

Abstract
Hematopoietic transcription factors GATA-1 and PU.1 bind each other on DNA to block transcriptional programs of undesired lineage during hematopoietic commitment. Murine erythroleukemia (MEL) cells that coexpress GATA-1 and PU.1 are blocked at the blast stage but respond to molecular removal (downregulation) of PU.1 or addition (upregulation) of GATA-1 by inducing terminal erythroid differentiation. To test whether GATA-1 blocks PU.1 in MEL cells, we have conditionally activated a transgenic PU.1 protein fused with the estrogen receptor ligand-binding domain (PUER), resulting in activation of a myeloid transcriptional program. Gene expression arrays identified components of the PU.1-dependent transcriptome negatively regulated by GATA-1 in MEL cells, including CCAAT/enhancer binding protein α (Cebpa) and core-binding factor, β subunit (Cbfb), which encode two key hematopoietic transcription factors. Inhibition of GATA-1 by small interfering RNA resulted in derepression of PU.1 target genes. Chromatin immunoprecipitation and reporter assays identified PU.1 motif sequences near Cebpa and Cbfb that are co-occupied by PU.1 and GATA-1 in the leukemic blasts. Significant derepression of Cebpa and Cbfb is achieved in MEL cells by either activation of PU.1 or knockdown of GATA-1. Furthermore, transcriptional regulation of these loci by manipulating the levels of PU.1 and GATA-1 involves quantitative increases in a transcriptionally active chromatin mark: acetylation of histone H3K9. Collectively, we show that either activation of PU.1 or inhibition of GATA-1 efficiently reverses the transcriptional block imposed by GATA-1 and leads to the activation of a myeloid transcriptional program directed by PU.1.
Introduction
During hematopoiesis, precise levels of specific transcription factors regulate lineage determination, and changes in their levels block or divert this process (1–3). PU.1 (Sfpi1, Spi-1) and GATA-1 are two lineage-specific transcription factors that play key roles in determining the fate of multipotential progenitors (4). PU.1 is an Ets family member that dose-dependently guides the development and differentiation of granulocyte-macrophage and common lymphoid progenitors by interacting with lineage-specific cofactors on DNA (5). Differentiation into myeloid precursors also involves CCAAT/enhancer binding protein α (Cebpa) and core-binding factor, β subunit (Cbfb), which cooperate with PU.1 in the further specification and maturation of cells (6, 7). PU.1 levels below a certain threshold (~20%) cause a block of hematopoietic differentiation accompanied with accelerated proliferation (8, 9). Mutations of PU.1 and some of its target genes, including Cebpa and Cbfb, are associated with a differentiation block in human acute leukemias (10). GATA-1 is a zinc finger transcription factor regulating erythro-megakaryopoiesis by sequence-specific targeting as well as cooperating with lineage-specific cofactors in chromatin, such as nuclear factor [erythroid-derived 2 (Nfe2)] and Friend of GATA-1 (Zfpm1; refs. 11, 12). Mutations affecting either the length of GATA-1 protein or its interactions with Zfpm1 are also associated with acute leukemias (13–15).
Murine erythroleukemia (MEL) cells are acute leukemia blasts blocked from further erythroid differentiation by deregulated expression of PU.1 (16). Removal of PU.1 (17) or addition of GATA-1 (18) causes erythroid differentiation of MEL cells, the incompleteness of which, however, suggests an involvement of additional factors. In MEL cells, PU.1 physically interacts with GATA-1 (19–21) and silences the transcription of its target genes by creating a repressive chromatin structure (22, 23) that forms when PU.1 binding on GATA-1 involves deacetylation of histone H3 lysine 9 (H3K9) and its trimethylation (22, 23). Ectopic expression of PU.1 also blocks chemically induced erythroid differentiation of MEL cells (24). Previous studies showed that the Ets domain of PU.1 interacts with and mediates the repression of GATA-1 (19, 21) without altering DNA binding (22, 23). Conversely, the C finger of GATA-1 can interact with and mediate the repression of PU.1 (20, 21).
We asked whether GATA-1 blocks PU.1 in MEL cells, and whether by increasing the PU.1/GATA-1 level ratio we could induce PU.1 target genes and drive non-erythroid differentiation. Our data show that, indeed, activated PUER induces non-erythroid differentiation of MEL proerythroblasts into cell cycle–arrested non–erythroid-like cells. Using gene expression arrays, we have identified a specific set of genes regulated positively by PU.1 and inhibited by GATA-1 that contains two key hematopoietic transcription factors, Cebpa and Cbfb, both required for proper myeloid development. Mutations in CEBPA and Cbfb in human acute leukemias block this process at the blast stage (10, 25–29). Our data, supported by chromatin immunoprecipitation (ChIP) and reporter analyses, indicate that Cebpa and CBFB are repressed by the inhibitory activity of GATA-1 on PU.1 in MEL cells, and that increase in PU.1 levels could reverse this repression and lead to MEL cell differentiation.
Results
PUER Activation Results in Non-Erythroid Differentiation of MEL Cells
Previous work from our laboratory showed that activation of ectopic GATA-1-estrogen receptor (GER) induces GATA-1 target genes in MEL cells by a mechanism overriding the repressive block imposed by PU.1 on DNA (23). To determine whether PU.1, apart from its repressive function on GATA-1, could also activate its target genes directly on DNA in MEL cells, we first tested stable transfectants containing PU.1 cDNA driven by strong EF1α promoter (24) and observed mRNA upregulation of known PU.1 target genes, Itgam, Il7r, and Rag1 (data not shown). Second, we used MEL cells stably transfected with vector encoding inducible form of PU.1 fused to the ligand-binding domain of the estrogen receptor (PUER; ref. 18), in which the control MEL cells contained stable GER transgene (18).
Quantitative reverse transcription-PCR analysis confirmed our initial observation of activation of PU.1 target genes (Itgam, Cd14, Mpo, Cebpa, and Cbfb) in MELPUER cells stimulated for 4 and 16 hours by 17β-estradiol (Fig. 1A). Stimulation of MELGER cells with 17β-estradiol at identical time points resulted in rapid upregulation of GATA-1 target genes: Nfe2, Zfpm1 (Fig. 1C), Hba-a1, Hbb-b1, and Klf1 (Supplementary Fig. S1). Induced MELGER cells overtly hemoglobinized between 72 and 120 hours, whereas MELPUER cells remained pale in the floating cell fraction. Both MELPUER and MELGER cells significantly inhibited their proliferation rates following 48 hours of induction (Fig. 1D and E). Flow cytometry analysis of MELPUER cells revealed significant induction of myeloid surface markers Itgam (CD11b), Ptprc (CD45), and Ly6g (Gr-1) at indicated time points after 17β-estradiol treatment (Fig. 1B). These surface markers were not induced in MELGER cells stimulated for the same periods of time (data not shown). The MEL cells lacking a transgene, either untreated or treated with 17β-estradiol, grew exponentially with a similar doubling time (18). 17β-Estradiol did not affect PU.1 or GATA-1 expression levels in MEL cells indicating that, indeed, either the activated PUER or the activated GER transgene mediates the specific effects (18).6
FIGURE 1.

Conditional activation of PUER and GER in MEL cells restarts myeloid and erythroid program, respectively, and inhibits cell proliferation. MELPUER (A) and MELGER (C) cells were cultured in the presence or absence of 10−7 mol/L of 17β-estradiol (E) for the indicated time periods. Total RNA was purified and subjected to quantitative reverse transcription-PCR as described in Materials and Methods. Y-axis, mRNA expression of indicated genes relative to housekeeping gene Gapdh. B. Aliquots of MELPUER cells stimulated for the indicated time periods were immunostained using antibodies against Itgam, Ptprc, and Ly6g and control isotypic antibodies, as described in Materials and Methods. Y-axis, percentage of immunostained cells by flow cytometry analysis. Bars, SE calculated for two independent experiments. Proliferation of MELPUER (D) and MELGER (E) cells unstimulated or stimulated by 17β-estradiol for 96 h. Y-axis, maximum of cell number (%); X-axis, time (days).
The presence of specific PU.1-dependent transcriptional responses and of protein markers associated with non-erythroid differentiation of MEL cells led us to examine the morphology of the cells following PUER induction (Fig. 2). Parental MEL cells (clone DS19) represent a heterogeneous cell population (diameter, 15–25 μm) of proerythroblasts (26%) and partially differentiated basophilic erythroblasts (74%), detected by May-Grünwald-Giemsa staining. A similar appearance of these respective populations was found in unstimulated MELPUER (23% and 77%, respectively,) and MELGER cells (53% and 47%). As expected, erythroid differentiation induced within 72 hours in MELGER cells by 17β-estradiol induction resulted in a significant shift of the cell proportions toward the basophilic erythroblast stage (4% and 96%) which was also associated with the lack of observable mitoses. In turn, the cell line with 17β-estradiol–activated PUER (MELPUER) for 72 hours displayed a significant decrease in proerythroblasts (only 8%), little or no change in proportion of basophilic erythroblasts (64%), and lack of mitoses. Importantly, we also observed a new population of atypical cells (28%) with irregularly shaped nuclei and with mildly basophilic and azurophilic granular cytoplasm (see Fig. 2 and Supplementary Fig. S2 for summary and details; dashed arrows). These atypical cells stained positive for α-naphthyl butyrate esterase, and unlike MEL and MELGER cells, they poorly destained, retaining diffuse granular positivity after the addition of sodium fluoride (data not shown). This observation indicates that these cells display similarities with monocytes.
FIGURE 2.
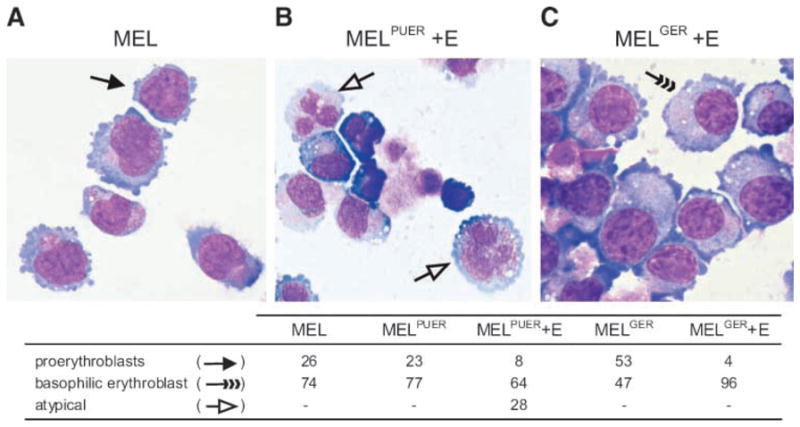
Distinct cell types are induced by PUER and GER transgenes from MEL cell line. MEL (A), MELPUER (B), and MELGER (C) cells were cultured in the presence of 10−7 mol/L of 17β-estradiol for 72 h, fixed, cyto-spinned, and stained using May-Grünwald-Giemsa. Microscopy (Olympus BX-51 apparatus, E-410 Camera) was done under ×1,000 magnification according to the manufacturer’s recommendations. Black arrow, proerythroblasts; dashed arrows, basophilic erythroblasts; empty arrows, atypical cells. The table displays the distribution of these cell types in MEL, MELPUER, MELPUER induced with 17β-estradiol for 3 d, MELGER, and MELGER cells induced with 17β-estradiol for 3 d.
Induced PUER Activates Its Endogenous Target Genes and Represses GATA-1 Targets in MEL Cells
As shown above, MEL cells can be differentiated by conditional GER and PUER activation into two distinct cell cycle–arrested populations within 96 hours. The mRNA and protein analyses revealed the induction of respective erythroid and non-erythroid expression programs. To identify complete profiles of PUER and GER activation in MEL cells, we used oligonucleotide expression arrays in biologically replicated profiling of MELGER or MELPUER cells stimulated with 10−7 mol/L of 17β-estradiol for the following time periods: 0, 2, 4, 8, 12, 16, 20, and 24 hours. The expression profiling data analysis identified 3,109 significant genes positively regulated by PU.1 and negatively regulated by GATA-1 (Supplementary Fig. S3A; Fig. 3A, left) and 4,292 significant genes positively regulated by GATA-1 and negatively regulated by PU.1 (Supplementary Fig. S3B; Fig. 3A, right). Subgroups of these genes encode critical lineage-specific regulatory molecules: transcription factors that are repressed in MEL cells and rapidly induced in MELPUER cells, but not in MELGER cells, including Cebpa, Cbfb, and also its established partner Runx1 (Fig. 3B; ref. 30). Runx1 cooperates with Cbfb on DNA and its expression is required for normal myelopoiesis, whereas its mutations predispose to leukemia (31–33). We also observed specific PU.1-dependent upregulation of Meis1, a known heterodimeric partner of Hoxa9, which is involved in myelopoiesis and also in leukemias harboring translocations with the mixed lineage leukemia gene (34, 35). PUER, but not GER, upregulated inhibitor of DNA binding 2, a known modulator of PU.1 and GATA-1 activities via interaction with transcription factor PU.1 (36); PUER also induced Ets1, another Ets family protein. We have also identified a subgroup of genes regulated by PUER, but not by GER, which belong to previously characterized differentiation-associated hematopoietic markers (Fig. 3B, bottom). Examples include markers of monocytes (Cd14 and Itgam), granulocytes (Mpo and Mmp9), and lymphocytes (Il7r and Thy1). An expanded set of the hematopoietic candidate targets of PU.1 and GATA-1 that are mutually inversely regulated is shown in Supplementary Fig. S3C to G. In addition, expression analysis also indicates that PU.1 and GATA-1 regulate two distinct sets of cell cycle genes involved in transcription factor–induced cell cycle arrest (Supplementary Fig. S3C–G). As shown in Fig. 3B, the majority of GATA-1 targets are already expressed in MEL cells, whereas the PU.1 targets are generally not expressed in MEL cells, indicating that the transcriptional program of PU.1 is markedly more inhibited than that of GATA-1, a fact also supported by the phenotypic appearance of MEL proerythroblasts (see Fig. 2; Supplementary Fig. S2).
FIGURE 3.

Gene expression of MEL cells could be reprogrammed either by PU.1 into mixed non-erythroid or by GATA-1 into erythroid transcriptional programs. MELPUER and MELGER cells were cultured for 24 h (X-axis) in the presence of 17β-estradiol in a duplicate experiment. Total RNA was purified and the fluorescently labeled cRNA probe was synthesized according to Affymetrix protocol for subsequent hybridization on the expression chip (MG-430A 2.0) containing 22602 probes. High-throughput data analyses were done using GeneSpring (Agilent Technologies) and MeV4 softwares. A. Box plots of gene expression patterns of indicated MEL cell lines stimulated for 24 h (X-axis; N, number of probes) identified by expression arrays (for details, see Materials and Methods and Supplementary Fig. S3A and B). Y-axis, relative expression of mRNAs relative to unstimulated cells. B. Cluster analysis of selected expression patterns (transcription factors and hallmarks of lineage differentiation) from gene expression arrays documenting the involvement of recognized lineage-specific mRNAs (for details, see Materials and Methods and Supplementary Fig. S3).
Activated PUER Induces Active Chromatin Structure Near Cebpa and Cbfb Genes, whereas Activated GER Inhibits This Effect
Our data indicated that PU.1 activates a large set of genes in MEL cells and inhibits a set of GATA-1 target genes. In contrast, activated GER induced genes inhibited by PUER (Fig. 3B). Among these genes were transcription factors previously associated with normal and leukemia cell differentiation. We have focused on key hematopoietic transcription factors Cbfb and Cebpa to determine the mechanism by which they are targeted and regulated by PU.1 and GATA-1. First, we measured the co-occupancy of PU.1 and GATA-1 by quantitative ChIP (qChIP) assay that covered 1-kb intervals of the selected genes (ranging from 10 kb upstream to 10 kb downstream relative to the transcription start site). Our data show co-occupancy of PU.1 and GATA-1 near their PU.1 binding sites at both Cbfb (Fig. 4A) near the PU.1 binding site at +1531 kb downstream of the transcription start site and Cebpa (Fig. 4B) near the PU.1 binding site at −2977 kb upstream of the transcription start site (see Tables 1 and 2; Supplementary Fig. S4E and F). Co-occupancy of PU.1 and GATA-1 near these early response gene targets, Cbfb and Cebpa, indicated that PU.1 and GATA-1 might regulate the expression of these target genes directly by a mechanism that involves regulatory changes in chromatin structure. This notion is also supported by studies showing that GATA-1 does not block binding of PU.1 to its DNA binding site (20, 21). To test this hypothesis, we have determined the levels of histone H3K9 acetylation near the candidate genes as this dynamic modification of chromatin was previously correlated with mutual antagonism of GATA-1 and PU.1 and with its disruption (23). As shown in Fig. 4A and B, PUER stimulation resulted in an increase of H3K9 acetylation near Cbfb and Cebpa genes (up to 2-fold and 3- to 4-fold, respectively) in comparison with GER stimulation or unstimulated MEL cells (data not shown). The direction of the H3K9 acetylation pattern correlated with the expression levels of the genes, and the chromatin change was not observed in other portions of the selected genes, indicating that the regulatory regions responded specifically to the activated (PUER and GER) transgenes.
FIGURE 4.

PU.1 and GATA-1 colo-calization near Cbfb and Cebpa genes and chromatin H3K9 hyperacetylation induced by ectopic PUER activation. ChIP was carried out on cross-linked chromatin as described in Materials and Methods using the following antibodies: anti-PU.1, anti–GATA-1 (both gray columns), anti–acetylated histone H3K9 (black columns), and control anti-rabbit IgG antibody (white columns, letter C on the X-axis). A and B. Occupancy of PU.1 and GATA-1 proteins at indicated positions (relative to transcription start site, in kilobases) near Cbfb and Cebpa was determined in unstimulated MELPUER cells. Levels of H3K9 hyperacetylation in these cells in the presence of 10−7 mol/L of 17β-estradiol for 24 h (third graphs in A and B) was determined relative to acetylation determined in stimulated MELGER cells under the same conditions. Primary binding sites of PU.1 were functional in reporter assays: 1.4 × 105 MELPUER cells were lipofected with Cbfb(+1531) (A, right) or Cebpa(−2978) (B, right) reporter plasmids (1.7 μg each). Cells remained unstimulated (−E) or treated with 17β-estradiol at 24 h (+E). Luciferase activity was determined 72 h after trans-fection (for details, see Materials and Methods). C and D. DNA regions (50–60 bp) near Cebpa(−2978) (no. 1 on X-axis) and Cbfb(+1531) (no. 2) were cloned together with a Cebpa(−2978) mutant (no. 3) into the reporter plasmid pGL3 and transfected into MELPUER (C) and MELGER (D) cells stimulated with 17β-estradiol for 72 h. Transfected qChIP technique using antibodies to PU.1, GATA-1, and antirabbit IgG antibody was done as described in Materials and Methods. PU.1 significantly stimulated luciferase activity of PU.1 binding site nos. 1 and 2, but not in site no. 3 (data not shown). The occupancy of PU.1 near Cebpa (−3.2 kb; E) and Cbfb (+2 kb; F) genes was tested by ChIP in either stimulated (48 h; +E) or unstimulated (−E) MELPUER cells and in MELPUER cells with knockdown of GATA-1 (si). All ChIP bars in C to F indicate the fold change (Y-axis) of DNA fragment in specific immunoprecipitates above the immunoprecipitates using control antirabbit IgG antibody. Bars, SE of at least two independent experiments.
Table 1.
GATA-1 and PU.1 Binding Sites Near Zfpm1, Nfe2, Cbfb, and Cebpa Genes
| Gene Name | Amplicon (+/− bp Relative to TSS) | Binding Site (+/− bp Relative to TSS) |
|---|---|---|
| Zpfm1 | 3537 to 3630 | +3579, AGATAA |
| Nfe2 | −2233 to −2111 | −1589, AGATAA |
| −1502 to −1390 | −1532, AGATAG | |
| −1084 to −966 | −1525, AGATAA | |
| −475 to −359 | −1052, AGATAG | |
| 22 to 194 | −804, AGATAG | |
| Cbfb | 1812 to 1913 | +1531, GAGGAACT |
| Cebpa | −3260 to −3170 | −2978, GAGGAAGT |
NOTE: Numbers indicate their relative position to the transcription start site.
Table 2.
Oligonucleotide Sequences of the Cloned Regions of Mouse Zfpm1, Nfe2, Cbfb, and Cebpa Genes
| Gene | Oligo Sequence | Relative Position from TSS |
|---|---|---|
| Zpfm1 | CTCTTTGAAATAAGATCAGCTGAGATAAGCATTCCGGGCTACAGGAAGCC GAGAAACTTTATTCTAGTCGACTCTATTCGTAAGGCCCGATGTCCTTCGG |
+3557 to +3606 |
| Nfe2 | TGAATTATCTGTAATCTGATATTATGTTACTATCTCTTATCTCCTATTCTGCCTATCTTATT ACTTAATAGACATTAGACTATAATACAATGATAGAGAATAGAGGATAAGACGGATAGAATAA |
Three segments: −1583 to −1559, −1537 to −1515, and −1055 to −1042 |
| ATTGATACGGCGTTAGATAGACCGTCTGATACAGAAAAGGATACCTCCTCAGATACCTG TAACTATGCCGCAATCTATCTGGCAGACTATGTCTTTTCCTATGGAGGAGTCTATGGAC |
Five segments: −828 to −818, −807 to −796, −687 to −676, −428 to −417, and −327 to −316 | |
| Cbfb | TTGAAAAATATCGAAGTGATCTAGTTCCTCTCTTGCCTCTTCGTTATCCTC AACTTTTTATAGCTTCACTAGATCAAGGAGAGAACGGAGAAGCAATAGGAG |
+1509 to +1559 |
| Cebpa | CCTCTGTAGCCGCTCCTGGAAGAGGAAGTGGGGTTGAAACAAGTCCTTTTG GGAGACATCGGCGAGGACCTTCTCCTTCACCCCAACTTTGTTCAGGAAAAC |
−2999 to −2949 |
NOTE: Numbers indicate their relative position to the transcription start site.
We then asked whether these PU.1 binding/response regions were functional in reporter assays. The PU.1 binding sites (see Materials and Methods) were subcloned into pGL3 reporter vector (see Table 2) encoding luciferase gene (Promega), transfected into MELPUER cells, and stimulated with 17β-estradiol for 48 hours. As shown in Fig. 4A and B (right), activated PUER in MEL cells stimulates the luciferase activity of these vectors, indicating that they respond to PU.1. The reporters were transfected into MELPUER (Fig. 4C) and MELGER (Fig. 4D) and stimulated for 72 hours, followed by analysis of luciferase activity (data not shown) and by transfected qChIP (details in Materials and Methods) using antibodies to PU.1 and GATA-1. The data indicate that PU.1 binds and transcriptionally stimulates the specific PU.1 DNA regions downstream of PU.1 binding sites Cbfb(+1531) and Cebpa(−2978) cloned in reporter plasmids. The data also indicate that GATA-1 could occupy these regions in the presence of PU.1. However, induction of MELPUER results in the depletion of GATA-1 from its association with PU.1 on DNA (Fig. 4E and F, right) followed by derepression of luciferase expression (data not shown).
Next, we used synthetic trimeric and pentameric PU.1 binding sites cloned into luciferase vectors (PU3x and PU5x) that were previously used to show PU.1 activation as well as its repression by GATA-1 in reporter assays (21). Supplementary Fig. S4A to D shows that the reporter vectors PU3x and PU5x are stimulated by PU.1 factor cotransfected into HeLa cells (Supplementary Fig. S4A–D) or induced as PUER in MEL cells by 17β-estradiol (Supplementary Fig. S4B). Supplementary Fig. S4C and D show that GATA-1 can inhibit PU.1 on its binding site in PU3x or PU5x and override the effect of transcription factor c-Jun, a known coactivator of PU.1 on DNA (20).
Activated GER Induces Active Chromatin Structure Near Zfpm1 and Nfe2 Genes, whereas Activated PUER Inhibits This Effect
The GATA-1 binding region near Nfe2 gene contains five putative GATA-1 binding sites (−1589, −1532, −1525, −1052, and −804 bp relative to the transcription start site; see Tables 1 and 2; Supplementary Fig. S5E). This GATA-1 binding region was previously shown to be negatively regulated by PU.1 or GATA-1 on DNA, which resulted in significant depletion of acetylated H3K9 (23). We used this region as a positive control (Fig. 5B) and tested whether another putative GATA-1 target gene, Zfpm1, was directly repressed by PUER and directly activated by GER. A qChIP assay done at a relatively large portion of the Zfpm1 gene revealed significant co-occupancy of GATA-1 and PU.1 (Fig. 5A) near the GATA-1 binding site at the 3′ portion of Zfpm1 gene (+3579; see Tables 1 and 2; Supplementary Fig. S5D). Furthermore, induction of MELPUER cells resulted in marked deacetylation near Zfpm1 and Nfe2 genes. An inverse pattern was observed at Nfe2 and Zfpm1 genes after the induction of GER (Fig. 5A and B). Subcloned GATA-1 binding sites were then tested using reporter assays in both MELGER and HeLa cells, and these experiments revealed that they are indeed induced by GATA-1 and that its effect was blocked dose-dependently by PU.1 (see Fig. 5, right; Tables 1 and 2). As controls, we used luciferase reporter constructs containing the chicken α-globin promoter fragment αD3 and its mutant αD4 (see Supplementary Fig. S5A–C). Significant co-occupancy of GATA-1 and PU.1 near GATA-1 binding sites near Zfpm1 and Nfe2 genes, induction of specific chromatin modification outcome, and the specific response of the DNA binding sites in reporter assays collectively indicated that the mutual repressive mechanisms between PU.1 and GATA-1 exist near the coding regions of the two critical hematopoietic transcription factors, Zfpm1 and Nfe2, and that these target genes may cooperate with GATA-1 during leukemia differentiation.
FIGURE 5.

Colocalization of GATA-1 and PU.1 proteins near Zfpm1 and Nfe2 genes and chromatin H3K9 hyperacetylation induced by ectopic GATA-1 activation. ChIP was carried out on cross-linked chromatin as described in Materials and Methods and in the legend to Fig. 4. Antibodies used were anti-PU.1, anti–GATA-1 (gray columns), anti–acetylated histone H3K9 (black columns), and antirabbit IgG antibody (letter C on the X-axis). A and B. Occupancy of PU.1 and GATA-1 proteins at the indicated positions (relative to transcription start site, in kilo-bases) near Zfpm1 and Nfe2 was determined in unstimulated MELGER cells. Levels of H3K9 hyperacetylation in these cells in the presence of 10−7 mol/L of 17β-estradiol for 24 h (third graphs) was determined relative to acetylation determined in stimulated MELPUER cells under the same conditions. Primary binding sites of GATA-1 are functional in reporter assays. A. Right, HeLa cells were transfected with reporter plasmid [Zfpm1, 0.25 μg (R)] and cDNA constructs [pXM-GATA-1, 0.125 μg (G) and pXM-PU.1, 0.125 μg (P); 0.375 μg (3P); 1.125 μg (9P)]. B. Right, MELGER cells were lipofected with Nfe2 reporter plasmid (1.7 μg) and either un-stimulated (−E) or further stimulated with 17β-estradiol after 24 h (+E) followed by measurement of luciferase activity at 72 h (for details, see Materials and Methods). C. MELPUER cells were either stimulated with 17β-estradiol (+E) or treated with PU.1-inhibiting siRNAs (si) for 48 h, and GATA-1 and PU.1 occupancy was detected by qChIP near Nfe2 (0 kb) and Zfpm1 (+3.5 kb) genes. All ChIP bars in C indicate the fold change (Y-axis) of DNA fragment in specific immunoprecipitates above the immunoprecipitates using control antirabbit IgG antibody. Bars, SE of at least two independent experiments.
To test if GATA-1 occupancy at GATA-1 binding sites near erythroid genes Nfe2 and Zfpm1 depends on PU.1, we did the following experiment. MELPUER cells were either stimulated with 17β-estradiol or treated with PU.1-inhibiting small interfering RNAs (siRNA) for 48 hours as described previously (17). Figure 5C shows that GATA-1 occupancy is detectable near Nfe2 (0 kb) and Zfpm1 (+3.5 kb) genes independently of PU.1 activation, a finding supported and complemented by previous findings on Nfe2 gene in MELGER cells (23).
Inhibition of GATA-1 Results in Non-Erythroid Differentiation of MEL Cells and Does Not Affect the Binding of PU.1 to Cebpa and Cbfb
Our data indicate that the transcription program of PU.1 is inhibited by GATA-1 on DNA, and that on activation of PU.1 by using PUER transgenes, we achieved efficient non-erythroid differentiation. To test whether GATA-1 is responsible for the inhibition of PU.1, we used siRNA oligonucleotides and inhibited GATA-1 expression and measured selected PU.1 target gene expression after 48 hours. As shown in Fig. 6A, inhibition of GATA-1 levels below 5% in MEL cells resulted in efficient derepression of the PU.1 target genes. Derepression of Cbfb and Cebpa genes seems to be more efficient after GATA-1 siRNA–mediated knockdown compared with stimulation of MELPUER cells by 17β-estradiol, supporting our observation of GATA-1–mediated repression of PU.1 target genes in MEL cells. In addition, derepression of Itgam and Cd14 genes after GATA-1 inhibition was achieved with slightly decreased efficiency compared with stimulation of PUER, indicating that full activation of Itgam and Cd14 might require additional PU.1 cofactors (such as Cebpa and Cbfb) at later time points. Conversely, PU.1-specific siRNA treatment resulted in the accumulation of mRNAs from GATA-1 target genes, Zfpm1 and Nfe2 (Fig. 6B). Our data indicate that GATA-1 is required in MEL cells to hold the repressive state of PU.1 target genes (see also Fig. 3), that both mechanisms of PU.1 repressing GATA-1 and of GATA-1 repressing PU.1 are functional in MEL cells, and that leukemia differentiation could be achieved by manipulating either of these two factors.
FIGURE 6.

Inhibition of GATA-1 derepresses PU.1 target genes and inhibition of PU.1 derepresses GATA-1 targets in MEL cells. A. MELPUER cells (8 × 104) were transfected either with siRNA oligos inhibiting GATA-1 (si) or with negative control oligo (CO) using Lipofectamine. The cells were cultured either in the absence (CO) or in the presence of 10−7 mol/L of 17β-estradiol (+E). B. MEL cells (8 × 104) were transfected either with siRNA oligos inhibiting PU.1 (si) or with negative control oligo (CO) using Lipofectamine and cultured for 72 h. Total RNA was purified and subjected to quantitative reverse transcription-PCR as described in Materials and Methods. Y-axis, mRNA expression of indicated genes relative to housekeeping gene Hprt1. Bars, SE of two independent experiments.
To test the mechanism by which PUER overcomes GATA-1–mediated repression on DNA, the occupancy of PU.1 near Cebpa and Cbfb genes was tested in either stimulated MELPUER cells (48 hours) or unstimulated MELPUER cells with knockdown of GATA-1. The qChIP technique using antibodies to PU.1 and GATA-1 was carried out as described in Materials and Methods. Activation of PUER (see Fig. 4E, +E on X-axis) or inhibition of GATA-1 by siRNA resulted in loss of GATA-1 occupancy at the PU.1 binding sites near Cebpa and Cbfb genes. Manipulation of GATA-1 or PU.1 did not result in a loss of PU.1 occupancy at these sites. These data strongly suggest that PUER overrides GATA-1–mediated repression of Cebpa and Cbfb by binding to DNA and that GATA-1 is not able to further associate with these regions. In addition, siRNA knockdown–mediated depletion of GATA-1 and its decreased occupancy near Cebpa and Cbfb genes results in transcriptional activation of Cebpa and Cbfb genes.
Discussion
PU.1 and GATA-1 are key transcription factors controlling the myelo-erythroid and myelo-lymphoid development of hematopoietic multipotential progenitors (4). PU.1 re-expression in PU.1−/− multipotential progenitors is sufficient to repress the program of GATA-1 by recruiting a repressive protein complex on its target genes (23). The next step of PU.1-dependent differentiation involves interactions with the PU.1-cooperating transcription factors Cebpa, Nab2, Gfi1, and Egr2 to progress further into the granulocyte-macrophage progenitor stage (5). In contrast, induction of erythroid differentiation in multipotential progenitors is followed by downregulation of PU.1 (37), a process that is likely prevented from completion in MEL cells by sustained low expression of PU.1 (16).
Our data support the hypothesis that PU.1-mediated block of GATA-1 in MEL cells resembles the block of erythroid differentiation that mediates PU.1 during early myeloid commitment of multipotential progenitors (23). The data show that MEL cells retain their multipotentiality and responsiveness to PU.1 and GATA-1 (see Fig. 3; Supplementary Fig. S4), and that PU.1 activity at the lineage commitment crossroads could redirect the erythroid commitment decision and drive the non-erythroid (monocyte-like and mixed) differentiation and cell cycle arrest of MEL cells. The manipulation of transcription factors PU.1 or GATA-1 that are directly involved in commitment decisions thus harbors a therapeutic potential for treating human leukemias. Our other data show that retroviral PU.1 rescue of several cellular systems of human acute leukemias that retained PU.1 and GATA-1 coexpression (NB4, K562 cell lines) or lacking their expression (U937) successfully induced differentiation and cell cycle arrest concomitant with the re-expression of multiple PU.1 target genes.6 These data are supported by experiments reported in other human leukemia cell systems (38, 39).
We report a strategy of using MEL cells stimulated with mutually antagonizing transcription factors PU.1 or GATA-1 that enabled us to identify mutually opposite gene expression programs regulating leukemia differentiation in a lineage-specific manner. These distinct programs are regulated at the chromatin DNA level, and whether and how these programs are propagated is determined by molecular interaction of GATA-1 and PU.1 at specific DNA-binding sites distributed near genes encoding transcription factors required for lineage specification such as Nfe2, Zfpm1, Cebpa, and Cbfb. This is in agreement with the studies indicating the critical and indispensable roles of Cebpa, Cbfb, Nfe2, and Zfpm1 genes for normal hematopoietic lineage specification (25–28). Furthermore, Cebpa and Cbfb are often mutated (10, 25–29) or epigenetically dysregulated (40) in human acute leukemias. Our data collectively indicate that leukemia differentiation in blocked leukemic MEL blasts is decided by a stoichiometric balance between PU.1 and GATA-1, the individual levels of which are sufficient to initiate specific changes of chromatin structure within their target genes that retained flexible responsiveness. The mechanisms of PU.1 and GATA-1, in a leukemic state and upon leukemia differentiation, involve the following putative steps: in myeloid genes such as Cebpa, PU.1 binds directly to DNA but is repressed by GATA-1 that binds directly to PU.1 molecules on DNA. Activation of PUER and stable levels of GATA-1 create excess of available PU.1, which is not paired by available GATA-1 on DNA, thus allowing gene activation. Similarly, in erythroid genes such as Nfe2, GATA-1 is bound to DNA but is repressed by PU.1 that binds to this GATA-1 molecule. Activation of GER creates an excess of available GATA-1, which is not paired on DNA by available PU.1, also allowing gene activation. Our mechanistic study implicates that transcription factor manipulation, such as inhibition of GATA-1 or activation of PU.1 in erythroleukemias, may represent an efficient tool of inducing leukemic blasts to differentiate.
Materials and Methods
Cell Cultures
MEL and HeLa cells were cultured in DMEM supplemented with 10% fetal bovine serum and antibiotics as described previously (22–24). The PUER and GER conditional activation in MEL cells containing these transgenes (MELPUER and MELGER) was induced by 10−7 mol/L of 17β-estradiol.
Extraction of Total RNA
Total cellular RNA was purified with modified TRIzol reagent (Invitrogen) involving enhanced precipitation by adding 1 vol of isopropanol to the extracted aqueous phase, precipitating at −20°C overnight, and centrifuging the RNA for 30 min at 14,000 rcf at 4°C. The concentration, purity, and integrity of total RNA were determined by NanoDrop ND-1000 and Agilent 2100 Bioanalyzer.
Microarray mRNA Profiling and Data Analysis
mRNA profiling in MELPUER and MELGER cells in a biologically duplicate experiment was done using Affymetrix Mouse Genome arrays MG-430A 2.0 containing 22,694 probes, following the one-cycle labeling protocol and standardized array processing procedures recommended by Affymetrix. The raw data (CEL files) were normalized using robust multi-chip average algorithm in GeneSpring GX software and filtered in the TM4 suite using significance analysis of microarrays with the false discovery rate set to 1%. The microarray data are deposited at the European Molecular Biology Laboratory’s European Bioinformatics Institute (ArrayExpress accession: E-MTAB-125).
ChIPs for qChIP
Chromatin from 3 × 107 cells from MELPUER and MELGER cells expressing PU.1-estrogen receptor or GATA-1-estrogen receptor (PUER and GER) fusion protein (in the absence or presence of 10−7 mol/L 17β-estradiol for 24 h) was cross-linked with 1% formaldehyde for 10 min at room temperature. Subsequently, cells were lysed by a set of lysis buffers to isolate the nuclei from cells that were resuspended in 2 mL of low-salt buffer and sonicated (45% intensity, 500 cycles of 2 s, in an ice-ethanol cooling bath) with a Branson Sonic Dismembrator model 500 equipped with a microtip to yield 200 to 400 bp DNA fragments (22, 23). ChIP was done using antibodies against PU.1 (T21, Santa Cruz), GATA-1 (N6, Santa Cruz), and acetyl-histone H3 (Lys9; Upstate); normal rabbit IgG (Cal-biochem) was used as a control nonspecific antibody. DNA extracted from the immunoprecipitates was used as the template for SYBR Green–based quantitative real-time PCR reactions as described below.
Transfected ChIP
Transient transfections into MELPUER and MELGER cells (105/mL) were carried out with the Lipofectamine 2000 reagent (Invitrogen) and the cells were simultaneously treated with 10−7 mol/L of 17β-estradiol. After 48 h, chromatin from ~106 cells was immunoprecipitated as described above.
Quantitative Real-time PCR
The quantity of specific DNA fragments in immunoprecipitates was determined by quantitative real-time PCR reactions. We used 7900 HT SDS PCR cycler with a 396-well configuration (Applied Biosystems). For the qChIP assays, 0.5 to 2 ng of immunoprecipitated DNA were amplified in each 1× SYBR Green Master Mix reaction (Applied Biosystems). All quantitative real-time PCR amplifications were done in 8-μL reaction volumes and consisted of 40 cycles of 10 s at 95°C, 20 s at 60°C, and 30 s at 72°C or in the presence of TaqMan probe (Roche) with the recommended protocol of 40 cycles of 15 s at 95°C and 1 min at 60°C. Fluorescence was read at both the annealing and polymerization steps. For each individual primer pair, a standard curve was generated using serial dilutions of the input DNA. The dissociation curve was determined for each PCR reaction to ensure the production of a single and specific product. Fluorescence was read in the exponential amplification phase (in case of SYBR Green reaction mix), and the raw data were expressed as CT values. Using a standard curve equation for each PCR primer pair, the CT values were transformed into DNA copy numbers. The copy number of a specific DNA fragment in each immunoprecipitate was compared with the copy number of that fragment in 1/100 dilution of the DNA obtained from the input sample used for immunoprecipitation (1% input DNA), and the “percentage of the input” was calculated. Percentage of input was also determined for each DNA fragment in immunoprecipitates using appropriate control antibodies (background), and these values were subtracted from the values obtained with the specific antibodies (details are also given in refs. 22, 23).
Immunoblotting
MELPUER and MELGER cells (3 × 106) were cultured in medium containing 10−7 mol/L of 17β-estradiol for 3 d and lysed for immunoblotting analysis using radioimmunoprecipitation assay buffer supplemented with proteinase inhibitors. Denatured lysates (20 μg protein per lane) were resolved on 10% PAGE. The gels were stained with Coomassie dye for load control and wet-blotted onto polyvinylidene difluoride membranes. Blots were blocked in 7.5% nonfat milk-PBS-Tween 20. Primary antibodies were used at 1:500 dilution [anti-PU.1 (sc-352), anti–GATA-1 (sc-265; Santa Cruz), and anti–estrogen receptor α (ab31949, Abcam)]. After washing with PBS-Tween 20, the membranes were stained with secondary horseradish peroxidase–labeled antibodies, followed by luminescence detection on X-ray films.
Transient Transfections and Reporter Assays
The putative GATA-1 binding sites in murine Zfpm1 (+3579, AGATAA) and Nfe2 (−1588 and −1524 AGATAA; −1531, −1051 and −803 AGATAG) genes and the PU.1 binding sites in murine Cbfb (+1531, GAGGAACT) and Cebpa (−2977, GAGGAAGT) genes and their deletion mutants were subcloned into the pGL3-basic vector (Promega). Transient transfections into MELPUER, MELGER, and HeLa cells were carried out with the Lipofectamine 2000 reagent (Invitrogen). Firefly luciferase activity was measured ~48 h after transfection using the Steady-Glo Luciferase Assay System (Promega) and shown as fold activation over background. Individual transfection experiments were done in duplicate and the results are reported as mean firefly fold activation ± SD.
Flow Cytometry
MELPUER and MELGER cells (1 × 105) induced with 17β-estradiol for the indicated time points were incubated with 2 μL of phycoerythrin-conjugated mouse monoclonal CD45 antibody (clone 30-F11, PharMingen), biotin-conjugated monoclonal CD11b antibody (clone M1/70, PharMingen), or biotin-conjugated monoclonal Gr-1 antibody (clone RB6-8C5, PharMingen). Biotinylated CD11b and Gr-1 antibodies were visualized with 2 μL of streptavidin-phycoerythrin (PharMingen). Flow cytometry analyses were done on an Aria cell sorter (BD).
SiRNA Inhibition
MELPUER cells were cultured in DMEM supplemented with 10% fetal bovine serum. SiRNA (Ambion) complementary to GATA-1 or negative control siRNA (Ambion) was transfected using the Lipofectamine 2000 reagent (Invitrogen). After 48 h, cells were harvested and total RNAs were isolated and analyzed by quantitative real-time PCR as described above. Custom siRNA (Qiagen) complementary to PU.1 (5′-ggaggugucugauggagaa-3′) with 3′ dTdT overhang.
Supplementary Material
Acknowledgments
We thank the NYU Cancer Institute Genomics Facility for superb technical assistance with the gene expression microarray work; Drs. J. Vargova, V. Pospisil, and K. Vargova for help in conducting experiments; Drs. L. Sefc and F. Savullidi for their expert assistance with flow cytometry analyses; and Drs. E. Necas and M. Trneny for stimulating discussions and mentorship.
Grant support: Grant Agency of the Czech Republic (301/06/1093), Internal Grant Agency of Ministry of Health (NR9021-4, 10310-3), and Grant Agency of Ministry of Education, Czech Republic (NPVII 2B06077, MSM 0021620806, and LC 06044). A.I. Skoultchi was supported by NIH grant HL 78381.
Footnotes
Stopka et al., unpublished observation.
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
The author contributions are as follows: P. Burda performed the ChIP and mRNA expression analyses and wrote the manuscript; N. Curik cloned the reporter plasmids and carried out the flow cytometry and transfection experiments; J. Kokavec cooperated in the microarray and DNA mapping analyses; P. Basova performed the reporter analyses; D. Mikulenkova was responsible for cell microscopy and cytochemistry; A.I. Skoultchi established the cell lines models; J. Zavadil designed and conducted mRNA profiling experiments; and T. Stopka designed and interpreted the experiments and wrote the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Graf T. Immunology: blood lines redrawn. Nature. 2008;452:702–3. doi: 10.1038/452702a. [DOI] [PubMed] [Google Scholar]
- 2.DeKoter RP, Kamath MB, Houston IB. Analysis of concentration-dependent functions of PU.1 in hematopoiesis using mouse models. Blood Cells Mol Dis. 2007;39:316–20. doi: 10.1016/j.bcmd.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dahl R, Simon MC. The importance of PU.1 concentration in hematopoietic lineage commitment and maturation. Blood Cells Mol Dis. 2003;31:229–33. doi: 10.1016/s1079-9796(03)00152-9. [DOI] [PubMed] [Google Scholar]
- 4.Arinobu Y, Mizuno S, Chong Y, et al. Reciprocal activation of GATA-1 and PU.1 marks initial specification of hematopoietic stem cells into myeloerythroid and myelolymphoid lineages. Cell Stem Cell. 2007;1:416–27. doi: 10.1016/j.stem.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Laslo P, Spooner CJ, Warmflash A, et al. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell. 2006;126:755–66. doi: 10.1016/j.cell.2006.06.052. [DOI] [PubMed] [Google Scholar]
- 6.Feng R, Desbordes SC, Xie H, et al. PU.1 and C/EBPα/β convert fibroblasts into macrophage-like cells. Proc Natl Acad Sci U S A. 2008;105:6057–62. doi: 10.1073/pnas.0711961105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang G, Zhang P, Hirai H, et al. PU.1 is a major downstream target of AML1 (RUNX1) in adult mouse hematopoiesis. Nat Genet. 2008;40:51–60. doi: 10.1038/ng.2007.7. [DOI] [PubMed] [Google Scholar]
- 8.Rosenbauer F, Wagner K, Kutok JL, et al. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat Genet. 2004;36:624–30. doi: 10.1038/ng1361. [DOI] [PubMed] [Google Scholar]
- 9.Metcalf D, Dakic A, Mifsud S, Di Rago L, Wu L, Nutt S. Inactivation of PU.1 in adult mice leads to the development of myeloid leukemia. Proc Natl Acad Sci U S A. 2006;103:1486–91. doi: 10.1073/pnas.0510616103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mueller BU, Pabst T. C/EBPα and the pathophysiology of acute myeloid leukemia. Curr Opin Hematol. 2006;13:7–14. doi: 10.1097/01.moh.0000190110.08156.96. [DOI] [PubMed] [Google Scholar]
- 11.Kim SI, Bresnick EH. Transcriptional control of erythropoiesis: emerging mechanisms and principles. Oncogene. 2007;26:6777–94. doi: 10.1038/sj.onc.1210761. [DOI] [PubMed] [Google Scholar]
- 12.Jing H, Vakoc CR, Ying L, et al. Exchange of GATA factors mediates transitions in looped chromatin organization at a developmentally regulated gene locus [see comment] Mol Cell. 2008;29:232–42. doi: 10.1016/j.molcel.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mueller BU, Pabst T, Osato M, et al. Heterozygous PU.1 mutations are associated with acute myeloid leukemia. Blood. 2002;100:998–1007. doi: 10.1182/blood.v100.3.998. [DOI] [PubMed] [Google Scholar]
- 14.Wechsler J, Greene M, McDevitt MA, et al. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet. 2002;32:148–52. doi: 10.1038/ng955. [DOI] [PubMed] [Google Scholar]
- 15.Freson K, Thys C, Wittewrongel C, Vermylen J, Hoylaerts MF, Van Geet C. Molecular cloning and characterization of the GATA1 cofactor human FOG1 and assessment of its binding to GATA1 proteins carrying D218 substitutions. Hum Genet. 2003;112:42–9. doi: 10.1007/s00439-002-0832-1. [DOI] [PubMed] [Google Scholar]
- 16.Moreau-Gachelin F, Tavitian A, Tambourin P. Spi-1 is a putative oncogene in virally induced murine erythroleukaemias. Nature. 1988;331:277–80. doi: 10.1038/331277a0. [DOI] [PubMed] [Google Scholar]
- 17.Papetti M, Skoultchi AI. Reprogramming leukemia cells to terminal differentiation and growth arrest by RNA interference of PU.1. Mol Cancer Res. 2007;5:1053–62. doi: 10.1158/1541-7786.MCR-07-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choe KS, Radparvar F, Matushansky I, Rekhtman N, Han X, Skoultchi AI. Reversal of tumorigenicity and the block to differentiation in erythroleukemia cells by GATA-1. Cancer Res. 2003;63:6363–9. [PubMed] [Google Scholar]
- 19.Rekhtman N, Radparvar F, Evans T, Skoultchi AI. Direct interaction of hematopoietic transcription factors PU.1 and GATA-1: functional antagonism in erythroid cells. Genes Dev. 1999;13:1398–411. doi: 10.1101/gad.13.11.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang P, Behre G, Pan J, et al. Negative cross-talk between hematopoietic regulators: GATA proteins repress PU.1. Proc Natl Acad Sci U S A. 1999;96:8705–10. doi: 10.1073/pnas.96.15.8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nerlov C, Querfurth E, Kulessa H, Graf T. GATA-1 interacts with the myeloid PU.1 transcription factor and represses PU.1-dependent transcription. Blood. 2000;95:2543–51. [PubMed] [Google Scholar]
- 22.Rekhtman N, Choe KS, Matushansky I, Murray S, Stopka T, Skoultchi AI. PU.1 and pRB interact and cooperate to repress GATA-1 and block erythroid differentiation. Mol Cell Biol. 2003;23:7460–74. doi: 10.1128/MCB.23.21.7460-7474.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stopka T, Amanatullah DF, Papetti M, Skoultchi AI. PU.1 inhibits the erythroid program by binding to GATA-1 on DNA and creating a repressive chromatin structure. EMBO J. 2005;24:3712–23. doi: 10.1038/sj.emboj.7600834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao G, Rekhtman N, Cheng G, Krasikov T, Skoultchi AI. Deregulated expression of the PU.1 transcription factor blocks murine erythroleukemia cell terminal differentiation. Oncogene. 1997;14:123–31. doi: 10.1038/sj.onc.1200807. [DOI] [PubMed] [Google Scholar]
- 25.Kirstetter P, Schuster MB, Bereshchenko O, et al. Modeling of C/EBPα mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells [see comment] Cancer Cell. 2008;13:299–310. doi: 10.1016/j.ccr.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 26.Miller J, Horner A, Stacy T, et al. The core-binding factor β subunit is required for bone formation and hematopoietic maturation. Nat Genet. 2002;32:645–9. doi: 10.1038/ng1049. [DOI] [PubMed] [Google Scholar]
- 27.Shivdasani RA, Orkin SH. Erythropoiesis and globin gene expression in mice lacking the transcription factor NF-E2. Proc Natl Acad Sci U S A. 1995;92:8690–4. doi: 10.1073/pnas.92.19.8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsang AP, Visvader JE, Turner CA, et al. FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor GATA-1 in erythroid and megakaryocytic differentiation. Cell. 1997;90:109–19. doi: 10.1016/s0092-8674(00)80318-9. [DOI] [PubMed] [Google Scholar]
- 29.Shigesada K, van de Sluis B, Liu PP. Mechanism of leukemogenesis by the inv(16) chimeric gene CBFB/PEBP2B-MHY11. Oncogene. 2004;23:4297–307. doi: 10.1038/sj.onc.1207748. [DOI] [PubMed] [Google Scholar]
- 30.Wang Q, Stacy T, Miller JD, et al. The CBFβ subunit is essential for CBFα 2 (AML1) function in vivo. Cell. 1996;87:697–708. doi: 10.1016/s0092-8674(00)81389-6. [DOI] [PubMed] [Google Scholar]
- 31.Kundu M, Compton S, Garrett-Beal L, et al. Runx1 deficiency predisposes mice to T-lymphoblastic lymphoma. Blood. 2005;106:3621–4. doi: 10.1182/blood-2005-04-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ganly P, Walker LC, Morris CM. Familial mutations of the transcription factor RUNX1 (AML1, CBFA2) predispose to acute myeloid leukemia. Leuk Lymphoma. 2004;45:1–10. doi: 10.1080/1042819031000139611. [DOI] [PubMed] [Google Scholar]
- 33.de Bruijn MF, Speck NA. Core-binding factors in hematopoiesis and immune function. Oncogene. 2004;23:4238–48. doi: 10.1038/sj.onc.1207763. [DOI] [PubMed] [Google Scholar]
- 34.Faber J, Krivtsov AV, Stubbs MC, et al. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood. 2008;113:2375–85. doi: 10.1182/blood-2007-09-113597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin G, Yamazaki Y, Takuwa M, et al. Trib1 and Evi1 cooperate with Hoxa and Meis1 in myeloid leukemogenesis. Blood. 2007;109:3998–4005. doi: 10.1182/blood-2006-08-041202. [DOI] [PubMed] [Google Scholar]
- 36.Ji M, Li H, Suh HC, Klarmann KD, Yokota Y, Keller JR. Id2 intrinsically regulates lymphoid and erythroid development via interaction with different target proteins. Blood. 2008;112:1068–77. doi: 10.1182/blood-2008-01-133504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nutt SL, Metcalf D, D’Amico A, Polli M, Wu L. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. J Exp Med. 2005;201:221–31. doi: 10.1084/jem.20041535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Durual S, Rideau A, Ruault-Jungblut S, et al. Lentiviral PU.1 overexpression restores differentiation in myeloid leukemic blasts. Leukemia. 2007;21:1050–9. doi: 10.1038/sj.leu.2404645. [DOI] [PubMed] [Google Scholar]
- 39.Mueller BU, Pabst T, Fos J, et al. ATRA resolves the differentiation block in t (15;17) acute myeloid leukemia by restoring PU.1 expression. Blood. 2006;107:3330–8. doi: 10.1182/blood-2005-07-3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jost E, do ON, Wilop S, Herman JG, Osieka R, Galm O. Aberrant DNA methylation of the transcription factor C/EBPα in acute myelogenous leukemia. Leuk Res. 2008;33:443–9. doi: 10.1016/j.leukres.2008.07.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.