

Abstract
The nucleosome complex of DNA wrapped around a histone protein octamer organizes the genome of eukaryotes and regulates the access of protein factors to the DNA. We performed molecular dynamics simulations of the nucleosome in explicit water to study the dynamics of its histone-DNA interactions. A high-resolution histone-DNA interaction map was derived that revealed a five-nucleotide periodicity, in which the two DNA strands of the double helix made alternating contacts. On the 100-ns timescale, the histone tails mostly maintained their initial positions relative to the DNA, and the spontaneous unwrapping of DNA was limited to 1–2 basepairs. In steered molecular dynamics simulations, external forces were applied to the linker DNA to investigate the unwrapping pathway of the nucleosomal DNA. In comparison with a nucleosome without the unstructured N-terminal histone tails, the following findings were obtained: 1), Two main barriers during unwrapping were identified at DNA position ±70 and ±45 basepairs relative to the central DNA basepair at the dyad axis. 2), DNA interactions of the histone H3 N-terminus and the histone H2A C-terminus opposed the initiation of unwrapping. 3), The N-terminal tails of H2A, H2B, and H4 counteracted the unwrapping process at later stages and were essential determinants of nucleosome dynamics. Our detailed analysis of DNA-histone interactions revealed molecular mechanisms for modulating access to nucleosomal DNA via conformational rearrangements of its structure.
Introduction
The nucleosome is the fundamental unit of chromatin in eukaryotes (1). It contains two copies each of histone proteins H2A, H2B, H3, and H4, and 146/147 basepairs (bp) of DNA (1,2). The DNA stably contacts the surface of the histone protein octamer core in a left-handed superhelix of almost two turns. Histone proteins consist of a globular part formed by three well-structured α-helices and the histone tails. These are long protruding N-terminal or, in the case of histone H2A, C-terminal extensions that lack secondary structure and adopt variable conformations. Removal of the histone tails leads to some increase of nucleosome flexibility and affects the binding of other proteins to the nucleosome and/or its associated DNA (3–5). The interactions of the unstructured histone N-terminal tails are modulated by posttranslational modifications like acetylation, methylation, and phosphorylation at numerous sites mostly in H3 and H4. These are set or removed in a dynamic manner by specific enzymes (6,7). Histone tails can serve as binding sites for protein domains that specifically interact with the posttranslationally modified histone state. In addition, acetylation of histone lysines has a direct effect on the stability of the nucleosome core particle, and on its higher-order interactions because the positively charged lysine is neutralized in the acetylated state (8).
The dynamics of the nucleosome in terms of spontaneous DNA unwrapping or breathing were studied by various approaches. It was investigated both experimentally and theoretically how proteins like restriction enzymes or RNA polymerases can access nucleosomal DNA and depend on the spontaneous unwrapping of DNA (9–11). The transition between the closed and partially or fully unwrapped open state was dissected in a number of single-molecule studies (12–15). These yielded lifetimes of several seconds for the closed state that were interrupted by open periods of a few tenths of a second, in which up to 80 bp of nucleosomal DNA were exposed. Finally, the unwrapping of nucleosomal DNA is also a crucial part of the activity of a specific class of enzymes referred to as remodeling complexes. They mediate the ATP-dependent translocations of nucleosomes along the DNA via formation of a DNA loop that is propagated around the histone octamer to change the translational position of the nucleosome (16,17). Thus, understanding the energetics of the DNA interaction with the histone octamer is essential for dissecting cellular processes that control unwrapping of nucleosomal DNA and nucleosome positioning. The strength of the interaction between the DNA and histone protein can be directly investigated in force spectroscopy experiments (18–23).
To relate the experimentally observed nucleosome dynamics with its high-resolution structure, all-atom molecular dynamics (MD) studies are ideally suited (24–27). They allow for a detailed comparison of free DNA with DNA bound to the histone octamer, and an analysis in terms of dynamic DNA double helix features and superhelix configuration (24,26,28). MD studies of systems in the size of the nucleosome already require large computational resources for simulations of tens of nanoseconds. However, with respect to nucleosome dynamics, many processes occur at the microsecond to second timescale (13,15). By applying an external force in so-called steered molecular dynamics (SMD) simulations, the transition of the system into a certain state can be induced and compared to results from single-molecule force spectroscopy experiments (29–31). Here, we have performed all-atom MD and SMD simulations of a solvated nucleosome with and without the N-terminal histone tails to investigate the process of unwrapping DNA from the histone octamer. Our findings reveal details of the dynamic protein-DNA interactions at atomic resolution that govern the accessibility of nucleosomal DNA. This has a number of implications for the competitive binding of transcription factors to DNA sites occupied by a nucleosome.
Materials and Methods
MD and SMD simulations were based on the structure of nucleosome and linker DNA in the tetranucleosome crystal structure 1ZBB (32) that had undergone minimization and had been solvated and equilibrated for 2 ns or 50 ns (33) (see structure NUC, Fig. S1 A, in the Supporting Material). For simulations of the tailless nucleosome structure the N-terminal tails were removed from the 50-ns equilibrated complete nucleosome structure so that only amino acids 41–135 (H3), 25–102 (H4), 17–128 (H2A), and 32–122 (H2B) remained (structure NUCΔtail). DNA sequence effects were studied in comparison to an additional nucleosome crystal structure with an adenine dA16·dT16 insert (based on coordinates PDB 2fj7, and termed structure NUC_A16) In addition, a structure referred to as NUC_loop was evaluated that had a central DNA loop of 20 bp (see Fig. S1 B). Simulations were conducted with the NAMD 2.6 (34) and AMBER 10.0 (35) software packages. Details of the MD and SMD simulations and data analysis are described in the Methods section in the Supporting Material.
Results
DNA-histone interactions occur with a five-nucleotide periodicity
To investigate the interactions between DNA and histone proteins at high resolution, we conducted two MD simulations over a time of ∼20 ns in explicit water at physiological concentrations of monovalent ions. In Fig. 1 A, the corresponding time-dependent DNA-histone interactions are shown. Additionally, the dynamics of DNA-histone contacts during a 120-ns MD simulation of a nucleosome structure with an internal DNA loop were investigated (see Fig. S2). For the NUC structure, the Fourier spectrum of the time-averaged interactions of the histone octamer and the DNA sequence was calculated (Fig. 1 B). It revealed a peak at 0.19/bp, corresponding to a ∼5.1-bp periodicity (Fig. 1 C). When evaluating the interactions of the two DNA strands of the double-helix separately, a ∼10-nucleotide periodicity for each DNA strand with a ∼5.5-nucleotide phase shift between the strands was apparent. Thus, contacts of the histone protein core with nucleotides occur with alternating DNA strands. To study effects of the DNA sequence on the observed periodicity, the Fourier spectra of a nucleosome with an insert of dA16 at bp −35 was also calculated. This yielded the same results for the interaction periodicity, albeit with weaker histone-DNA interactions in the region of the adenine tract (see Fig. S3).
Figure 1.
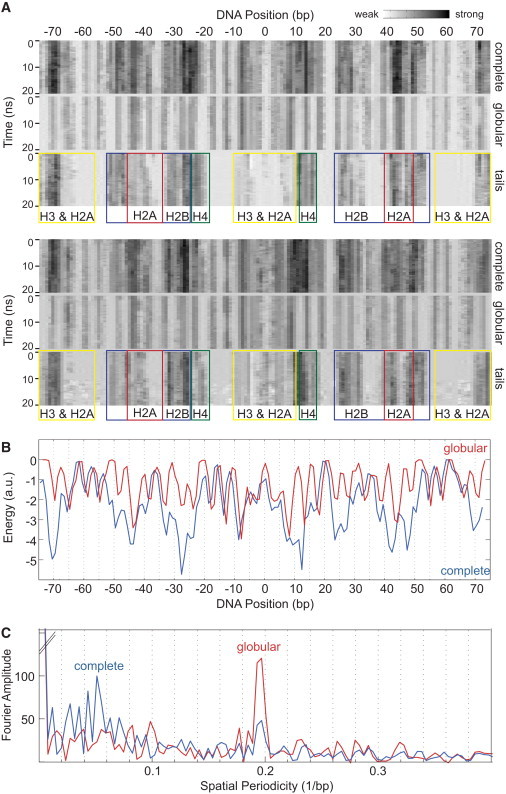
High resolution DNA-histone interaction maps. (A) Temporal evolution of the histone-DNA electrostatic and van der Waals interactions in a nucleosome calculated for two 20-ns MD simulation trajectories. The interaction strength increases (from white to black). The DNA position numbering refers to the central base pair 0 at the nucleosomal dyad axis. The three panels in each of the two MD simulations differ with respect to the histone residues that were taken into account for calculating interaction energies with the DNA. They show the complete nucleosome (complete, top panel), the interactions without the N-terminal tails (globular, middle panel), and the isolated tail contributions (tails, bottom panel). The colored boxes indicate the specific histones that are involved in the DNA interactions in this region (H3 and C-terminal H2A, yellow; N-terminal H2A, red; H2B, blue; and H4, green). The first MD simulation shown (top panel) was conducted with a start configuration of a nucleosome that was derived from the crystal structure (32) but had been already equilibrated for 50 ns (33). The nucleosome start structure of the second MD simulation shown (bottom panel) was derived from the same crystal structure, minimized, and equilibrated for only 2 ns. (B) The temporal mean energy values of histone-DNA interactions for the entire MD simulation trajectory are plotted with respect to the DNA position for the complete (blue line) and the tailless (red line) nucleosome. Relative to the nucleosome dyad axis, the red line was much more symmetric than the blue one, demonstrating that the histone tails have a strong influence on the interaction pattern and the interaction symmetry within the nucleosome. (C) Fourier spectra for complete and tailless nucleosome interaction maps calculated with the data from panel B. When the unstructured histone tails were excluded, a pronounced frequency peak appeared at 0.19/bp that corresponds to a 5.1-bp spatial periodicity.
Histone tail interactions stabilize the nucleosome and can cause an asymmetric DNA interaction pattern
The images in Fig. 1 A illustrate the temporal evolution of interactions between DNA and histone proteins. From a comparison of the complete nucleosome with a nucleosome without the N-terminal histone tails, the contribution of the tails can be extracted. As expected, the interactions between DNA and the globular parts of the histone octamer were symmetric relative to the dyad axis. However, due to the variability of contacts made by the flexible tails, the interaction pattern became asymmetric. Thus, factors that direct the histone tails to certain DNA contact regions could induce a preference for unwrapping from one linker DNA side. It appears likely that the tail-DNA interaction pattern will become symmetric on longer timescales due to sampling over the different conformations. However, on the 100-ns timescale, no large rearrangements of the histone tails were observed in the MD simulation (Fig. 1 A, and see Fig. S2). The only exception was a relocation of the H2A C-terminus that is depicted in Fig. S4. The spontaneous opening of the DNA-histone interactions at the entry-exit site of the DNA in the nucleosome as well as at the start/end of the looped region comprised only 1–2 bp during the observation period (Fig. 1 A, and see Fig. S2, Fig. S5, Fig. S6, and Movie S1 in the Supporting Material).
Unwrapping nucleosomal DNA by applying an external force reveals five phases of DNA unwrapping with two main energy barriers at ±70 bp and ±45 bp
To investigate the unwrapping of DNA from the histone octamer, SMD simulations were conducted. One end of the nucleosomal DNA was fixed, and a harmonic potential moving at constant velocity was applied to the opposite DNA end (Fig. 2). Due to the limitations of the available computational resources, the SMD simulations had to be conducted at a speed that was several orders of magnitude higher than those used in the experimental setups that operate at ∼0.1 μm/s. Nevertheless, as discussed in the context of other SMD studies, important information on the most relevant energy barriers as well as the reaction pathway can be derived despite this limitation (29,36–38). The application of the SMD approach to dissect the DNA unwrapping process from the nucleosome identified five characteristic phases and two main energy barriers (Figs. 2 and 3, and see Movie S2 and Movie S3).
Figure 2.
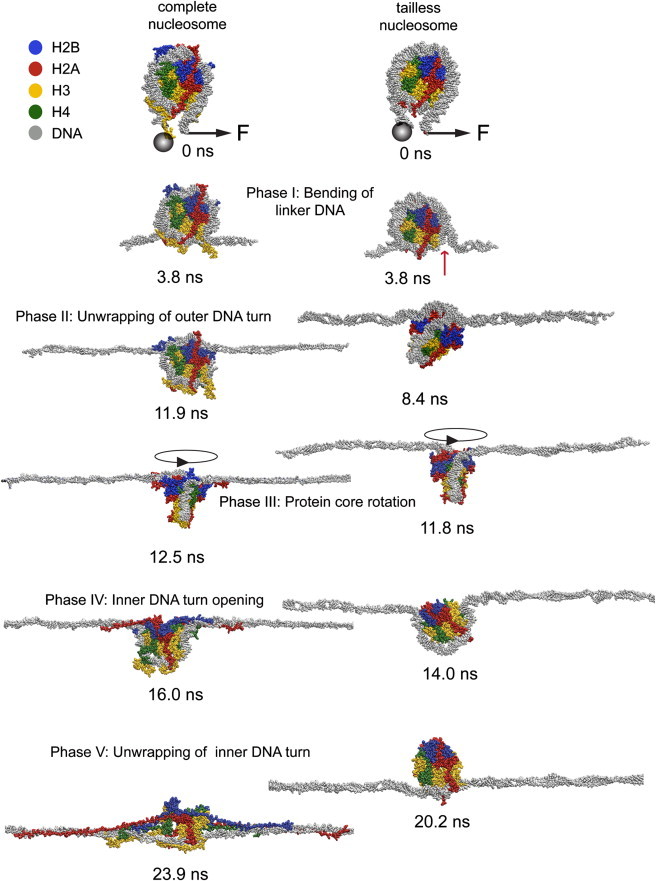
Intermediate structures formed during the DNA unwrapping process of a complete and a tailless nucleosome. The last nucleotide in the linker DNA on one side of the nucleosome was fixed (represented by the gray sphere), whereas the C1′ atom of the last nucleotide of the opposite linker DNA was moved along the indicated direction with an additional harmonic potential. Snapshots of the complete (left panel) and the tailless (right panel) nucleosomes at the indicated times of the SMD simulations are displayed. The complete trajectory is shown in Movie S2 and Movie S3. (Color code: H2B, blue; H2A, red; H3, yellow; H4, green; and DNA, gray.)
Figure 3.
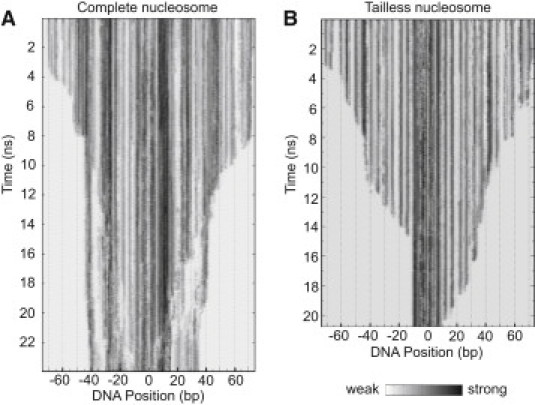
Temporal evolution of electrostatic and van der Waals interactions between DNA and histones during the unwrapping simulations. Changes of relative interaction strength from strong (black) to no-interaction (white) during the unwrapping simulations are shown. Contacts broke first between the histone protein residues and DNA bp closest to the DNA entry-exit site of the nucleosome. Regions with differences in disruption of interactions between complete and tailless nucleosomes were found around bp −18 to −25, −35 to −42, and 20 to 35 where the histone tails maintained contacts even with the completely unwrapped DNA. (A) Complete nucleosome. (B) Tailless NUCΔtail structure.
Phase I: bending of linker DNA
At the entry-exit site of the nucleosome, the DNA became bent into the direction of applied forces. Without breaking contacts with the histone residues, the nucleosome aligned and the DNA elongated by rearranging bases (tilting and untwisting). Interactions of the globular histones and the DNA at position ±70 bp were enhanced by binding of the C-terminal H2A and N-terminal H3 tails. These defined a first barrier toward unwrapping.
Phase II: unwrapping of the outer DNA turn
Upon further DNA unwrapping, the barrier at ±70 bp was broken and DNA segments were released in ∼5 bp steps until almost the entire outer DNA turn dissociated from the histone octamer core. The outer turn comprised ∼67 bp, i.e., the region from ±73 bp to ±40 bp.
Phase III: protein core rotation
To enable further DNA unwrapping, the protein core rotated around the dyad axis. The extension of the DNA ends resulted mostly from the rotation itself and from reorganization of already unwrapped DNA.
Phase IV: inner DNA turn opening
The DNA unwrapping process proceeded differently for the complete and tailless nucleosomes. For both structures, a second barrier for further unwrapping of DNA was apparent around bp position ±45. This was more pronounced for the NUC structure due to the DNA binding of H2A and H2B N-terminal tails. In addition, a partial disassembly of the histone core protein secondary structure occurred. It involved the disruption of interactions between the two H2A·H2B dimers due to changes of the relative location of α-helices in the histone fold and their partial unwinding. The latter process was apparent in both H2A histones in the first N-terminal α-helix (at ∼62 nm/∼75 nm extension, t = ∼13.4 ns/∼16.1 ns) as well as in the second (at ∼71 nm/∼78 nm extension, t = ∼15.1 ns/∼16.7 ns).
Both H2Bs displayed an opening of the angle between the two C-terminal α-helices at extension ∼92/∼113 nm and t = ∼19.5 ns/∼21.5 ns. This was more pronounced for the H2B histone that rearranged first, which also showed a subsequent widening of the angle between the first N-terminal α-helices (extension ∼96.5 nm, t = ∼21.4 ns). In addition, the H2A·H2B dimers were shifted slightly relative to the H3·H4 tetramer. The (H3·H4)2 complex conformation itself was much less affected. Only in one H3 histone the first N-α-helix unwound (extension ∼101 nm, t = ∼20.2 ns), whereas the other H3 and both H4s did not change their folding. For DNA unwrapping from the tailless nucleosome, no reorganization of the histone octamer structure was apparent. The opening of the inner DNA turn comprised the disruption of the DNA-core histone interactions at ±45 bp.
Phase V: unwrapping of inner DNA turn
DNA-core histone contacts were successively disrupted until the DNA was completely straightened along the direction of the applied force. Residual contacts between the globular histone domains and the stretched DNA remained only at the former dyad axis. In addition, some histone tail-DNA interactions persisted.
The N-terminal histone tails and the H2A C-terminal tail counteract DNA dissociation from the nucleosome
The H3 N-terminus and the H2A C-terminus contributed to the first barrier toward nucleosome unwrapping because they interacted with the DNA at the entry-exit site (see Fig. S4 and Fig. S7). Both tails were located in the minor groove and bind DNA via a variety of electrostatic and van der Waals interactions as well as hydrogen bonds. After linker DNA bending to ∼90° relative to the nucleosomal DNA, the structure extended due to DNA stretching with partial DNA unstacking. Because the latter process is energetically unfavorable, the H3 and H2A tails must provide a significant contribution to the histone DNA interaction that prevented DNA unwrapping during a ∼4-ns time period at the ±70 bp boundary (Fig. 3 A). At ±40 bp, the N-terminal H2A and H2B tails stabilized the inner DNA turn.
In response to the applied potential, the N-terminal α-helices of H2A started to break open after ∼16 ns. This transition allowed it to maintain DNA contacts with the H2A and H2B tails (Fig. 2, Fig. 3 A, and Movie S2). Interestingly, the histone tails remained associated with the DNA during the entire 24-ns simulation period up to the point when the DNA was fully unwound from the histone octamer core. Their flexible conformation accommodated the significant DNA extension in the SMD experiments. The above contributions of the histone tails can be identified from the behavior of the NUCΔtail structure in the SMD simulations (Fig. 2, Fig. 3 B, and Movie S3), which displayed the following differences as compared to the complete nucleosome: 1), The first breakage of histone-DNA contacts at −70 bp occurred before 3.8 ns (Fig. 2, indicated by the arrow). 2), After breaking the contacts at ±70, the DNA dissociated more continuously from the protein core. 3), The second energy barrier toward unwrapping at ±40 bp was much less pronounced. 4), DNA-histone interactions remained only over a small region of ∼20 bp in the unwrapped structure (Fig. 3 B).
Differences in the stability of the inner and outer DNA turn are partly due to additional histone tail-DNA interactions when the outer turn is unwrapped
In experimental studies, an apparent higher stability of the inner DNA turn has been reported (19,22,23,39). As proposed previously, this could include a contribution of the histone tails (39,40): While the tails are bound to both DNA turns in a complete nucleosome, they might reposition to the inner turn as soon as the outer turn is unwrapped. We calculated the DNA interactions of all histone tails separately over the unwrapping trajectory. An ∼20% fraction of histone tail interactions rearranged from the outer to the inner DNA turn during unwrapping of the outer DNA turn at ∼12 ns, resulting in a moderate stabilization of the inner DNA turn (Fig. S8). The most relevant histone tail rearrangements involved in this process took place for the N-terminal tails of H2A, H2B, and H4. For example, after ∼15 ns, the DNA interaction of the N-terminal tail of one histone H2A (top-left image in Fig. S8) increased as indicated by a change of the color coding to dark blue and relocated from a position at ∼+42 bp toward +30 bp. In addition, both H2B N-termini displayed significant movements in the >12-ns time regime, and to a somewhat lesser extent the same was observed for the H4 tails.
In addition to changes of the histone tail location, the two DNA turns might repel each other while being wrapped around the histone proteins, and the inner turn might be stabilized by a reduced repulsion when the outer DNA turn is unwrapped (39,40). Our analysis of the DNA-DNA strand repulsion revealed a nonhomogeneous relatively low repulsion of 1–10% of the total calculated interaction energy at only a few bp positions (see Fig. S9). If averaged over the entire nucleosome, this value was reduced to a minor fraction of the total DNA-histone interaction energy. Accordingly, the effect of DNA repulsion on destabilizing the outer DNA turn appears to be negligible.
The force extension curve of unwrapping nucleosomal DNA results from a complex overlay of stretching and disruption events
Fig. 4 shows force-extension curves computed from the unwrapping simulations of the complete and tailless nucleosomes. Some of the most prominent peaks in the simulation trajectory of the NUC structure can be assigned to specific intermediates. The peaks at an extension of 20 nm, 37 nm, or 54 nm clearly originated from breaking DNA-histone contacts. The subsequent relaxation event at the 57–66 nm extension reflected the rotation of the protein core. The positive slope at 66–78 nm extension was due to the breaking of interactions between histone-dimers. The large force increase at extension ∼85 nm resulted from numerous single disruption events that arose from a collapse of secondary structure interactions between two H2A histone and the N-terminal α-helices of one H2B histone. The final binding site opening event within the outer turn in the simulations correlated with the first disruption event observed experimentally (23) (see Fig. S10).
Figure 4.
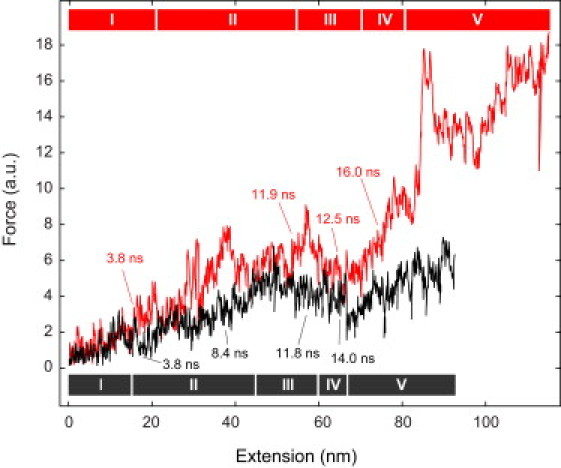
Virtual force spectroscopy of nucleosome unwrapping. From the SMD simulations, force spectroscopy curves were computed. The assignment of the five unwrapping phases and intermediates according to the indicated simulation times corresponds to that given in Fig. 2. The complete nucleosome structure (red line) is shown in comparison to the NUCΔtail structure (black line).
An unexpected contribution to the force extension curve was the persistence of histone tail contacts with the inner DNA turn throughout the stretching simulations. These induced a partial resolution of secondary structure and opening of the histone octamer complex. Because this process was overlaid with breaking of DNA-histone contacts, the identification of distinct intermediates of the unwrapping process becomes increasingly difficult (see Fig. S10). This contribution has, so far, not been considered in the interpretation of force spectroscopy experiments that do not provide sufficient resolution to dissect structural transformations on the nanometer lengthscale. In the virtual force spectroscopy curve of the NUCΔtail structure, the peaks that preceded the breakage of DNA-histone interactions were less pronounced. The unwrapping of the outer DNA turn occurred significantly faster, consistent with the lacking contributions from the H3 N-terminus and the H2A C-terminus. A corresponding effect was observed at the ±40 bp energy barrier that was largely reduced in the absence of histone tails.
Discussion
The investigation of biologically relevant large-scale motions of the nucleosome is a challenging task because molecular dynamics simulations are currently limited to the nanosecond timescale (26,28,29). In contrast, experimental data suggest that processes such as nucleosome breathing, linker DNA opening, and sliding of nucleosomes occur spontaneously at the microsecond timescale and beyond (13,14). One approach to close this gap between experiments and theory is the use of coarse-grained models (33,41,42). However, these require some a priori knowledge of the system features to devise a description that preserves the characteristic properties to be studied. Alternatively, the technique of steered molecular dynamics can be applied to direct motions via an externally applied force, so that the associated conformational rearrangements can be investigated within a time period that is accessible to MD simulations.
So far, SMD has been used primarily to visualize force-induced protein unfolding events of small systems with roughly 200 amino acids (29,43–45). Larger systems investigated with SMD approaches focused mostly on local structural changes by translocating a relatively small structure with respect to a bigger macromolecule (46–49). For example, in a study by Ishida and Hayward (49), a small peptide was pulled to move through the ribosome. It is noted that the magnitude and fluctuations of the forces applied in our constant-velocity SMD simulations depended on the parameters used for stretching velocity and spring constant. As discussed previously, a limitation of the SMD simulations is it that the currently available computational resources impose the use of stretching velocities that are orders-of-magnitude faster than those used in the experimentally conducted force spectroscopy experiments (29,36–38). Accordingly, it is expected that random structural fluctuations on a larger timescale than that observed here could largely facilitate the unwrapping process at lower stretching speed. Furthermore, force peak values larger than those recorded in experiments, and a loss of details on conformational changes that occur during the force-induced transitions, are to be expected. Nevertheless, the currently available cases of SMD simulations in comparison with experimental data demonstrate that valuable information can be obtained that was later validated in experimental studies (29).
Here, we applied a combination of MD and SMD to dissect the details of DNA-histone protein interactions, and to investigate the process of unwrapping the DNA from the histone octamer core. Our MD and SMD simulations revealed nucleosome dynamics for all-atom structures to identify the biologically relevant structural transitions at a resolution that cannot be reached in the experimental studies. The molecular dynamics of the nucleosome in an approximately physiological aqueous environment provided a DNA-histone interaction map at bp resolution (Fig. 1 A and see Fig. S2). From the analysis, a five-nucleotide periodicity pattern emerged that originated from contacts alternating between the two DNA strands of the double-helix. This is in agreement with conclusions from recent force spectroscopy experiments (22). Previously published histone-DNA interaction maps based on the crystal structure of the nucleosome (1,2) or on stretching experiments (22,23,50) identified a ∼10-bp periodicity of interactions of the DNA with the histone proteins. These were assigned to 14 main interaction sites at regions where the minor groove faces inwards. Our MD analysis indicates that each of these interaction sites can be considered as comprising two ∼5-bp separated contacts between each of the two individual DNA strands and the histone octamer.
The main origin of variations between different MD trajectories arose from the interactions of the histone tails with the DNA. Due to their flexibility they can bind to DNA in numerous conformations. This is reflected by the lack of defined structural information on their location in the available crystal structures. In addition, it is clear from the asymmetric and different location of the tails observed in our simulations that various positions are equally possible. At this point of time, we are unable to evaluate the effect of the conformational flexibility of the tails in the simulations in a systematic manner due to the high demands in computation time.
Despite these limitations, two important conclusions can be drawn from the MD simulations: Except for the relocation of the H2A C-terminus (see Fig. S4) the histone N-terminal tails are relatively stably attached to a certain region of the DNA in the 100-ns time regime. Furthermore, on this timescale, the degree of spontaneous disruption of DNA-histone interactions at the entry-exit site of the DNA in the nucleosome as well as at the start/end of the looped region in the NUC and NUC_loop structures was limited to 1–2 bp. This is in agreement with experimental studies, concluding that this almost completely wrapped state has a lifetime of several seconds that are interrupted by open periods of a few tenths of a second, in which up to 80 bp of nucleosomal DNA are spontaneously exposed (13–15). For this process, the energy barrier at ±70 bp, which is stabilized by interactions of the H3 tail and the C-terminus of H2A, has to be broken.
The comparative SMD analysis conducted here revealed a large influence of the unstructured histone tails on the stability and mobility of nucleosomes (Figs. 2–4 and see Movie S2 and Movie S3). This is supported by previous MD simulations of nucleosome dynamics (28) and the MD simulations presented here. Our SMD analysis is in good agreement with force spectroscopy experiments (21). These show that the amount of outer-turn DNA wrapping was reduced by ∼60% if the histone tails were removed. The primary contribution to this effect was from the H3 and H4 N-terminal tails. This agrees with our conclusion on the H3 tail interactions as opposing unwrapping of outer turn DNA. In addition, the removal of the H2A/H2B tails reduced histone-DNA interactions at ∼±36 bp in the experiments, which is in line with our findings on the contribution of the H2A tail in the SMD simulations. In the cell, the H3 and H4 interactions are subject to regulation via acetylation or methylation of lysine residues.
In the in vitro experiments, the acetylation of histones H3 and H4 is clearly apparent in the experimental force spectroscopy curves as a factor that weakens histone-DNA interactions (21). Furthermore, numerous proteins recognize specifically modified histone tails as binding signals and would disrupt the tail-DNA interactions in their bound state. These include, for example, heterochromatin protein HP1 binding the H3 tail via its chromodomain (51), the interactions of the PCAF, Brd2, Brd4, and BRDT bromodomains with acetylated H4 tails (52), and the binding of the H2A C-terminus to linker histone H1 (5). Our SMD studies revealed large contributions from the N-termini of H2A, H2B, H3, and H4 and the C-terminal histone H2A tail to DNA binding within the nucleosome. In particular, the N-terminus of H3 and the C-terminus of H2A counteracted the initiation of DNA unwrapping by stabilizing DNA interactions at positions ∼±70 bp (Figs. 2 and 3 A). Binding of these tails by the protein factors mentioned above would prevent their DNA association and could represent a mechanism to facilitate unwrapping of a certain nucleosome. The second barrier opposing unwrapping of nucleosomal DNA was mapped in our simulations to the ±45 bp positions.
These two regions were identified also in experimental studies as sites of strong DNA-histone interactions (22,23). A direct comparison of the simulated force-extension curves with the data from Mihardja et al. (23) is shown in Fig. S10 and depicts two distinct transitions in both the simulated and experimental curves: The removal of the outer DNA turn is reflected as a reduction in the force increase. This state has been referred to as the “unwrapping event of the outer DNA turn” in the experimental analysis (23), and a good agreement between the two curves is apparent. A second significant decrease of force was observed in our analysis that was assigned to the “unwrapping event of the inner DNA turn” in the experiments (23). However, although this part of nucleosomal DNA dissociation occurred at about the same DNA extension in both the experiments and simulations, it did not represent a defined unwrapping transition in the simulations. The SMD trajectories revealed numerous local dissociation events, DNA bending, and conformational rearrangements that took place over a relatively large force regime as opposed to a sudden release of the outer DNA turn. In general, an unwrapping step might not be apparent in the experimental force-extension curves due to the complex superposition of several processes: 1), linker DNA bends into the direction of the applied force, 2), DNA stretches, 3), DNA-histone contacts open, 4), the histone octamer rotates, and 5), its secondary and tertiary structure partially disassembles. We conclude that the assignment of peaks to a single conformational transition in the experimental force-extension curve is fraught with difficulties. In contrast, the trajectory of the SMD simulations revealed all details of the unwrapping process. From the complex response of the nucleosome conformation to the applied force, we identified a sequence of five main phases of the unwrapping process that are depicted in Fig. 2: Phase 1), Bending of linker DNA. Phase 2), Unwrapping of the outer DNA turn. Phase 3), Protein core rotation. Phase 4), Inner turn opening with partial breakage of the octamer structure if histone tails are present. Phase 5), The unwrapping of the inner DNA turn. It is noted that within each of these five phases, the flexibility of both the DNA and histones is considerable so that these regimes partly overlap.
Our MD and SMD simulations elucidate how histone-DNA interactions determine access to the DNA sequence information. The results are relevant in the context of modeling competitive transcription factor binding and the conformational properties of the nucleosome chain. The energy potential of the DNA unwrapping process was described in a previous study by a spool (representing the histone octamer) from which the adhesive DNA tube was removed when tension was applied (39,40). This coarse-grained model explained several features of the unwrapping process, and was applied to improve force spectroscopy simulations of nucleosome chains (53). However, it lacks details on the shape of the nonhomogeneous histone-DNA potential along the nucleosome surface revealed here. In particular, a histone tail-dependent widening of the histone core during the unwrapping process has not been considered previously. This opening occurred mostly via conformational changes in the two H2A·H2B dimers, which is consistent with previous experimental findings. In these a hexasome particle was identified as an intermediate during nucleosome (dis)assembly that lacked one H2A·H2B dimer (54). Thus, the H2A·H2B dimers appear to be the least stable part of the nucleosome. Their conformational flexibility could make a significant contribution to the histone-DNA interaction dynamics.
In summary, the MD and SMD simulations conducted here identify several features of the mechanism by which DNA unwraps from the histone octamer core that need to be considered in coarse-grained descriptions of the nucleosome. This leads to integrative modeling approaches as for example applied in a recent study on the competitive binding of transcription factors to nucleosomal DNA (11). Furthermore, it is anticipated that SMD simulations of the nucleosome along the lines described here will provide insights into the molecular mechanisms by which chromatin remodeling complexes translocate nucleosomes along the DNA.
Acknowledgments
We are grateful to Karin Voltz for coordinates of an equilibrated nucleosome structure, Robert Schöpflin for critical reading of the manuscript, and Marc Kirchner and Stefan Fischer for valuable discussions.
This work was supported by Deutsche Forschungsgemeinschaft grant Ri 1283/8-1 and within project EpiGenSys by the Federal Ministry of Education and Research (BMBF), partners of the ERASysBio+ initiative supported under the European Union ERA-NET Plus scheme in FP7. Computations were conducted within project mvb00007 of the North German Supercomputing Alliance (HLRN) and BMBF grant 01IG07015G (Services@MediGRID).
Supporting Material
References
- 1.Luger K., Mäder A.W., Richmond T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Davey C.A., Sargent D.F., Richmond T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 3.Polach K.J., Lowary P.T., Widom J. Effects of core histone tail domains on the equilibrium constants for dynamic DNA site accessibility in nucleosomes. J. Mol. Biol. 2000;298:211–223. doi: 10.1006/jmbi.2000.3644. [DOI] [PubMed] [Google Scholar]
- 4.Vitolo J.M., Thiriet C., Hayes J.J. The H3-H4 N-terminal tail domains are the primary mediators of transcription factor IIIA access to 5S DNA within a nucleosome. Mol. Cell. Biol. 2000;20:2167–2175. doi: 10.1128/mcb.20.6.2167-2175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogler C., Huber C., Schneider R. Histone H2A C-terminus regulates chromatin dynamics, remodeling, and histone H1 binding. PLoS Genet. 2010;6:e1001234. doi: 10.1371/journal.pgen.1001234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campos E.I., Reinberg D. Histones: annotating chromatin. Annu. Rev. Genet. 2009;43:559–599. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 7.Lee B.M., Mahadevan L.C. Stability of histone modifications across mammalian genomes: implications for ‘epigenetic’ marking. J. Cell. Biochem. 2009;108:22–34. doi: 10.1002/jcb.22250. [DOI] [PubMed] [Google Scholar]
- 8.Wang X., Hayes J.J. Physical methods used to study core histone tail structures and interactions in solution. Biochem. Cell Biol. 2006;84:578–588. doi: 10.1139/o06-076. [DOI] [PubMed] [Google Scholar]
- 9.Anderson J.D., Widom J. Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol. 2000;296:979–987. doi: 10.1006/jmbi.2000.3531. [DOI] [PubMed] [Google Scholar]
- 10.Hodges C., Bintu L., Bustamante C. Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II. Science. 2009;325:626–628. doi: 10.1126/science.1172926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teif V.B., Ettig R., Rippe K. A lattice model for transcription factor access to nucleosomal DNA. Biophys. J. 2010;99:2597–2607. doi: 10.1016/j.bpj.2010.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park Y.J., Dyer P.N., Luger K. A new fluorescence resonance energy transfer approach demonstrates that the histone variant H2AZ stabilizes the histone octamer within the nucleosome. J. Biol. Chem. 2004;279:24274–24282. doi: 10.1074/jbc.M313152200. [DOI] [PubMed] [Google Scholar]
- 13.Li G., Levitus M., Widom J. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 2005;12:46–53. doi: 10.1038/nsmb869. [DOI] [PubMed] [Google Scholar]
- 14.Tomschik M., Zheng H., Leuba S.H. Fast, long-range, reversible conformational fluctuations in nucleosomes revealed by single-pair fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA. 2005;102:3278–3283. doi: 10.1073/pnas.0500189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koopmans W.J., Buning R., van Noort J. spFRET using alternating excitation and FCS reveals progressive DNA unwrapping in nucleosomes. Biophys. J. 2009;97:195–204. doi: 10.1016/j.bpj.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strohner R., Wachsmuth M., Längst G. A ‘loop recapture’ mechanism for ACF-dependent nucleosome remodeling. Nat. Struct. Mol. Biol. 2005;12:683–690. doi: 10.1038/nsmb966. [DOI] [PubMed] [Google Scholar]
- 17.Bowman G.D. Mechanisms of ATP-dependent nucleosome sliding. Curr. Opin. Struct. Biol. 2010;20:73–81. doi: 10.1016/j.sbi.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennink M.L., Leuba S.H., Greve J. Unfolding individual nucleosomes by stretching single chromatin fibers with optical tweezers. Nat. Struct. Biol. 2001;8:606–610. doi: 10.1038/89646. [DOI] [PubMed] [Google Scholar]
- 19.Brower-Toland B.D., Smith C.L., Wang M.D. Mechanical disruption of individual nucleosomes reveals a reversible multistage release of DNA. Proc. Natl. Acad. Sci. USA. 2002;99:1960–1965. doi: 10.1073/pnas.022638399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pope L.H., Bennink M.L., Marko J.F. Single chromatin fiber stretching reveals physically distinct populations of disassembly events. Biophys. J. 2005;88:3572–3583. doi: 10.1529/biophysj.104.053074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brower-Toland B., Wacker D.A., Wang M.D. Specific contributions of histone tails and their acetylation to the mechanical stability of nucleosomes. J. Mol. Biol. 2005;346:135–146. doi: 10.1016/j.jmb.2004.11.056. [DOI] [PubMed] [Google Scholar]
- 22.Hall M.A., Shundrovsky A., Wang M.D. High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nat. Struct. Mol. Biol. 2009;16:124–129. doi: 10.1038/nsmb.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mihardja S., Spakowitz A.J., Bustamante C. Effect of force on mononucleosomal dynamics. Proc. Natl. Acad. Sci. USA. 2006;103:15871–15876. doi: 10.1073/pnas.0607526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bishop T.C. Molecular dynamics simulations of a nucleosome and free DNA. J. Biomol. Struct. Dyn. 2005;22:673–686. doi: 10.1080/07391102.2005.10507034. [DOI] [PubMed] [Google Scholar]
- 25.Korolev N., Nordenskiöld L. H4 histone tail mediated DNA-DNA interaction and effects on DNA structure, flexibility, and counterion binding: a molecular dynamics study. Biopolymers. 2007;86:409–423. doi: 10.1002/bip.20749. [DOI] [PubMed] [Google Scholar]
- 26.Bishop T.C. Geometry of the nucleosomal DNA superhelix. Biophys. J. 2008;95:1007–1017. doi: 10.1529/biophysj.107.122853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruscio J.Z., Onufriev A. A computational study of nucleosomal DNA flexibility. Biophys. J. 2006;91:4121–4132. doi: 10.1529/biophysj.106.082099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roccatano D., Barthel A., Zacharias M. Structural flexibility of the nucleosome core particle at atomic resolution studied by molecular dynamics simulation. Biopolymers. 2007;85:407–421. doi: 10.1002/bip.20690. [DOI] [PubMed] [Google Scholar]
- 29.Sotomayor M., Schulten K. Single-molecule experiments in vitro and in silico. Science. 2007;316:1144–1148. doi: 10.1126/science.1137591. [DOI] [PubMed] [Google Scholar]
- 30.Li P.-C., Makarov D.E. Simulation of the mechanical unfolding of ubiquitin: probing different unfolding reaction coordinates by changing the pulling geometry. J. Chem. Phys. 2004;121:4826–4832. doi: 10.1063/1.1778152. [DOI] [PubMed] [Google Scholar]
- 31.Gräter F., Shen J., Grubmüller H. Mechanically induced titin kinase activation studied by force-probe molecular dynamics simulations. Biophys. J. 2005;88:790–804. doi: 10.1529/biophysj.104.052423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schalch T., Duda S., Richmond T.J. X-ray structure of a tetranucleosome and its implications for the chromatin fiber. Nature. 2005;436:138–141. doi: 10.1038/nature03686. [DOI] [PubMed] [Google Scholar]
- 33.Voltz K., Trylska J., Smith J. Coarse-grained force field for the nucleosome from self-consistent multiscaling. J. Comput. Chem. 2008;29:1429–1439. doi: 10.1002/jcc.20902. [DOI] [PubMed] [Google Scholar]
- 34.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Case D.A., Darden T.A., Kollman P.A. University of California; San Francisco, CA: 2008. AMBER 10. [Google Scholar]
- 36.Harris S. The physics of DNA stretching. Contemp. Phys. 2003;45:11–30. [Google Scholar]
- 37.Balsera M., Stepaniants S., Schulten K. Reconstructing potential energy functions from simulated force-induced unbinding processes. Biophys. J. 1997;73:1281–1287. doi: 10.1016/S0006-3495(97)78161-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gullingsrud J., Braun R., Schulten K. Reconstructing potentials of mean force through time series analysis of steered molecular dynamics simulations. J. Comput. Phys. 1999;151:190–211. [Google Scholar]
- 39.Kulić I.M., Schiessel H. DNA spools under tension. Phys. Rev. Lett. 2004;92:228101. doi: 10.1103/PhysRevLett.92.228101. [DOI] [PubMed] [Google Scholar]
- 40.Kulic I.M., Schiessel H. Opening and closing DNA: theories on the nucleosome. In: Dias R.S., Lindman B., editors. DNA Interactions with Polymers and Surfactants. Wiley; London, UK: 2008. pp. 173–208. [Google Scholar]
- 41.Sharma S., Ding F., Dokholyan N.V. Multiscale modeling of nucleosome dynamics. Biophys. J. 2007;92:1457–1470. doi: 10.1529/biophysj.106.094805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arya G., Schlick T. Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model. Proc. Natl. Acad. Sci. USA. 2006;103:16236–16241. doi: 10.1073/pnas.0604817103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krammer A., Lu H., Vogel V. Forced unfolding of the fibronectin type III module reveals a tensile molecular recognition switch. Proc. Natl. Acad. Sci. USA. 1999;96:1351–1356. doi: 10.1073/pnas.96.4.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Genchev G.Z., Källberg M., Lu H. Mechanical signaling on the single protein level studied using steered molecular dynamics. Cell Biochem. Biophys. 2009;55:141–152. doi: 10.1007/s12013-009-9064-5. [DOI] [PubMed] [Google Scholar]
- 45.Gao M., Craig D., Schulten K. Identifying unfolding intermediates of FN-III(10) by steered molecular dynamics. J. Mol. Biol. 2002;323:939–950. doi: 10.1016/s0022-2836(02)01001-x. [DOI] [PubMed] [Google Scholar]
- 46.Cuendet M.A., Michielin O. Protein-protein interaction investigated by steered molecular dynamics: the TCR-pMHC complex. Biophys. J. 2008;95:3575–3590. doi: 10.1529/biophysj.108.131383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grubmüller H., Heymann B., Tavan P. Ligand binding: molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 1996;271:997–999. doi: 10.1126/science.271.5251.997. [DOI] [PubMed] [Google Scholar]
- 48.Lüdemann S.K., Lounnas V., Wade R.C. How do substrates enter and products exit the buried active site of cytochrome P450cam? 2. Steered molecular dynamics and adiabatic mapping of substrate pathways. J. Mol. Biol. 2000;303:813–830. doi: 10.1006/jmbi.2000.4155. [DOI] [PubMed] [Google Scholar]
- 49.Ishida H., Hayward S. Path of nascent polypeptide in exit tunnel revealed by molecular dynamics simulation of ribosome. Biophys. J. 2008;95:5962–5973. doi: 10.1529/biophysj.108.134890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brower-Toland B., Wang M.D. Use of optical trapping techniques to study single-nucleosome dynamics. Methods Enzymol. 2004;376:62–72. doi: 10.1016/S0076-6879(03)76005-4. [DOI] [PubMed] [Google Scholar]
- 51.Maison C., Almouzni G. HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 2004;5:296–304. doi: 10.1038/nrm1355. [DOI] [PubMed] [Google Scholar]
- 52.Zeng L., Zhou M.M. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 2002;513:124–128. doi: 10.1016/s0014-5793(01)03309-9. [DOI] [PubMed] [Google Scholar]
- 53.Kepper N., Ettig R., Rippe K. Force spectroscopy of chromatin fibers: extracting energetics and structural information from Monte Carlo simulations. Biopolymers. 2011;95:435–447. doi: 10.1002/bip.21598. [DOI] [PubMed] [Google Scholar]
- 54.Mazurkiewicz J., Kepert J.F., Rippe K. On the mechanism of nucleosome assembly by histone chaperone NAP1. J. Biol. Chem. 2006;281:16462–16472. doi: 10.1074/jbc.M511619200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.