (Chla) and PA is Chla′, the C132 epimer of Chla (Fig. 1 a) (1,2). A second Chla pair next to PA/PB is the accessory Chla A−1A/A−1B. In addition, there are two additional distant Chla (A0), two phylloquinones (A1), and one iron-sulfur cluster (FX) in the PsaA/PsaB subunits. Similar to PSII, these six Chla and two phylloquinones are arranged in two electron-transfer branches (A and B), which adopt a pseudo-C2 symmetry with the rotation axis passing through PA/PB and FX. In PSII, only one of the two branches serves as an electron-transfer active branch; this is in contrast to PSI, in which two branches are electron-transfer active (3), as shown by the biphasic forward electron transfers from P700 to A1 or from A1 to FX observed in kinetic studies.
Figure 1.
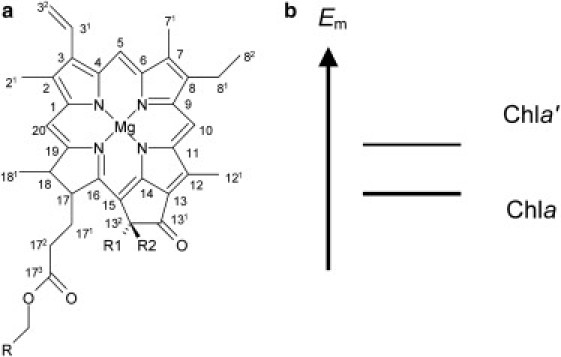
(a) Geometry of Chla (R1 = CH3COO−, R2 = H) and the C132 epimer, Chla′ (R1 = H, R2 = CH3COO−). (b) Em values of Chla and Chla′ in the reference system (e.g., aprotic solvents).
The primary process of charge separation culminates with a hole on the PA/PB pair in PSI or on the PD1/PD2 pair in PSII. In PSII, the cationic state over PD1/PD2 is predominantly observed in the Chla of the electron active branch PD1, with a PD1•+/PD2•+ ratio of ∼80:20 (4,5). For PSI, the PA•+/PB•+ ratios or the corresponding spin-density distributions were obtained experimentally by Fourier transform infrared (FTIR) spectroscopy (6) or electron paramagnetic resonance (EPR) (4,7,8) studies, respectively. FTIR studies indicated that the PA•+/PB•+ ratios were in the range of 50:50–33:67 (6). On the other hand, the spin-density distribution over PA/PB is 25:75 (4) and 25:75–20:80 in PSI from spinach (7) and 15:85 in PSI from Thermosynechococcus elongatus (8) (reviewed in Webber and Lubitz (9)).
To understand the role of the amino-acid residues or cofactors of PSI in the energetics of P700, influences of all amino-acid residues and redox-active cofactors on the energetics of PA/PB Chla monomers as well as the P700 Chla dimer are, for the first time to our knowledge, here elucidated. We present the following: 1), The computational results of the influence of the PSI protein environment on the redox potentials, Em(PA) and Em(PB), are presented on the basis of the PSI crystal structure (2) in the presence of all protein subunits and cofactors. The linear Poisson-Boltzmann equation is solved by considering the protonation states of all titratable sites in PSI. 2), The PA•+/PB•+ ratio for the PA/PB pair is calculated using a large-scale quantum chemical/molecular mechanical (QM/MM) approach with explicit treatment of the complete PSI atomic coordinates divided into two subsystems; the QM region contains the PA/PB dimer and is treated by quantum mechanics (unrestricted DFT/B3LYP and LACVP∗ level) and the remaining protein subunits and cofactors are treated with the MM force field. The computational conditions and procedures used in studies on the Thermosynechococcus vulcanus PSII (10) (using the 1.9 Å structure (11)) were consistently used in our study. This can facilitate the direct analysis and comparison of the influences of protein environments on Chla in both PSI and PSII.
Experimental Procedures
In this article, we employed the following systematic modeling procedure:
First, we constructed a realistic molecular model of the whole PSI protein-pigment complex using a high-resolution crystal structure. Based on this atomistic model, we evaluated the redox potential of PA/PB by solving the linear Poisson-Boltzmann equation with explicit consideration of the protonation states for all titratable residues.
Second, to gain deeper insights into the electronic structure of PA/PB Chla′/Chla heterodimer, we performed large-scale QM/MM calculations for the entire PSI protein-pigment complex.
Third, and finally, after confirming the validity of the computational results through comparison with available experimental data, we discussed the atomistic origin that determines the asymmetric distribution of the cationic state of the PA/PB pair. Technical details of each modeling procedure are summarized as follows.
Coordinates
The atomic coordinates were taken from the x-ray structure of the PSI protein-pigment complex from T. elongatus at 2.5 Å resolution (Protein Data Bank code: 1JB0) (11). Hydrogen atoms were generated and energetically optimized using the CHARMM force field (12). The positions of all nonhydrogen atoms were fixed, and all titratable groups were kept in their standard protonation states; i.e., acidic groups were ionized and basic groups were protonated. For the QM/MM calculations, we added additional counterions to neutralize the whole system.
Atomic partial charges
Atomic partial charges of the amino acids were adopted from the all-atom CHARMM22 (13) parameter set. The charges of the protonated acidic O atoms were increased successively by +0.5 units to implicitly account for the presence of a proton. Similarly, instead of removing a proton to generate the deprotonated state, the charges of all of the protons of the basic groups of Arg and Lys were successively diminished to a total of 1 unit of charge. For residues for which the protonation states were not available in the CHARMM22 parameter set, appropriate charges were computed (14). For the atomic charges of Chla (except for PA/PB) and quinones, we used atomic partial charges from previous studies on PSI (15–17); the charges were determined by fitting the surrounding electrostatic potential of these molecules using the RESP procedure (18). The charges of equivalent H atoms (e.g., three H atoms of methyl group) were averaged. The electronic wave functions were calculated after geometry optimization with the DFT module in JAGUAR (19) (B3LYP/LACVP∗) (see Table S1 in the Supporting Material).
Computation of redox potential Em(Chla)
Our computation was based on the electrostatic continuum model, where the linear Poisson-Boltzmann equation was solved using the MEAD program (20). To facilitate direct comparisons with previous computational results, identical computational conditions and parameters such as atomic partial charges and dielectric constants were used (21,22). The redox states of all other cofactors (e.g., A–1 and A0 Chla, and A1 quinone) were kept in their neutral charge states during the redox titration of each Chla. We considered FX, FA, and FB in the oxidized charge state [Fe4S4(SCH3)4]2– for the wild-type PSI as in previous studies (15–17). The ensemble of the protonation patterns was sampled using the Monte Carlo method with the Karlsberg program (http://agknapp.chemie.fu-berlin.de/karlsberg/(1999)) (23). The dielectric constants were set to εp = 4 inside the protein and εw = 80 for water.
All computations were performed at 300 K, pH 7.0, and an ionic strength of 100 mM. The linear Poisson-Boltzmann equation was solved using a three-step grid-focusing procedure at resolutions of 2.5, 1.0, and 0.3 Å. The Monte Carlo sampling for a redox-active group yielded the probabilities [Aox] and [Ared] of the two redox states of molecule A. The shifts in Em(Chla′) and Em(Chla) due to the PSI protein environment were evaluated using the Nernst equation. A bias potential was applied to obtain an equal amount of both redox states ([Aox] = [Ared]), yielding the redox midpoint potential Em as the resulting bias potential. For convenience, the computed Em was represented in millivolt accuracy and the last digit was not considered to be significant.
QM/MM calculations
In all QM/MM calculations reported here, we employed the so-called electrostatic embedding QM/MM scheme. In all the QM/MM calculations, we used the QSITE (24) program code. The electrostatic and steric effects created because of the complex PSI architecture were explicitly considered in all calculations. Owing to the large system size of PSI, the QM region was limited to the PA/PB Chla′/Chla heterodimer for simplicity, whereas the other protein units and all cofactors were approximated by the MM force field. Because the atomic partial charges were optimized (e.g., A–1 and A0 Chla, and A1 quinone), the QM/MM partition was accurate enough to describe the electronic structure of the [PA/PB]•+ Chla′/Chla heterodimer. To reliably determine the cationic character of the [PA/PB]•+ heterodimer, we employed the unrestricted DFT method with the B3LYP functional and LACVP∗ basis sets. The [PA/PB]•+ heterodimer geometry was refined by the constrained QM/MM optimizations; for the surrounding MM protein environment, the atomistic coordinates were exactly fixed with the original x-ray coordinates. After obtaining the stable geometry of the QM fragment, we determined the ESP charges for the cationic state of the [PA/PB]•+ heterodimer in the presence of the entire group of PSI atomic coordinates (see Table S1).
Results and Discussion
PA•+/PB•+ ratio and spin-density distribution in wild-type PSI
The PA•+/PB•+ ratio was calculated to be 27.9:72.1 (Table 1), demonstrating that the cationic state is stabilized more in PB than in PA: this should result in the value of Em(PB) being lower than that of Em(PA). The PA•+/PB•+ ratio of 27.9:72.1 was considerably close to the charge-distribution ratio of 33:67 obtained from FTIR studies of PSI from Synechocystis sp. PCC 6803 (6). The calculated spin-density distribution over PA/PB was 22.4:77.6 (Table 1), which shows greater asymmetry than the ratio of the charge distribution (i.e., PA•+/PB•+), a fact already pointed out previously in PSII (25,26). The obtained value was in good agreement with the experimental values of 25:75 (4) and 25:75–20:80 of PSI from spinach (7) and 15:85 of PSI from T. elongatus (8) (note: according to another interpretation of the T. elongatus data (8), the ratio of 25:75–30:70 may be possible (9)).
Table 1.
Values of Em(PA), Em(PB) (in mV), PA•+/PB•+ ratios, and spin-density distributions in the PSI protein (in %)
| (Factor) |
Em shift (versus reference) |
Charge |
Spin |
||||
|---|---|---|---|---|---|---|---|
| PA | PB | PA•+ | PB•+ | PA | PB | ||
| Wild-type | 29∗ | 27 | 27.9 | 72.1 | 22.4 | 77.6 | |
| (1) | T743V [H-bond] | 20∗ | 28 | 32.2 | 67.8 | 26.9 | 73.1 |
| (2) | CH3COO− deleted [Epimer] | 32.2 | 67.8 | 29.7 | 70.3 | ||
| (3) | Δ(Arg-A750/Ser-B734) [PsaA/PsaB pair] | 30.6 | 69.4 | 25.0 | 75.0 | ||
| (4) | Δ(A−1A/A−1B) [Cofactor] | 24.0 | 76.0 | 18.7 | 81.3 | ||
The symbol Δ stands for deletion of atomic charges of the residues/groups.
Because Em(Chla′) in the reference model system is unknown, only the Em shift was calculated. An actual Em(Chla′) may be higher than Em(Chla) because Chla′ is thermodynamically less stable than Chla (9).
The PSI protein environment shifted the Em values of PA/PB only marginally from those of the reference model system (Table 1). The shifts were essentially the same for PA and PB. However, the resulting Em values for the PA/PB molecules must be different because of the difference between Em(Chla′) and Em(Chla) in the reference model system. Although the difference between Em(Chla′) and Em(Chla) is not known so far, we infer that the former may be higher than the latter because Chla′ is thermodynamically less stable than Chla (Fig. 1 b) (9).
Influence of H-bond on the PA•+/PB•+ ratio
The −OH group of Thr-A743 can form an H-bond with the 131-keto group of PA, whereas the corresponding H-bond is absent in PB. A mutation of Thr-A743 to Val would result in the loss of the H-bond (27,28). Using the wild-type PSI crystal structure, we modeled the T(A743)V PSI by substituting the −OH side chain group of Thr with –CH3 (followed by geometry optimization of the A743 side chain whereas all of the other parts remained fixed).
Em(PA) was lowered by ∼10 mV upon the T(A743)V mutation whereas Em(PB) remained unchanged (Table 1). The marginal downshift in Em(PA) was in agreement with a relatively weak H-bond of Thr-A743, as pointed out in spectroscopic studies (29). As Em(PA) was lowered (i.e., PA•+ was stabilized), the calculated PA•+/PB•+ ratio was significantly shifted and the population of PA•+ increased from 27.9 in the wild-type PSI to 32.2 in the T(A743)V PSI (Table 1). This observed PA•+ population shift is in agreement with 1), the previous proposal that deletion of the H-bond leads to a downshift in Em(PA) (27), and 2), the experimentally observed relocation of ∼14–18% of the cationic state from PB to PA upon T(A743)V mutation in PSI from Chlamydomonas reinhardtii (28). The slightly smaller relocation of the cationic state (i.e., an increase in PA•+ by 5%) in our study is possibly due to 1), the difference between the used T(A743)V PSI model structure where the −OH group of Thr was replaced with –CH3, without altering the other parts of the atomic coordinates (to avoid an uncertainty of prediction of the protein structure), or 2), the difference of the PSI proteins between T. elongatus PSI (corresponds to the crystal structure (2)) and the Chlamydomonas reinhardtii PSI. As Em(P700) differs by up to ∼70 mV among different species (30), the PA/PB moiety may be slightly different among different species.
The individual values of Em(PA) and Em(PB) are not known experimentally due to the presence of their coupling. It is particularly difficult to determine Em(PA) because of the small amount of PA•+ compared with PB•+. Our study demonstrates that shifts in Em(PA) alter the distribution of the cationic (spin) state over PA/PB as reported in mutational studies (27,28).
Influence of C132 epimer Chla′ on PA•+/PB•+ ratio
The asymmetric charge distribution over PA/PB may also result from the asymmetry of the molecular geometry in PA/PB. The methyl-ester (CH3COO−) groups in Chla′ and Chla are oppositely orientated with respect to the chlorin plane. Except for this methyl-ester orientation difference, the molecular structures of PA and PB are essentially identical (2).
To investigate this orientation difference on the PA•+/PB•+ ratio, it would have been more relevant to replace Chla′ of PA with Chla. Unfortunately, a replacement of Chla′ of PA with Chla leads to unreasonable contacts with Tyr-A603, Gly-A739, Thr-A742, and Thr-A743. As an alternative, we substituted the CH3COO− groups with H in both PA and PB (deletion of CH3COO−), and calculated the PA•+/PB•+ ratio. Upon deletion of the CH3COO− groups the PA•+ population increases by ∼5% relative to the wild-type PSI, resulting in a PA•+/PB•+ ratio of 32.2:67.8 (Table 1). Thus, the orientation difference in Chla′/Chla significantly influences the PA•+/PB•+ ratio in the PSI protein environment.
Under vacuum conditions, the PA/PB heterodimer yielded a PA•+/PB•+ ratio of 41.5:58.5 (Table 2), which indicates a considerably delocalized cationic state distribution over PA/PB in the absence of the PSI protein environment. The deletion of CH3COO− from PA/PB delocalized the cationic state, giving a PA•+/PB•+ ratio of 45.5:54.5 (Table 2); i.e., the cationic state is almost equally distributed over PA/PB. It can be concluded that in the absence of the PSI protein environment, the orientation of the CH3COO− group in PA is predominantly responsible for the PA•+/PB•+ ratio (Table 3).
Table 2.
Values of PA•+/PB•+ ratios and spin-density distributions in vacuum (in %)
| Charge |
Spin |
|||
|---|---|---|---|---|
| PA•+ | PB•+ | PA | PB | |
| Wild-type | 41.5 | 58.5 | 38.1 | 61.9 |
| CH3COO− deleted | 45.5 | 54.5 | 46.2 | 53.8 |
Table 3.
Analysis of factors (listed in Table 1) that differentiate the populations of PA•+ and PB•+ in the PSI protein environment (in %)
| Charge |
Spin |
|||
|---|---|---|---|---|
| PA•+ | PB•+ | PA | PB | |
| Wild-type | 27.9 | 72.1 | 22.4 | 77.6 |
| (1) + (2) | 37.0 | 63.0 | 35.1 | 64.9 |
| (1) + (3) | 35.6 | 64.4 | 30.5 | 69.5 |
| (1) + (2) + (3) | 40.5 | 59.5 | 39.1 | 60.9 |
Factors (1) T743V (deletion of a H-bond with PA), (2) CH3COO− substitution with –H (deletion of the Chla′/Chla difference), and (3) deletion of charges of PsaA/PsaB residue pairs responsible for the Em(PA)/Em(PB) difference.
Influence of PsaA/PsaB symmetrical residue pairs on the PA•+/PB•+ ratio
The protein environment, in particular the charged residues, contribute to Em shifts of the redox active site significantly. Because the Em difference Em(PA) – Em(PB) (= ΔEm) is a key factor that can alter the PA•+/PB•+ ratio (27) (Fig. 2), it is important to clarify the contributions of residues, in particular those located in the PsaA/PsaB heterodimer subunits.
Figure 2.
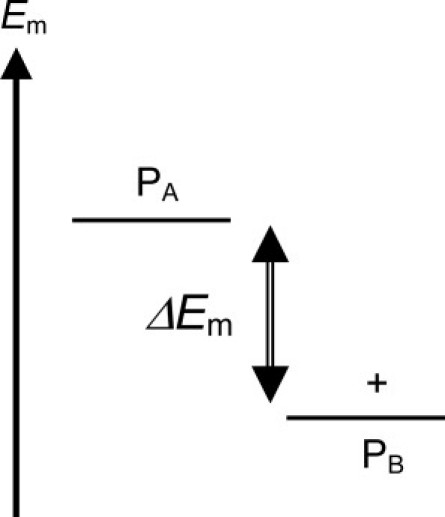
Relationship between Em and population of the cationic state over the PA/PB pair. The + sign indicates a population of the energetically distributed cationic state owing to the Em levels. ΔEm = Em(PA) – Em(PB).
Arg-A750, located on the lumenal side (Fig. 3 a), upshifted Em(PA) the most by 65 mV, due to its positively charged protonated state (Table 4). The Arg-A651 on the lumen side and Arg-A728 near FX, both upshifted Em(PA) by 35 mV. Arg-A728 is ∼20 Å away from PA/PB, but its electrostatic influence on Em(PA) and Em(PB) is significant because of the complete absence of other titratable residues in the region between PA/PB and FX (Fig. 3 d).
Figure 3.
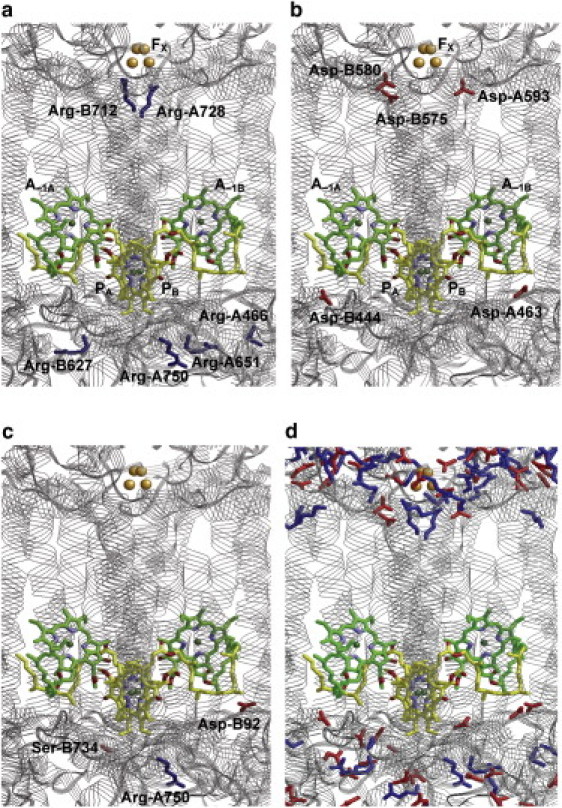
Arrangement of residues in PsaA/PsaB of PSI. (Blue and red sticks) Side chains of basic and acidic residues, respectively. (a) Residues increasing Em(PA) or Em(PB). (b) Residues decreasing Em(PA) or Em(PB). (c) Residue pairs enhancing Em(PA) > Em(PB). (d) All acidic and basic residues in PsaA/PsaB.
Table 4.
Residues that shift Em(PA) and Em(PB) in the PSI protein environment (mV)
| Residue |
Em(PA) shift |
Residue |
Em(PB) shift |
||||
|---|---|---|---|---|---|---|---|
| Side chain | Backbone | Total | Side chain | Backbone | Total | ||
| Increasing Em(PA) and Em(PB) | |||||||
| Arg-A750 | 53 | 12 | 65 | Arg-A651 | 60 | −5 | 55 |
| Arg-A651 | 42 | −7 | 35 | Arg-A750 | 30 | 5 | 36 |
| Arg-A728 | 39 | −4 | 35 | Arg-A466 | 32 | 4 | 35 |
| Arg-B627 | 36 | −3 | 33 | Arg-B712 | 36 | −4 | 32 |
| Arg-B712 | 34 | −3 | 31 | Arg-A728 | 35 | −2 | 32 |
As both Arg-A651 and Arg-A728 concomitantly increase Em(PA) and Em(PB) with almost the same magnitude (Table 4), these residues were not responsible for the asymmetric distribution of PA•+/PB•+. Furthermore, according to the protein sequence alignment (with the CLUSTAL program (31)), Arg-A651 and Arg-A728 in PsaA are conserved as Arg-B627 and Arg-B712 in PsaB, respectively, which also upshifts Em(PA) and Em(PB) (Table 4).
The same argument also holds true for the Em downshifting residues, which are mainly acidic residues (Table 5). Asp-B444 significantly downshifted Em(PA) by 90 mV and Em(PB) by 63 mV; and its counterpart in PsaA, Asp-A463, downshifted Em(PA) by 67 mV and Em(PB) by 98 mV. Thus, the influence of Asp-B444 on ΔEm is completely compensated for by the influence of the counterpart Asp-A463.
Table 5.
Residues that shift Em(PA) and Em(PB) in the PSI protein environment (mV)
| Residue |
Em(PA) shift |
Residue |
Em(PB) shift |
||||
|---|---|---|---|---|---|---|---|
| Side chain | Backbone | Total | Side chain | Backbone | Total | ||
| Decreasing Em(PA) and Em(PB) | |||||||
| Asp-B444 | −94 | 4 | −90 | Asp-A463 | −102 | 4 | −98 |
| Asp-A463 | −70 | 3 | −67 | His-B660 | −70 | 6 | −64 |
| His-A680 | −72 | 5 | −67 | Asp-B444 | −66 | 3 | −63 |
| A–1A | −65 | FX | −47 | ||||
| FX | −48 | Asp-B575 | −42 | 0 | −42 | ||
| Asp-B575 | −41 | 0 | −42 | A–1B | −41 | ||
| Asp-A593 | −33 | −2 | −35 | Asp-B580 | −31 | −2 | −33 |
| His-B660 | −31 | 0 | −31 | His-A680 | −31 | −1 | −32 |
| Asp-B580 | −28 | −2 | −30 | Asp-A593 | −29 | −2 | −31 |
To clarify how the PSI protein environment differentiates PA•+/PB•+, it is not sufficient to search for the residue that induces large changes in Em values, instead it is important to find a residue pair of PsaA/PsaB that increases ΔEm (Fig. 2). Bearing this in mind, we found only a few residue pairs that contribute to nonzero values of ΔEm. The residue pair, Arg-A750/Ser-B734, increased ΔEm by 17 mV (Table 6). Note that the residue pair is fully conserved in cyanobacteria and green plants (Fig. 4). The Arg-A750/Ser-B734 pair is located near PA/PB on the lumen side (Fig. 3 c). Indeed, Arg-A750 was already mentioned as the residue that upshifted Em(PA) the most among all residues in PSI (Tables 4 and 5) whereas its counterpart is an uncharged residue, Ser.
Table 6.
PsaA/PsaB residue pairs responsible for ΔEm (= Em(PA) – Em(PB), see Fig. 2) (mV)
| PsaA |
PsaB |
|||||
|---|---|---|---|---|---|---|
| Residue | Em(PA) | Em(PB) | Residue | Em(PA) | Em(PB) | ΔEm |
| ΔEm increasing pairs (i.e., pairs that contribute to accumulation of PB•+) | ||||||
| Arg-A750 | 65 | 36 | Ser-B734 | 9 | 21 | 17 |
| — | — | — | Asp-B92 | −15 | −24 | 9 |
Contribution to ΔEm was obtained as [influence of PsaA on Em(PA)] + [influence of PsaB on Em(PA)] – [influence of PsaA on Em(PB)] – [influence of PsaB on Em(PB)]. PsaA/PsaB residue pairs in each line were generated from the protein sequence alignment performed with the CLUSTAL program (31) (Fig. 4).
Figure 4.

Amino-acid sequences of the PsaA and PsaB subunits of PSI from T. elongatus, Synechocystis PCC 6803 (S.6803), Chlamydomonas reinhardtii (C. reinhardtii), and spinach. PsaA/PsaB residue pairs in each line were generated from the protein sequence alignment performed with the CLUSTAL program (31).
In the absence of the atomic charges of Arg-A750/Ser-B734, we obtained the PA•+/PB•+ ratio of 30.6:69.4; in which the PA•+ population has increased by ∼3%, with respect to wild-type PSI (Table 1). This result indicates that, in turn, the Arg-A750/Ser-B734 pair contributes to increase the PB•+ population in the wild-type PSI by increasing ΔEm.
The Arg-A750/Ser-B734 residue pair contributes the most to ΔEm among all PsaA/PsaB residue pairs; however, it increases ΔEm only by 17 mV (Table 6). The second most contributing residue pair —/Asp-B92, in which the corresponding residue in PsaA is absent among cyanobacteria and green plants (Fig. 4), contributes 9 mV to ΔEm (Table 6). Hence, in comparison with the much larger influence of a number of D1/D2 residue pairs on the Em(PD1)/Em(PD2) difference in PSII (10), the protein environments of PsaA and PsaB in PSI are quite similar in their electrostatic characters.
The residue pairs Arg-A750/Ser-B734 and —/Asp-B92 are responsible for ΔEm in wild-type PSI. Mutations of these residues would produce other protonation patterns of the titratable residues of PSI to compensate for the induced changes (e.g., charges or protein conformations). Unless the protonation pattern of the titratable residues and the protein conformation remain unchanged upon the corresponding mutations, mutational studies do not directly correspond to our results of the wild-type PSI.
Differences in the two accessory Chla pairs A−1A and A−1B
There are also pairs that contribute to decrease ΔEm and facilitate an accumulation of PB•+. Unexpectedly, the most influential was the accessory Chla pair, A−1A and A−1B, which decreased ΔEm by 28 mV (Table 7). It was surprising, because both A−1A and A−1B are identical molecules, Chla, and the methyl-ester groups of PA/PB, i.e., the region causing the Chla′/Chla difference, is sufficiently far from the interface between PA/PB and A−1A/A−1B.
Table 7.
PsaA/PsaB residue pairs responsible for ΔEm (= Em(PA) – Em(PB), see Fig. 2) (in mV)
| PsaA |
PsaB |
|||||
|---|---|---|---|---|---|---|
| Residue | Em(PA) | Em(PB) | Residue | Em(PA) | Em(PB) | ΔEm |
| ΔEm decreasing pairs (i.e., pairs that contribute to accumulation of PA•+) | ||||||
| A–1A | −65 | −23 | A–1B | −19 | −41 | −28 |
| Gln-A657 | −4 | −2 | Asn-B633 | 18 | 26 | −14 |
Contribution to ΔEm was obtained as [influence of PsaA on Em(PA)] + [influence of PsaB on Em(PA)] – [influence of PsaA on Em(PB)] – [influence of PsaB on Em(PB)]. PsaA/PsaB residue pairs in each line were generated from the protein sequence alignment performed with the CLUSTAL program (31) (Fig. 4).
As expected from the ΔEm value, eliminating the atomic charges of A−1A and A−1B resulted in the decrease in the PA•+ population, shifting the PA•+/PB•+ ratio to 24.0:76.0 (Table 1). Thus, the influence of the accessory Chla pair on the PA•+/PB•+ ratio as well as ΔEm appears to be significant.
For the first time to our knowledge, in this work it has been found that the difference in the energy contribution of A−1A/A−1B to PA/PB results from the orientation of the methyl-ester groups of A−1A/A−1B, which are orientated at an angle of ∼180° to each other across the C132-C133 axis (Fig. 5). Because the methyl-ester groups are situated on the same side of the chlorin plane, the A−1A/A−1B pair consist of methyl-ester rotamers and not methyl-ester epimers. The carbonyl O atom of A−1A is 5.4 Å away from the Mg atom of PA whereas the corresponding distance is 7.1 Å between A−1B and PB (Fig. 5). Because the carbonyl O atom is polarized more than the ester O atom, the methyl-ester group of A−1A can stabilize the cationic PA•+ state more effectively than that of the A−1B for the PB•+ state, thus decreasing Em(PA) relative to Em(PB). The possible role of the methyl-ester orientation of a Chla in altering the Em of another Chla in the neighborhood was originally reported in PSII (32), and our results indicate that the same phenomenon holds true for PA/PB in PSI. Note that a clear discrimination between the –O–CH3 and O edges of the ester-group in the electron density map was possible at a resolution of ∼2.5 Å (W. Saenger, Free University of Berlin, personal communication, 2011).
Figure 5.
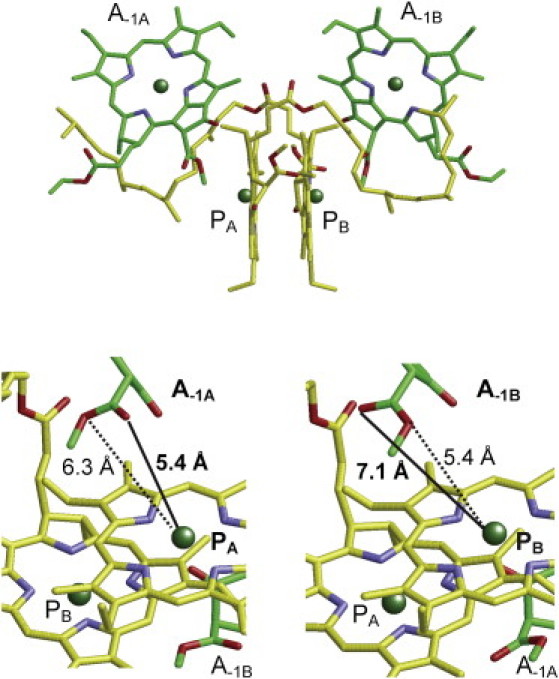
Geometry of the accessory Chla A−1A and A−1B. (Solid and dotted lines) Distances between the Mg atom and the O atoms of the carbonyl and ester groups, respectively.
The H-bonding partner for any group that specifies the orientation of the methyl-ester groups of A−1A/A−1B or cause steric hindrance is absent. By rotating each methyl-ester group in A−1A/A−1B that is itself rotated by 180° along the C132–C133 axis, the PA•+/PB•+ ratio changed from 27.9:72.1 (wild-type) to 22.4:77.6 (rotated methyl-ester: A−1A/A−1B, Table 8) and the corresponding spin-density distributions in PA/PB were shifted from 22.4:77.6 to 15.1:84.9, respectively. The spin-density distribution of 15.1:84.9 was considerably shifted from the wild-type PSI value. A similar value of the spin-density distribution (15:85) was reported from EPR studies of PSI from T. elongatus (8). As the energy required for rotation of the methyl-ester group is not unusually large (due to the sp3 carbons) and the H-bonding partner that specifies the orientation of the methyl-ester groups of A−1A/A−1B is absent, it is reasonable to assume that the PA•+/PB•+ ratio and the spin-density distribution over PA/PB fluctuates within the ranges given in Table 8. Hence, the orientation of the methyl-ester group in A−1A/A−1B can affect the PA/PB ΔEm and the PA•+/PB•+ ratio.
Table 8.
Influence of the orientation of the methyl-ester group of A−1A/A−1B on the PA•+/PB•+ ratio and spin-density distribution calculated in the PSI protein (in %)
| Charge |
Spin |
|||
|---|---|---|---|---|
| PA•+ | PB•+ | PA | PB | |
| Wild-type | 27.9 | 72.1 | 22.4 | 77.6 |
| [Rotated methyl-ester] | ||||
| A−1A | 26.6 | 73.4 | 19.7 | 80.3 |
| A−1B | 23.5 | 76.5 | 17.4 | 82.6 |
| A−1A/A−1B | 22.4 | 77.6 | 15.1 | 84.9 |
Conclusions
The PA•+/PB•+ ratio of the PA/PB dimer calculated over the entire PSI protein was 28:72, which was close to the charge-distribution ratio of 33:67 obtained in the FTIR studies of PSI from Synechocystis sp. PCC 6803 (6). The calculated spin density, which was asymmetrically distributed over the PA/PB dimer with a ratio of 22:78, was in good agreement with the experimental values of 25:75 (4) and 20:80 in EPR studies from spinach (7). The Chla′/Chla difference for PA/PB was revealed to be partially responsible for the larger population of the PB•+ state, from the methyl-ester deletion simulations. Removal of the H-bond between PA and Thr-A743 in T(A743)V mutation resulted in a slight decrease in Em(PA) and a partial movement of the cationic state from PA to PB, as suggested by mutational studies (27,28).
Conserved PsaA/PsaB pairs, Arg-A651/Arg-B627 and Arg-A728/Arg-B712 upshifted both Em(PA) and Em(PB) by ∼30 mV, and Asp-A463/Asp-B444 and Asp-A593/Asp-B580 residue pairs downshifted both Em(PA) and Em(PB) in the range ∼30–60 mV. The residue pair that led to a notable value of ΔEm (i.e., Em(PA) > Em(PB)) was Arg-A750/Ser-B734, with a ΔEm of 17 mV, thus contributing to an increase in the PB•+ population (Note that a simple mutation of the residues would cause a change in the protonation states of the titratable residues in PSI, compensating for changes in the net charges of the mutated sites.). On the other hand, the most influential pair enhancing Em(PA) < Em(PB) was the accessory A-1A/A-1B Chla pair, with a ΔEm of 28 mV; this pair contributed to an increase in the PA•+ population in the original PSI crystal structure. The different influence of A−1A and A−1B on Em(PA) and Em(PB) was due to the differences in their methyl-ester orientations. Rotation of the methyl-ester groups alters the PA/PB spin-density distributions from 22:78 to 15:85. The latter value was in agreement with the value of 15:85 measured by EPR for PSI from T. elongatus (8).
Acknowledgments
We are grateful to Dr. Wolfram Saenger and Dr. Ernst-Walter Knapp for useful discussions.
This research was supported by the Japan Science and Technology Agency PRESTO program (to H.I.); Grant-in-Aid (21770163 to H.I. and 22740276 to K.S.) for Science Research from the Ministry of Education, Science, Sport and Culture of Japan; Special Coordination Fund (to H.I.) for Promoting Science and Technology of MEXT; Takeda Science Foundation (to H.I.); and Kyoto University Step-up Grant-in-Aid for young scientists (to H.I.).
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Watanabe T., Kobayashi M., Murata N. Evidence that a chlorophyll a′ dimer constitutes the photochemical reaction center 1 (P700) in photosynthetic apparatus. FEBS Lett. 1985;191:252–256. [Google Scholar]
- 2.Jordan P., Fromme P., Krauss N. Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature. 2001;411:909–917. doi: 10.1038/35082000. [DOI] [PubMed] [Google Scholar]
- 3.Guergova-Kuras M., Boudreaux B., Redding K. Evidence for two active branches for electron transfer in photosystem I. Proc. Natl. Acad. Sci. USA. 2001;98:4437–4442. doi: 10.1073/pnas.081078898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rigby S.E.J., Nugent J.H.A., O'Malley P.J. ENDOR and special triple resonance studies of chlorophyll cation radicals in photosystem 2. Biochemistry. 1994;33:10043–10050. doi: 10.1021/bi00199a031. [DOI] [PubMed] [Google Scholar]
- 5.Diner B.A., Schlodder E., Chisholm D.A. Site-directed mutations at D1-His198 and D2-His197 of photosystem II in Synechocystis PCC 6803: sites of primary charge separation and cation and triplet stabilization. Biochemistry. 2001;40:9265–9281. doi: 10.1021/bi010121r. [DOI] [PubMed] [Google Scholar]
- 6.Breton J., Nabedryk E., Leibl W. FTIR study of the primary electron donor of photosystem I (P700) revealing delocalization of the charge in P700+ and localization of the triplet character in 3P700. Biochemistry. 1999;38:11585–11592. doi: 10.1021/bi991216k. [DOI] [PubMed] [Google Scholar]
- 7.Davis I.H., Heathcote P., Evance M.C.W. Modulation analysis of the electron spin echo signals of in vivo oxidized primary donor 14N chlorophyll centers in bacterial, P870 and P960, and plant photosystem I, P700, reaction centers. Biochim. Biophys. Acta. 1993;1143:183–189. [Google Scholar]
- 8.Kass H., Fromme P., Lubitz W. Orientation and electronic structure of the primary donor radical cation P700+ in photosystem I: a single crystals EPR and ENDOR study. J. Phys. Chem. B. 2001;105:1225–1239. [Google Scholar]
- 9.Webber A.N., Lubitz W. P700: the primary electron donor of photosystem I. Biochim. Biophys. Acta. 2001;1507:61–79. doi: 10.1016/s0005-2728(01)00198-0. [DOI] [PubMed] [Google Scholar]
- 10.Saito K., Ishida T., Ishikita H. Distribution of the cationic state over the chlorophyll pair of photosystem II reaction center. J. Am. Chem. Soc. 2011;133:14379–14388. doi: 10.1021/ja203947k. [DOI] [PubMed] [Google Scholar]
- 11.Umena Y., Kawakami K., Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature. 2011;473:55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 12.Brooks B.R., Bruccoleri R.E., Karplus M. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 13.MacKerell A.D., Jr., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 14.Rabenstein B., Ullmann G.M., Knapp E.-W. Calculation of protonation patterns in proteins with structural relaxation and molecular ensembles—application to the photosynthetic reaction center. Eur. Biophys. J. 1998;27:626–637. [Google Scholar]
- 15.Ishikita H., Knapp E.-W. Redox potential of quinones in both electron transfer branches of photosystem I. J. Biol. Chem. 2003;278:52002–52011. doi: 10.1074/jbc.M306434200. [DOI] [PubMed] [Google Scholar]
- 16.Ishikita H., Stehlik D., Knapp E.W. Electrostatic influence of PsaC protein binding to the PsaA/PsaB heterodimer in photosystem I. Biophys. J. 2006;90:1081–1089. doi: 10.1529/biophysj.105.069781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karyagina I., Pushkar Y., Golbeck J.H. Contributions of the protein environment to the midpoint potentials of the A1 phylloquinones and the FX iron-sulfur cluster in photosystem I. Biochemistry. 2007;46:10804–10816. doi: 10.1021/bi700846z. [DOI] [PubMed] [Google Scholar]
- 18.Bayly C.I., Cieplak P., Kollman P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- 19.Schrödinger . Schrödinger, LLC; New York, NY: 2008. JAGUAR, v. 7. [Google Scholar]
- 20.Bashford D., Karplus M. pKa's of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry. 1990;29:10219–10225. doi: 10.1021/bi00496a010. [DOI] [PubMed] [Google Scholar]
- 21.Ishikita H., Saenger W., Knapp E.W. How photosynthetic reaction centers control oxidation power in chlorophyll pairs P680, P700, and P870. Proc. Natl. Acad. Sci. USA. 2006;103:9855–9860. doi: 10.1073/pnas.0601446103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishikita H., Biesiadka J., Knapp E.W. Cationic state of accessory chlorophyll and electron transfer through pheophytin to plastoquinone in photosystem II. Angew. Chem. Int. Ed. Engl. 2006;45:1964–1965. doi: 10.1002/anie.200503804. [DOI] [PubMed] [Google Scholar]
- 23.Rabenstein B., Knapp E.W. Calculated pH-dependent population and protonation of carbon-monoxy-myoglobin conformers. Biophys. J. 2001;80:1141–1150. doi: 10.1016/S0006-3495(01)76091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schrödinger . Schrödinger, LLC; New York, NY: 2010. QSITE, v. 5.6. [Google Scholar]
- 25.Plato M., Krauss N., Lubitz W. Molecular orbital study of the primary electron donor P700 of photosystem I based on a recent x-ray single crystal structure analysis. Chem. Phys. 2003;294:483–499. [Google Scholar]
- 26.Okubo T., Tomo T., Noguchi T. Perturbation of the structure of P680 and the charge distribution on its radical cation in isolated reaction center complexes of photosystem II as revealed by Fourier transform infrared spectroscopy. Biochemistry. 2007;46:4390–4397. doi: 10.1021/bi700157n. [DOI] [PubMed] [Google Scholar]
- 27.Witt H., Schlodder E., Lubitz W. Hydrogen bonding to P700: site-directed mutagenesis of threonine A739 of photosystem I in Chlamydomonas reinhardtii. Biochemistry. 2002;41:8557–8569. doi: 10.1021/bi025822i. [DOI] [PubMed] [Google Scholar]
- 28.Li Y., Lucas M.-G., Redding K. Mutation of the putative hydrogen-bond donor to P700 of photosystem I. Biochemistry. 2004;43:12634–12647. doi: 10.1021/bi036329p. [DOI] [PubMed] [Google Scholar]
- 29.Hastings G., Ramesh V.M., Webber A. Primary donor photo-oxidation in photosystem I: a reevaluation of (P700+-P700) Fourier transform infrared difference spectra. Biochemistry. 2001;40:12943–12949. doi: 10.1021/bi0155753. [DOI] [PubMed] [Google Scholar]
- 30.Nakamura A., Suzawa T., Watanabe T. Species dependence of the redox potential of the primary electron donor P700 in photosystem I of oxygenic photosynthetic organisms revealed by spectroelectrochemistry. Plant Cell Physiol. 2011;52:815–823. doi: 10.1093/pcp/pcr034. [DOI] [PubMed] [Google Scholar]
- 31.Higgins D.G., Thompson J.D., Gibson T.J. Using CLUSTAL for multiple sequence alignments. Methods Enzymol. 1996;266:383–402. doi: 10.1016/s0076-6879(96)66024-8. [DOI] [PubMed] [Google Scholar]
- 32.Ishikita H., Loll B., Knapp E.W. Tuning electron transfer by ester-group of chlorophylls in bacterial photosynthetic reaction center. FEBS Lett. 2005;579:712–716. doi: 10.1016/j.febslet.2004.12.049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.