

Abstract
Pentameric ligand-gated ion channels are targets of general anesthetics. Although the search for discrete anesthetic binding sites has achieved some degree of success, little is known regarding how anesthetics work after the events of binding. Using the crystal structures of the bacterial Gloeobacter violaceus pentameric ligand-gated ion channel (GLIC), which is sensitive to a variety of general anesthetics, we performed multiple molecular dynamics simulations in the presence and absence of the general anesthetic isoflurane. Isoflurane bound to several locations within GLIC, including the transmembrane pocket identified crystallographically, the extracellular (EC) domain, and the interface of the EC and transmembrane domains. Isoflurane also entered the channel after the pore was dehydrated in one of the simulations. Isoflurane disrupted the quaternary structure of GLIC, as evidenced in a striking association between the binding and breakage of intersubunit salt bridges in the EC domain. The pore-lining helix experienced lateral and inward radial tilting motion that contributed to the channel closure. Isoflurane binding introduced strong anticorrelated motions between different subunits of GLIC. The demonstrated structural and dynamical modulations by isoflurane aid in the understanding of the underlying mechanism of anesthetic inhibition of GLIC and possibly other homologous pentameric ligand-gated ion channels.
Introduction
Nicotinic acetylcholine receptors (nAChRs), serotonin receptors, glycine receptors, and the γ-aminobutyric acid class A receptors are pentameric ligand-gated ion channels (pLGICs). They are also putative targets of general anesthetics (1,2). Anesthetics inhibit agonist-elicited cation conductivity in nAChRs and serotonin receptors, but potentiate anion conductivity in γ-aminobutyric acid class A and glycine receptors (1,2). Specific anesthetic binding to these proteins has been investigated intensively during recent decades. Multiple binding sites have been identified mainly through functional mutations of the proteins and anesthetic photolabeling to the proteins (3–8). Direct interaction and functional modulation of general anesthetics are evidenced, but the underlying mechanisms of anesthetic action on these channel proteins remain unclear.
pLGICs are molecular machines. Each of the five subunits in a pLGIC has an extracellular (EC) domain and four transmembrane domains (TM1–TM4). The intracellular linker for TM3 and TM4 varies in length, ranging from a few residues in bacteria to over a hundred residues in eukaryotic pLGICs. Agonist binding at the orthosteric sites in the EC domains allosterically activates receptors. Signal transduction upon agonist binding has been described vividly as a conformational wave based on studies with nAChRs (9,10). It is conceivable that if anything, such as anesthetic binding, modifies normal waving of the agonist-binding signals, it will alter the channel function. Anesthetic potentiation of channel activation elicited by agonists occurs via allosteric action (5), but inhibition may occur either through allosteric inhibition or direct channel occlusion. Direct occlusion of a channel pore easily explains anesthetic inhibition. Some experimental and computational data support the channel occlusion mechanism (11–13). Anesthetics can also act as negative allosteric modulators (14–17) to stabilize a nonconducting conformation or to increase the rate of channel desensitization. Binding sites for negative allosteric modulators, including anesthetics, have been found at the EC-TM interface (18,19), in nonorthosteric sites at the subunit interfaces in the EC domain (19,20), and in an intrasubunit pocket formed by four TM helices (21). Inhibition by ligands binding other than in pore lumen is allosteric inhibition. Understanding how binding at a remote site affects channel behavior remains a great challenge.
The role of electrostatic interactions in modulating pLGIC gating has been recognized in the past (10,22–24). Overall clusters of positively and negatively charged residues, instead of specific amino acids, mediate interactions between the agonist binding domain and channel region, and assist the agonist-elicited channel opening. It has been shown that electrostatic interaction for signal transmitting is not limited within a subunit. The intersubunit electrostatic coupling has been found critically important for normal activation of glycine receptors (25). Disruption of a salt bridge between adjacent subunits via mutations of charged residues caused a profound change in channel function (25). It has not yet been investigated whether anesthetics affect the functionally imperative electrostatic interactions that will further affect channel function.
To fill the knowledge gap between anesthetic binding and channel inhibition or potentiation, high-resolution structural information of proteins in various functional states is highly desirable. For a long time, the model of the Torpedo nAChR from cryo-electron microscopy measurements (26) was the only structural reference for pLGICs. Recent crystal structures of the bacterial pLGICs from the Erwinia chrysanthemi (ELIC) (27) and Gloeobacter violaceus (GLIC) (28,29) provide valuable structural frames for understanding homologous mammalian pLGICs. In addition to their structural homology, GLIC and nAChRs show similar functional sensitivity to anesthetics. The GLIC cation current activated by protons can be inhibited by a number of anesthetics (30), including isoflurane that is used in this study. Multiple anesthetic binding sites in GLIC have been identified experimentally (19) or predicted computationally (13,19). More recently, the crystal structures of GLIC complexed with anesthetic desflurane or propofol provide valuable high-resolution information about the location of binding sites (21). A series of point mutations and functional measurements have validated the functional relevance of anesthetic binding in an intrasubunit pocket formed by four TM helices (21). The crystal structures of GLIC bound with or without anesthetics, however, are virtually identical (21,28), as if anesthetics had no impact on GLIC. The apparent structural insensitivity to anesthetics may result from a biased conformational preference under certain crystallization conditions. The same concern is also valid for the anesthetic sites revealed in the crystal structures, in which only a subset of binding sites were identified likely due to the low affinity of anesthetics.
In this study, we performed multiple molecular dynamics (MD) simulations on GLIC in the absence and presence of isoflurane. There are several advantages for choosing isoflurane for the study. First, it has demonstrated a functional relevance by inhibiting the GLIC ion current at a low concentration (30). Second, it differs by only one atom from desflurane, which has resolved binding sites in the GLIC crystal structure (21). The site for desflurane is a reliable reference for isoflurane. Third, the isoflurane parameterization, essential for MD simulations, has been completed (31). Our simulations demonstrated that isoflurane binds to multiple locations in GLIC, including the pore lumen. Isoflurane located between two adjacent subunits could disturb intersubunit salt bridges, either directly or allosterically, which caused profound quaternary structural and dynamics changes in GLIC. Insights into isoflurane modulation on the structure and dynamics of GLIC revealed in this study may have a general implication in anesthetic action on other pLGICs.
Methods
System preparation
Three systems were generated with the same preparation except isoflurane binding status: the control system containing no isoflurane; system X containing isoflurane at positions suggested by the x-ray structure of the GLIC-desflurane complex (21); and system Y containing isoflurane at computationally predicted sites. Details of isoflurane positions are shown in Fig. 1. For each system, the published crystal structure of GLIC (PDB code: 3EAM) (28) was inserted into an equilibrated binary mixture of bacterial lipids (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine/1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) in the ratio of 3:1) in a hexagonal prism, solvated, and ionized with VMD plugins (32). In an attempt to simulate an open channel, GLIC was set in a protonated state (33). The predicted pKa values for some titratable residues of GLIC (28) were used in the simulations. For residues without published values, we calculated their pKa values at a pH of 4.6 using PROPKA 2.0 (34,35). We also calculated a probability of protonation for each residue and determined how many of the equivalent residues in the five subunits should be protonated at the beginning of a simulation. Details of protonation for residues in each subunit are provided in Table S1, in the Supporting Material. The control system contains one GLIC, 167 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine, 54 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol), 48 Na+, 21 Cl−, and 24,486 TIP3 water molecules.
Figure 1.
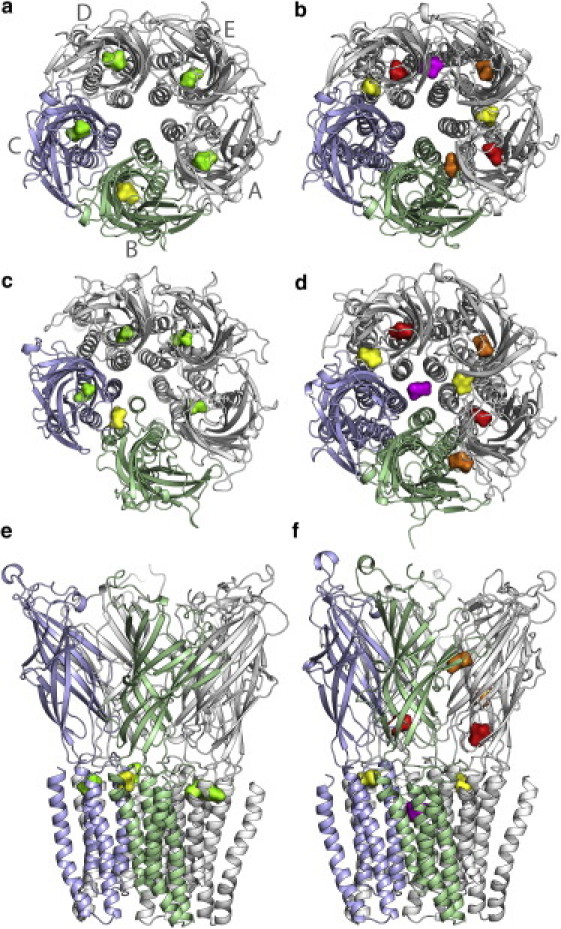
Isoflurane binding sites in system X and system Y at the beginning and end of each simulation. (a and b) are top views of systems X and Y at the beginning of each simulation, respectively. (c and e) and (d and f) are top and side views at the end of each simulation for systems X and Y, respectively. Subunit labels (gray letters) and colors are consistent throughout all figures in the manuscript. Isoflurane molecules are colored according to their final binding sites: yellow for those in the intersubunit sites in the TM domain; green for those in intrasubunit sites in the TM domain; red for those at or close to the EC/TM interface; purple for the one inside the pore; and orange for those in the EC domain.
MD simulations
NAMD 2.7b1 (36) and CHARMM-27 force field (37) were used for all simulations reported in this study. Each system was initially energy minimized with a fixed GLIC backbone for 10,000 steps and with no restraints for another 10,000 steps. The system was warmed from 0 to 310 K over 62 ps with 1 fs/step and 1 kcal/mol/Å2 backbone restraints on GLIC, which were gradually reduced and then removed after 600 ps of simulation. The equilibrated systems were simulated with a time step of 2 fs at a constant 1 atm of pressure. Langevin damping value was set to 10 ps−1 for the first 2 ns of simulation and reduced to 1 ps−1 for the following 100 ns of simulation. Particle mesh Ewald was used for long-range electrostatic interactions and a 12-Å cutoff was used for nonbonded interactions. Nonbonded interactions were evaluated at every step and full electrostatic interactions at every other step. The systems were simulated with periodic boundary conditions as hexagonal prisms that were ∼130 Å in the dimension parallel to the membrane normal, and 104 Å between two parallel sides of the hexagon. We performed two sets of production simulations for the control system (namely, cMD1 and cMD2), two sets for each isoflurane system (system X: xMD1 and xMD2; system Y: yMD1 and yMD2). Each simulation lasted for ∼102 ns.
Anesthetic binding positions
The geometry and parameters of R- or S-isoflurane were used as published (31). In system X, S-isoflurane was manually placed in the S-desflurane site revealed in the crystal structure of the desflurane-GLIC complex (21). Note that isoflurane differs from desflurane by only one atom. Each subunit contained one isoflurane molecule and there were a total of five isoflurane molecules in system X. In system Y, putative isoflurane binding sites were identified initially through docking and preliminary (∼16 ns) MD simulations. Only those isoflurane molecules that showed minimal displacements in the preliminary MD simulations were included in the production MD simulations. Isoflurane docking results were compiled from 500 runs using the Lamarckian genetic algorithm implemented in AutoDock 4.0 (38). A population size of 300, a maximum of 27,000 generations, and maximum of 15 million energy evaluations were used. For each isoflurane enantiomer, 15 of the most populated and lowest energy, nonoverlapping poses were selected for the preliminary 16-ns MD simulations. A total of 10 isoflurane molecules (five sites for each enantiomer) with the lowest displacements were selected for the production NPT MD simulations. Only the results from the production simulation are reported in this manuscript.
Gaussian network model and principal component analysis
To elucidate how anesthetics affect GLIC dynamics, we calculated the mean squared fluctuation (MSF) of GLIC using the Gaussian network model (GNM) (39,40). The three slowest modes of the GNM were used to capture low frequency motions that may be important to the protein function. We also performed principal component analysis (PCA) (41,42) of trajectories from our MD simulations. For each system, GLIC structural snapshots with a 20-ps interval were taken and a total of 5100 structures were used for each PCA. Principal component (PC) calculations were based on the Cα coordinates of GLIC. Visualization of the directions and extents of the principal motions of GLIC Cα atoms was done on VMD (32) and the NMWiz plugin (43). Each cross correlation map was generated based on all 5100 PCs of each simulation set.
Results and Discussion
Anesthetic binding sites
Fig. 1 summarizes the isoflurane locations in GLIC before and after over 100-ns MD simulations. In system X, isoflurane was initially in an intrasubunit pocket at the top of the TM helices, where desflurane was found in the crystal structure of the GLIC (21). The intravenous anesthetic propofol, which has a very different molecular structure from desflurane, was also found nearby the pocket in the cocrystal of GLIC and propofol (21). With only a one-atom difference between desflurane and isoflurane, it is reasonable to assume that isoflurane shares the same binding site as desflurane. Indeed, four of five isoflurane molecules remained close to their initial sites for the duration of the simulation. One isoflurane molecule, named isof-1, moved toward the EC domain early in the simulation and resided in an intersubunit pocket in the TM domain at the end of the simulation (Fig. 1, c and e), suggesting the existence of additional anesthetic binding sites that were not captured by the crystal structure. In the simulation of xMD2, isof-1, along with another isoflurane molecule, also moved out of their binding pocket (Fig. S1). Volatile general anesthetics, such as isoflurane and desflurane, are low affinity drugs and may have multiple binding modes. Some of the binding sites are probably not observable under certain crystallization conditions. Thus, it is necessary to have various approaches to identify other plausible anesthetic binding sites.
In system Y, seven isoflurane molecules are bound with GLIC over the 100-ns simulation. Two (colored yellow in Fig. 1 b) occupied intersubunit pockets that are homologous to the final location of isof-1 in system X; two (colored red in Fig. 1 b) were located at intrasubunit sites on the EC side of the EC/TM interface. The binding pattern for these four molecules remained in another independent simulation (yMD2) for system Y (Fig. S1), suggesting that the pockets at these two locations are favorable for isoflurane. It is worth mentioning that both locations were also identified for halothane binding in our previous experiments on GLIC using point mutations and fluorescence quenching (19). Halothane was found in proximity to W160 in one location and to N200 in the other location (19). The remaining three isoflurane molecules began the simulation in the EC domain. One (colored purple in Fig. 1 b) migrated from the interface between two subunits into the pore after ∼70 ns in the first simulation, but stayed in the EC domain in yMD2 (Fig. S1). The other two isoflurane molecules (colored orange in Fig. 1 b) showed a distinct difference: the one within the subunit remained in the pocket in both runs of simulations, but another behind loop C of subunit B showed a larger displacement in the first simulation (Fig. 1) and moved into bulk water at a late stage of the second simulation (Fig. S1). The homologous site was suggested for halothane binding in the previous MD simulations on α4β2 and α7 nAChRs (44–46) and [3H]azietomidate photolabeling at the α-γ interface of Torpedo nAChR (6).
Isoflurane bound to a dehydrated channel
Although there is presently no experimental evidence of isoflurane binding to the GLIC pore lumen, one of our simulations (yMD1) in this study and a previous simulation (13) indicate a possibility that isoflurane could go into the pore. Interestingly, we found that isoflurane entered the pore after the pore was no longer permeable to water. The hydrophobic region of the pore, involving residues from I240 (16′) to I233 (9′), was dehydrated after ∼20 ns of simulation (Fig. S2). But this particular isoflurane molecule was not near the extracellular pore entrance until ∼70 ns after the simulation began (Fig. S3). Once it moved close to I240, its trifluoromethyl group approached I240. It took only 0.4 ns for this isoflurane molecule to pass the hydrophobic ring of five I240 residues. This isoflurane molecule was then trapped between I240 (16′) and I233 (9′) for the rest of the simulation. The location of isoflurane inside the pore is consistent with the earlier observation that isoflurane had the most probable contact with A237 (13′) in the GLIC pore (13).
It is striking to see that isoflurane was able to pass the channel hydrophobic gate even when the gate was closed for water. Most general anesthetics are amphipathic. Their lipophilic groups, such as the trifluoromethyl group in isoflurane, could mediate close contact with pore-lining hydrophobic residues. If these residues are as flexible as I240 of GLIC, drug molecules may have an opportunity to pass the hydrophobic gate even though the pore size is not large enough for water passage. A recent x-ray study of GLIC complexed with bromo-lidocaine (47), a positively charged local anesthetic, suggested that bromo-lidocaine bound to the pore region. General anesthetics have a wide range of structures and pharmacological properties. We need to collect more experimental data to define the importance of the pore occlusion mechanism in functional inhibition of GLIC and other pLGICs.
Isoflurane binding weakened the quaternary structure of GLIC
Salt bridges play a critical role not only in stabilizing protein structures, but also in protein functions (48). It was evidenced that salt bridges in Cys-loop receptors were involved in the agonist elicited channel gating process (10,23,49). In GLIC, four residues in loop C (E177, D178, R179, and E181) were found to form intersubunit salt bridges. These residues contacted residues on the complementary subunit (R62, D91, R105, and K148) and formed salt bridges over the course of simulations (Fig. S4). To determine whether the presence of isoflurane affects formation of these salt bridges, we counted the number of salt bridges in frames collected every 20 ps and examined the entire simulation period for each system. The percentage of frames containing intersubunit salt bridges in all the examined frames was calculated for every intersubunit interface in all three systems. The results are summarized in Fig. 2 and Fig S5.
Figure 2.
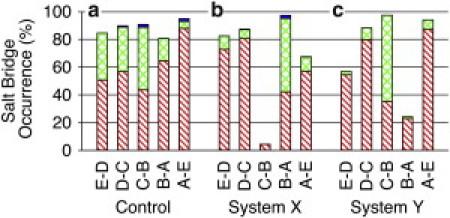
Stacked bar graphs representing the probability of the salt bridge occurrence between residues of loop C in the principal subunit and residues in the adjacent complementary subunit over the course of each simulation: (a) control system, (b) system X, and (c) system Y. Subunits for salt bridges at the interface are labeled accordingly. Number of simultaneous salt bridges in a single frame is indicated by single hash marks, a single salt bridge; double hash marks, two salt bridges; solid bar, three salt bridges. Salt bridges were examined in frames collected every 20 ps using the saltbr plugin of VMD (32). Salt bridges were considered as formed if the distance between the center of mass of the oxygen atoms in the acidic side chain and center of mass of the nitrogen atoms in the basic side chain was <4 Å.
For the control system (Fig. 2 a), every subunit interface of GLIC had at least one salt bridge for over 80% of the simulation. Two or three salt bridges existed simultaneously in some of the interfaces. Compared to the isoflurane systems, the control system had a relatively even distribution of salt bridges among five intersubunit interfaces.
In xMD1 of system X (Fig. 2 b), the salt bridges decreased dramatically at the interface of subunits B and C, whereas there was an obvious increase of salt bridge formation at the adjacent interface of subunits B and A. All these changes in salt bridges affected the quaternary structure of GLIC, as reflected in Fig. 1 c. Isof-1 seemed to be responsible for these changes. This isoflurane molecule was initially in a TM pocket of subunit B, and later migrated into an interfacial pocket between subunits B and C. isof-1 was the only one among five isoflurane molecules in xMD1 that moved from the intrasubunit pocket to being at the intersubunit interface. Apparently such a movement allosterically destabilized the salt bridges between subunits B and C in the EC domain. In xMD2, more subunits were involved in losing interfacial salt bridges due to an additional isoflurane molecule that moved out from the original binding site.
Similar disruptions by interfacial isoflurane were also observed in system Y. Significantly lower occurrence of salt bridges at the interface of subunits A and B (Fig. 2 c) could be ascribed to isoflurane movement at the same interface. The relocation of isoflurane from the interface of subunits D and E to the pore also perturbed the formation of salt bridges. Interestingly, when this isoflurane stayed steadily in the EC domain in the second set of simulation (yMD2), the occurrence of salt bridges went back to normal (Fig. S5 c), indicating that a lower occurrence of salt bridges in Fig. 2 c was indeed caused by disruptions of isoflurane movement. As also observed in system X, suppression of salt bridge formation at one interface was accompanied by promotion of salt bridges in the neighboring interface. The formation and disruption of these salt bridges were often accompanied by narrowed and widened intersubunit gaps that altered the quaternary structure and dynamics of GLIC.
TM2 tilting motion
It is evidenced that isoflurane disrupted the quaternary structure in the EC domain of GLIC, but what about the TM domain? Orientations of TM2 helices shape the pore geometry and consequently affect the ion-conducting status of a channel. We characterized the orientation of each TM2 helix using the lateral tilting angle (δ) and radial tilting angle (θ) as shown in Fig. S6 and defined previously (50). Trajectories of θ and δ for each TM2 helix in the control system and isoflurane systems over the course of the first set of simulations are illustrated in Fig. 3. The data for the repeated simulations are shown in Fig. S7. Compared to the starting orientation of five TM2 helices in GLIC that show a funnel-shaped pore opening with the most restricting part at the intracellular entry of the channel, the EC ends of TM2 helices in all systems had inward movement, as evidenced in smaller, or negative, θ values at the end of the simulations. Lateral tilting angles of TM2 also deviated from the initial structure. Degrees of deviation varied among five subunits in a given system. We calculated tilt angles of TM helices averaged over the first and last 5 ns of simulations for each individual system and summarized the data in Table S2 and Table S3. Several subunits in system Y show profound clockwise lateral tilting of TM helices (Fig. 3 c and Fig. S7 c), but the averaged tilting angles among all subunits in all simulations are not statistically different from those for the simulations of the control system.
Figure 3.
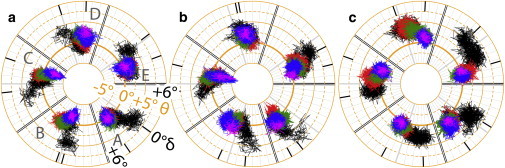
Polar plots of radial (θ) and lateral (δ) tilt angles of the TM2 helices for (a) control; (b) system X; and (c) system Y. The θ angle defines the inclination of the principal axis of a TM2 helix with respect to the direction of the channel symmetry axis. The δ angle measures the orientation of the principal axis of a TM2 helix projected onto a lateral plane that is perpendicular to the radial plane. Every 25-ns simulation trajectory is colored differently in a time order of black, red, green, blue, and magenta (last 2 ns). The averaged lateral angels for the first and last 5-ns simulations are marked by black ticks. The principal axis of each TM2 helix was calculated based on coordinates of backbone atoms of residues E222–I240.
The impact of the TM2 helix tilting on the channel opening became obvious when the structure of GLIC was compared with a closed-channel structure of a homologous protein, ELIC (27). The crystal structure of GLIC represents a more open channel, which has the EC end of TM2 tilted radially away from the channel axis (28) with θ of 6.5°, but a fairly small lateral tilting (δ = 0.7°). The crystal structure of ELIC (27), on the other hand, shows θ and δ values of −3.5° and −7.9°, respectively. Clearly, TM2 in ELIC has a clockwise lateral tilting in addition to inward radial movement. In simulations, however, the perfect symmetry among five subunits in the crystal structures was not sustained. TM2 helices in different subunits experienced different degrees of tilting at a given time point. Moreover, it seems that TM2 tilting in one or two subunits is sufficient to alter channel permeability to water (Fig. S2). The widest range of lateral tilting motion experienced by subunit B in both system X and system Y was concurrent with the most severe destruction of intersubunit salt bridges (Fig. 2, b and c), indicating that isoflurane disruption of intersubunit salt bridges may promote the clockwise lateral tilting of the TM2 helices.
Interplay between protein dynamics and isoflurane binding
Fig. 4 shows the MSF derived from the three slowest modes of GNM analysis for all three GLIC systems. The data convey at least two important messages. First, the isoflurane binding sites are almost exclusively restricted to residues near global hinge regions of GLIC, the majority of which are located in the EC/TM interface, areas of MSF minima. Those anesthetics placed in highly mobile regions at the beginning of the simulations were not retained there and quickly migrated into bulk water. Hinge regions often play critical roles in protein motion that are relevant to protein function (51). Anesthetic occupancy in these hinge regions, especially at or near the EC/TM interface, may trigger changes in global protein motions that could affect channel function. Second, the overall dynamics of GLIC changed upon isoflurane binding. In system X, there was a marked increase in dynamics of the EC domain in subunits B and C, where isof-1 bound to the intersubunit pocket in the TM domain (see regions marked with yellow diamond symbols in Fig. 4, B and C). The EC domains of the neighboring subunits, however, experienced overall decreased dynamical motions compared to the control system. A similar trend was also observed in system Y. A substantial number of residues in subunits C and D interacted with isoflurane bound to the intersubunit pocket, as highlighted by the yellow circles in Fig. 4, C and D. The EC domains of these two subunits also show higher MSF than the control system. Although it is almost impossible to rule out dynamical contributions of isoflurane bound to other regions, the increased MSF in the EC domain seems to correlate the best with isoflurane binding in the intersubunit TM pockets near the EC-TM interface.
Figure 4.
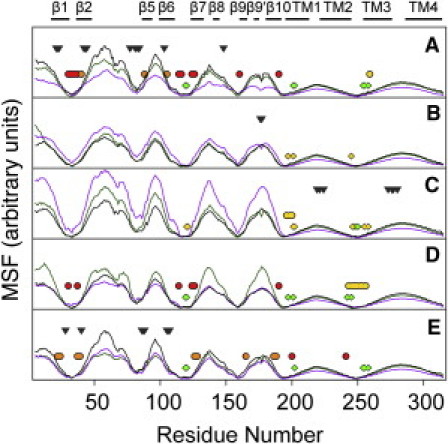
Mean squared fluctuation of the control system (black), system X (purple), and system Y (green) based on three slowest modes of GNM analysis (39,40). Structures after 102-ns simulations were used for GNM analysis. Each residue of GLIC within 3.5 Å of isoflurane for >30 ns in the last 40-ns simulation is marked with a diamond (system X) or a circle (system Y) that are colored the same as isoflurane molecules at each equivalent site shown in Fig. 1. Gray triangles indicate initial isoflurane positions where isoflurane migrated away from the protein during the simulations. The GLIC secondary structure is highlighted at the top of the figure.
PCA allows us to visualize the direction and extent of the principal motions of Cα atoms of GLIC in the absence and presence of isoflurane. The first two eigenvectors account for over 45% of the total variances observed in the 100 ns of the simulation trajectories in system X and system Y, but only ∼33% in the control system. Strong subunit repelling motion in EC domains was captured in PC1 of xMD1 (Fig. S8 b) and yMD2 (Fig. S9 c). However, such a motion was not found in the control simulations. Most TM1, TM3, and TM4 helices showed counterclockwise tilting motions in PC1 and PC2, but the TM2 helices exhibited more diverse motional directions. It appeared that the majority of TM2 helices experienced a clockwise lateral tilting motion, but some showed profound radial motion (Fig. S8 a and Fig. S9 c).
The influence of isoflurane on the correlated motions of GLIC was elucidated in cross correlation maps in Fig. 5. Without isoflurane (Fig. 5, a), correlated motions were observed within each subunit, particularly in EC domains. Anticorrelated motion was not obvious either within or between subunits in the control system. In contrast, anticorrelated motions between different subunits of GLIC became much more apparent in system X and system Y (Fig. 5, b and c). The correlated motions of TM4 and TM3 as well as TM1 also became stronger than those in the control system. These major differences between systems with and without isoflurane were validated in our repeated simulations (Fig. S10).
Figure 5.
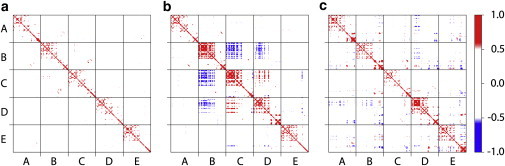
Correlated motions of GLIC. (a) Cross correlation map of GLIC in the control simulation cMD1 reveals residues within a subunit or between subunits that fluctuate in the same direction at the same time. (b) Cross correlation map of GLIC in the xMD1 simulation of the X system. (c) Cross correlation map of GLIC in the yMD1 simulation of the Y system. Note those motions that exist in b and c, but not in a: the strong anticorrelated motions in EC domains between different subunits and the strong correlated motions among TM4, TM3, and TM1. The color scale runs from blue (anticorrelated motion) to red (correlated motion).
Conclusions
Three conclusions can be drawn from the current study. First, isoflurane binds to the pore lumen and other allosteric sites of GLIC in addition to the sites identified crystallographically (21). This is consistent with the previous experimental and computational results that multiple anesthetic binding sites exist in GLIC and other homologous proteins (4,13,19,45). Second, the isoflurane binding at or near the EC-TM interface allosterically weakens the quaternary structure of GLIC, as evidenced by a striking association between the binding of isoflurane and the disruption of intersubunit salt bridges in the EC domain. Third, isoflurane binding not only alters the GLIC structure, but also the GLIC motion. Changes in the low frequency motion of GLIC as captured by GNM analysis, the opposite motion directions between adjacent subunits as captured by PCA, and the strong anticorrelated motions among different subunits as revealed in cross correlation maps, present possible dynamics characteristics of anesthetic inhibition to channel functions. We need more experimental evidence to further determine key elements that can account for the underlying mechanism of anesthetic action on GLIC or other homologous pLGICs.
Acknowledgments
We thank Ms. Sandra C. Hirsch for reading and editing the manuscript.
This research was supported in part by the National Science Foundation through TeraGrid resources (TG-MCB050030N, TG-MCB100047, and TG-MCB100069) that are hosted by Indiana University, Laboratory of Neuro Imaging, National Center for Atmospheric Research, National Center for Supercomputing Applications, National Institute for Computer Sciences, Oak Ridge National Laboratory, Pittsburgh Supercomputer Center, Purdue University, San Diego Supercomputer Center, Texas Advanced Computer Center, and University of California, Argonne National Laboratory. This research was supported by grants from the National Institutes of Health (R01GM066358, R01GM056257, and R37GM049202, and T32GM075770).
Supporting Material
References
- 1.Campagna J.A., Miller K.W., Forman S.A. Mechanisms of actions of inhaled anesthetics. N. Engl. J. Med. 2003;348:2110–2124. doi: 10.1056/NEJMra021261. [DOI] [PubMed] [Google Scholar]
- 2.Hemmings H.C., Jr., Akabas M.H., Harrison N.L. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol. Sci. 2005;26:503–510. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 3.Chiara D.C., Dangott L.J., Cohen J.B. Identification of nicotinic acetylcholine receptor amino acids photolabeled by the volatile anesthetic halothane. Biochemistry. 2003;42:13457–13467. doi: 10.1021/bi0351561. [DOI] [PubMed] [Google Scholar]
- 4.Chiara D.C., Hong F.H., Cohen J.B. Time-resolved photolabeling of the nicotinic acetylcholine receptor by [3H]azietomidate, an open-state inhibitor. Mol. Pharmacol. 2009;75:1084–1095. doi: 10.1124/mol.108.054353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li G.D., Chiara D.C., Cohen J.B. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J. Neurosci. 2006;26:11599–11605. doi: 10.1523/JNEUROSCI.3467-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ziebell M.R., Nirthanan S., Cohen J.B. Identification of binding sites in the nicotinic acetylcholine receptor for [3H]azietomidate, a photoactivatable general anesthetic. J. Biol. Chem. 2004;279:17640–17649. doi: 10.1074/jbc.M313886200. [DOI] [PubMed] [Google Scholar]
- 7.Eckenhoff R.G. An inhalational anesthetic binding domain in the nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA. 1996;93:2807–2810. doi: 10.1073/pnas.93.7.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forman S.A., Miller K.W. Anesthetic sites and allosteric mechanisms of action on Cys-loop ligand-gated ion channels. Can. J. Anaesth. 2011;58:191–205. doi: 10.1007/s12630-010-9419-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Purohit P., Mitra A., Auerbach A. A stepwise mechanism for acetylcholine receptor channel gating. Nature. 2007;446:930–933. doi: 10.1038/nature05721. [DOI] [PubMed] [Google Scholar]
- 10.Lee W.Y., Sine S.M. Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature. 2005;438:243–247. doi: 10.1038/nature04156. [DOI] [PubMed] [Google Scholar]
- 11.Forman S.A., Miller K.W., Yellen G. A discrete site for general anesthetics on a postsynaptic receptor. Mol. Pharmacol. 1995;48:574–581. [PubMed] [Google Scholar]
- 12.Wenningmann I., Barann M., Dilger J.P. The effects of isoflurane on acetylcholine receptor channels: 3. Effects of conservative polar-to-nonpolar mutations within the channel pore. Mol. Pharmacol. 2001;60:584–594. [PubMed] [Google Scholar]
- 13.Brannigan G., LeBard D.N., Klein M.L. Multiple binding sites for the general anesthetic isoflurane identified in the nicotinic acetylcholine receptor transmembrane domain. Proc. Natl. Acad. Sci. USA. 2010;107:14122–14127. doi: 10.1073/pnas.1008534107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arias H.R. Positive and negative modulation of nicotinic receptors. Adv. Protein Chem. Struct. Biol. 2010;80:153–203. doi: 10.1016/B978-0-12-381264-3.00005-9. [DOI] [PubMed] [Google Scholar]
- 15.Blanton M.P., Xie Y., Cohen J.B. The steroid promegestone is a noncompetitive antagonist of the Torpedo nicotinic acetylcholine receptor that interacts with the lipid-protein interface. Mol. Pharmacol. 1999;55:269–278. doi: 10.1124/mol.55.2.269. [DOI] [PubMed] [Google Scholar]
- 16.Gumilar F., Arias H.R., Bouzat C. Molecular mechanisms of inhibition of nicotinic acetylcholine receptors by tricyclic antidepressants. Neuropharmacology. 2003;45:964–976. doi: 10.1016/s0028-3908(03)00247-8. [DOI] [PubMed] [Google Scholar]
- 17.Francis M.M., Vazquez R.W., Oswald R.E. Subtype-selective inhibition of neuronal nicotinic acetylcholine receptors by cocaine is determined by the alpha4 and beta4 subunits. Mol. Pharmacol. 2000;58:109–119. doi: 10.1124/mol.58.1.109. [DOI] [PubMed] [Google Scholar]
- 18.Chen Z., White M.M. Forskolin modulates acetylcholine receptor gating by interacting with the small extracellular loop between the M2 and M3 transmembrane domains. Cell. Mol. Neurobiol. 2000;20:569–577. doi: 10.1023/a:1007011911611. [DOI] [PubMed] [Google Scholar]
- 19.Chen Q., Cheng M.H., Tang P. Anesthetic binding in a pentameric ligand-gated ion channel: GLIC. Biophys. J. 2010;99:1801–1809. doi: 10.1016/j.bpj.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moroni M., Vijayan R., Bermudez I. Non-agonist-binding subunit interfaces confer distinct functional signatures to the alternate stoichiometries of the alpha4beta2 nicotinic receptor: an alpha4-alpha4 interface is required for Zn2+ potentiation. J. Neurosci. 2008;28:6884–6894. doi: 10.1523/JNEUROSCI.1228-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nury H., Van Renterghem C., Corringer P.J. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature. 2011;469:428–431. doi: 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- 22.Xiu X., Hanek A.P., Dougherty D.A. A unified view of the role of electrostatic interactions in modulating the gating of Cys loop receptors. J. Biol. Chem. 2005;280:41655–41666. doi: 10.1074/jbc.M508635200. [DOI] [PubMed] [Google Scholar]
- 23.Gay E.A., Yakel J.L. Gating of nicotinic ACh receptors; new insights into structural transitions triggered by agonist binding that induce channel opening. J. Physiol. 2007;584:727–733. doi: 10.1113/jphysiol.2007.142554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gleitsman K.R., Kedrowski S.M., Dougherty D.A. An intersubunit hydrogen bond in the nicotinic acetylcholine receptor that contributes to channel gating. J. Biol. Chem. 2008;283:35638–35643. doi: 10.1074/jbc.M807226200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Todorovic J., Welsh B.T., Mihic S.J. Disruption of an intersubunit electrostatic bond is a critical step in glycine receptor activation. Proc. Natl. Acad. Sci. USA. 2010;107:7987–7992. doi: 10.1073/pnas.1001845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J. Mol. Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 27.Hilf R.J., Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- 28.Bocquet N., Nury H., Corringer P.J. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- 29.Hilf R.J., Dutzler R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature. 2009;457:115–118. doi: 10.1038/nature07461. [DOI] [PubMed] [Google Scholar]
- 30.Weng Y., Yang L., Sonner J.M. Anesthetic sensitivity of the Gloeobacter violaceus proton-gated ion channel. Anesth. Analg. 2010;110:59–63. doi: 10.1213/ANE.0b013e3181c4bc69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hénin J., Brannigan G., Klein M.L. An atomistic model for simulations of the general anesthetic isoflurane. J. Phys. Chem. B. 2010;114:604–612. doi: 10.1021/jp9088035. [DOI] [PubMed] [Google Scholar]
- 32.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 33.Bocquet N., Prado de Carvalho L., Corringer P.J. A prokaryotic proton-gated ion channel from the nicotinic acetylcholine receptor family. Nature. 2007;445:116–119. doi: 10.1038/nature05371. [DOI] [PubMed] [Google Scholar]
- 34.Bas D.C., Rogers D.M., Jensen J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- 35.Li H., Robertson A.D., Jensen J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 36.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 38.Morris G.M., Goodsell D.S., Olson A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 39.Bahar I., Atilgan A.R., Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 40.Yang L.W., Liu X., Bahar I. iGNM: a database of protein functional motions based on Gaussian Network Model. Bioinformatics. 2005;21:2978–2987. doi: 10.1093/bioinformatics/bti469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ichiye T., Karplus M. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 1991;11:205–217. doi: 10.1002/prot.340110305. [DOI] [PubMed] [Google Scholar]
- 42.Amadei A., Linssen A.B., Berendsen H.J. Essential dynamics of proteins. Proteins. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 43.Bakan A., Meireles L.M., Bahar I. ProDy: protein dynamics inferred from theory and experiments. Bioinformatics. 2011;27:1575–1577. doi: 10.1093/bioinformatics/btr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu L.T., Haddadian E.J., Tang P. Higher susceptibility to halothane modulation in open- than in closed-channel alpha4beta2 nAChR revealed by molecular dynamics simulations. J. Phys. Chem. B. 2010;114:626–632. doi: 10.1021/jp908944e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu L.T., Willenbring D., Tang P. General anesthetic binding to neuronal alpha4beta2 nicotinic acetylcholine receptor and its effects on global dynamics. J. Phys. Chem. B. 2009;113:12581–12589. doi: 10.1021/jp9039513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mowrey D., Haddadian E.J., Tang P. Unresponsive correlated motion in alpha7 nAChR to halothane binding explains its functional insensitivity to volatile anesthetics. J. Phys. Chem. B. 2010;114:7649–7655. doi: 10.1021/jp1009675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hilf R.J., Bertozzi C., Dutzler R. Structural basis of open channel block in a prokaryotic pentameric ligand-gated ion channel. Nat. Struct. Mol. Biol. 2010;17:1330–1336. doi: 10.1038/nsmb.1933. [DOI] [PubMed] [Google Scholar]
- 48.Hertel B., Tayefeh S., Kast S.M. Salt bridges in the miniature viral channel Kcv are important for function. Eur. Biophys. J. 2010;39:1057–1068. doi: 10.1007/s00249-009-0451-z. [DOI] [PubMed] [Google Scholar]
- 49.Law R.J., Henchman R.H., McCammon J.A. A gating mechanism proposed from a simulation of a human alpha7 nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA. 2005;102:6813–6818. doi: 10.1073/pnas.0407739102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng X., Ivanov I., McCammon J.A. Nanosecond-timescale conformational dynamics of the human alpha7 nicotinic acetylcholine receptor. Biophys. J. 2007;93:2622–2634. doi: 10.1529/biophysj.107.109843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang L.W., Bahar I. Coupling between catalytic site and collective dynamics: a requirement for mechanochemical activity of enzymes. Structure. 2005;13:893–904. doi: 10.1016/j.str.2005.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.