

Abstract
We previously demonstrated that hearts from Brown Norway (BN) rats were more resistant to ischemic injury than hearts from Dahl S (SS) rats. Here we determined the susceptibility to LPS-induced cardiomyopathy in these rats and examined the involvement of inflammatory signaling. Both strains were treated with LPS (20 mg/kg) via i.p. injection for 6 h. Myocardial function was assessed by the Langendorff system, and proinflammatory cytokines were measured by the enzyme-linked immunosorbent assay. LPS significantly reduced left ventricular developed pressure in both strains. Interestingly, the decrease of left ventricular developed pressure in BN rat hearts was approximately 25% less than that in SS rat hearts. Furthermore, LPS significantly reduced the peak rate of contraction and the peak rate of relaxation in SS hearts but not in BN hearts. No differences in LPS-induced decreases in coronary flow rate were observed between BN and SS rats. In addition, LPS-induced increases in proinflammatory cytokines, TNF-α, IL-1β, and IL-6, were significantly lower in both plasma and hearts of BN rats compared with production in SS rats. LPS notably up-regulated the expression of proinflammatory enzymes, iNOS and cyclooxygenase 2, in SS hearts but not in BN hearts. Interestingly, LPS did not stimulate Toll-like receptor 4 or its adaptor myeloid differentiation factor 88 expression in the hearts of either strain but did increase IκB and P65 phosphorylation, less prominently in BN hearts than in SS hearts. These data indicate that reduced production of proinflammatory cytokines and diminished nuclear factor κB activation are major mechanisms by which BN hearts are more resistant to LPS-induced myocardial dysfunction than SS hearts.
Keywords: Myocardial dysfunction, Brown Norway rats, Dahl S rats, LPS, NF-κB pathway, proinflammatory cytokines
INTRODUCTION
Septic shock is a severe syndrome caused by decreased tissue perfusion and oxygen delivery due to infection and sepsis. In the United States, septic shock is one of the primary causes of death in the intensive care unit (1). Although a majority of late mortality from sepsis seems to be due to multisystem organ failure, early death has been attributed either to distributive shock or to a cardiogenic form of septic shock (1, 2). Myocardial depression by septic shock is characterized by impaired myocardial contractility and reduced ejection fraction (1). Further, 40% to 50% of patients with prolonged septic shock developed myocardial dysfunction (3).
Myocardial dysfunction caused by septic shock is due, in part, to the production of proinflammatory cytokines induced by the bacterial endotoxin LPS. Although the precise mechanism is incompletely understood, increasing evidence suggests that LPS-induced endotoxemia and myocardial dysfunction are caused by multiple proinflammatory mediators, such as TNF-α, IL-1β, and cytokine iNOS (4, 5). TNF-α is an early mediator of endotoxin-induced shock. IL-1β plays crucial roles in systemic immune responses and also depresses myocardial function by stimulating iNOS expression (6). IL-6 is consistently increased in patients with septic shock and is considered a “strong” biomarker of the severity of bacterial infection (7). Recent studies have suggested that the Toll-like receptor pathway (especially Toll-like receptor 4 [TLR-4]) signals LPS bioactivity (8). Toll-like receptor 4 binds LPS and mediates LPS signaling in conjunction with other molecules such as myeloid differentiation factor 88 (MyD88). Myeloid differentiation factor 88 is a universal adaptor protein that is used by all TLRs (except TLR-3) to activate nuclear factor κB (NF-κB) (9).
Although proinflammatory cytokines act directly or indirectly to cause cardiac myocyte injury, the etiology of and the strategies to prevent myocardial dysfunction in sepsis in humans remain elusive. Previously, we have shown that hearts from Brown Norway (BN) rats were more resistant to ischemia reperfusion injury than hearts from Dahl S (SS) rats (10). An important advantage in studying mechanisms of protection against and susceptibility to myocardial dysfunction in BN and SS rats is that an extensive body of information already exists regarding phenotypic differences in cardiovascular and renal function between these strains (http://www.rgd.mcw.edu/). However, no information about resistance to endotoxemia is available in BN and SS rats. In the present study, we investigated the mechanisms by which BN rats are more resistant to myocardial dysfunction induced by LPS than SS rats. We determined differences in resistance of hearts from BN and SS rats to LPS-induced cardiomyopathy and examined the involvement of proinflammatory mediators as well as TLR-4 pathways.
MATERIALS AND METHODS
Materials
LPS was purchased from Sigma (St Louis, Mo). Antibodies against phospho-P65, P65, phospho-IκB, and IκB were from Cell Signaling Technology (Boston, Mass). Antibodies against TLR-4, Myd88, iNOS, and GAPDH were from Santa Cruz (Santa Cruz, Calif). Anti–cyclooxygenase 2 (COX-2) was from Cayman Chemical (Ann Arbor, Mich).
Animal model
Eight-week-old BN and SS male rats were obtained from Charles River (Wilmington, Mass). All animal protocols were approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin. Rats used in this study received humane care in compliance with the Guide for the Care and Use of Laboratory Animals by the National Research Council.
Rats were maintained on a low salt (0.4% NaCl) diet with unlimited access to water. Environmental influences were minimized by maintaining rats in identical housing conditions. Rats were injected with either 20 mg/kg i.p. LPS or saline. Six hours later, animals were anesthetized with pentobarbital sodium (50 mg/kg i.p.), and hearts were rapidly excised and perfused for 30 min in the Langendorff mode. For analysis of the NF-κB pathway, rats were injected with 20 mg/kg i.p. LPS or saline for 30 min. The hearts were excised, flushed quickly with Krebs buffer, rapidly frozen in liquid nitrogen, and stored at −80°C until use.
Langendorff isolated heart preparation and measurements
Hearts from BN and SS rats were isolated and perfused as previously described (10, 11). Briefly, hearts were perfused in the Langendorff mode at a perfusion pressure equivalent to 80 mmHg. Perfusate and bath temperatures were maintained at 37.2 ± 0.1°C using a thermostatically controlled water circulator (Lauda E100; Lauda Dr. R. Wobser GMBH & CO KG, Pfarrstraβe, Germany). Left ventricular developed pressure (LVDP) was measured isovolumetrically with a transducer connected to a thin saline-filled latex balloon inserted into the left ventricle through the mitral valve from an incision in the left atrium. At the end of the experiments, the hearts were freeze clamped and stored at −80°C until use. Coronary flow rate was measured by timed collections of the coronary effluent from the right side of the heart into a graduated cylinder. Coronary flow rate was expressed as milliliters per minute.
Western blot analysis
Western blot analysis was performed as previously reported (12). Briefly, hearts were homogenated in lysis buffer, and 60 μg of the homogenate was loaded onto and separated by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The separated proteins were transferred to a nitrocellulose membrane, and the membrane was incubated with primary antibodies overnight and secondary antibody for 1 h. Bands of identity were then visualized with the Super Signal West Pico kit (Pierce Biotechnology, Rockford, Ill).
Enzyme-linked immunosorbent assay
Heart tissue was homogenized in ice-cold lysis buffer, and the supernatant was collected and used for ELISA and protein measurements. Levels of TNF-α, IL-1β, and IL-6 in plasma or heart homogenates were determined by murine-specific DuoSet ELISA development kits (R&D Systems, Minneapolis, Minn). Data are expressed as a function of protein (picogram or nanogram per milliliter in plasma, and picogram per milligram protein in heart tissue).
Statistical analysis
Data are expressed as mean ± SD and were analyzed by 1-way ANOVA or by 2-tailed t test. P < 0.05 was considered statistically significant.
RESULTS
Effects of LPS on myocardial function in BN and SS rats
To determine whether the inhibitory effects of LPS on myocardial function are different in hearts from BN and SS rats, we isolated the hearts and determined the myocardial function 6 h after rats were i.p. injected with LPS. Left ventricular developed pressure, which is systolic minus diastolic pressure, was used to assess myocardial function. In both untreated BN and SS rats, no differences in LVDP were observed; however, LPS treatments decreased LVDP compared with untreated controls in both strains (Fig. 1A, upper panel; P < 0.05). Interestingly, there was 25% less decrease in LVDP in BN hearts than in SS hearts (Fig. 1A, lower panel). Further, LPS significantly reduced the peak rate of contraction (+dP/dt) and the peak rate of relaxation (−dP/dt) in SS hearts but not in BN hearts (Fig. 1, B and C; P < 0.05). LPS did not affect heart rates in either strain (Fig. 1D, upper panel); however, LPS decreased coronary flow to the same extent in both BN and SS hearts (Fig. 1D, low panel; P < 0.05).
Fig. 1. Different resistance to LPS-induced endotoxemia in BN and SS rat hearts.

LVDP, +dP/dt and −dP/dt, heart rate, and coronary flow were determined after hearts from controls and LPS-treated rats (6 h) were isolated and perfused for 30 min in Langendorff mode. Values are presented as mean ± SD. *P < 0.05 versus control group without LPS; #P < 0.05 compared with SS rats with LPS (n = 6).
Effects of LPS on cytokine production in plasma and hearts in BN and SS rats
Six hours after injection, LPS significantly increased the release of inflammatory cytokines into the plasma of both BN and SS rats (Fig. 2, A–C), whereas these cytokines were undetectable in normal rat plasma. Consequently, cytokine levels in control rats were not included in Figure 2. LPS significantly increased TNF-α, IL-1β, and IL-6 in plasma of both BN and SS rats; however, the magnitude of the increase in BN rats was much smaller than that in SS rats (Fig. 2, A–C). Among these cytokines, TNF-α was elevated dramatically in SS rats (~158-fold higher than in BN rats). No significant differences in TNF-α, IL-1β, or IL-6 were observed in hearts from the control BN and SS rats. Similar to plasma data, LPS also increased cytokine expression in the hearts of both strains. One exception was TNF-α, which was not significantly increased in BN hearts after LPS treatments (Fig. 3, A–C). Compared with SS rats, BN rats produced much lower levels of TNF-α, IL-1 β, and IL-6 in their hearts after LPS treatment (P < 0.05, n = 6).
Fig. 2. Inflammatory cytokines in plasma from SS and BN rats challenged by LPS.
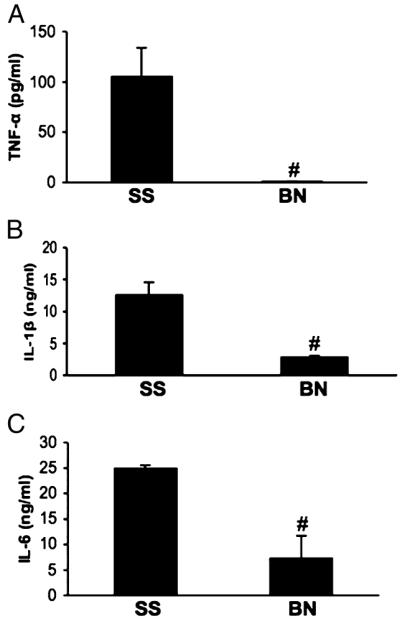
Plasma from SS and BN were assayed for TNF-α, IL-1β, and IL-6 by ELISA. A, TNF-α; B, IL-1β; C, IL-6. *P < 0.05 versus control without LPS; #P < 0.05 versus SS rats with LPS (n = 6).
Fig. 3. Inflammatory cytokines in hearts from SS and BN rats challenged by LPS.

Heart homogenates from SS and BN were assayed for TNF-α, IL-1β, and IL-6 by ELISA. A, TNF-α; B, IL-1β; C, IL-6. *P < 0.05 versus control without LPS; #P < 0.05 versus SS rats with LPS (n = 6).
Effects of LPS on iNOS and COX-2 expression in hearts
iNOS and COX-2 are classic proinflammatory enzymes that have been shown to dramatically increase in response to LPS. As shown in Figure 4, iNOS and COX-2 were almost undetectable in hearts from control rats in both strains. However, after 6 h, LPS strongly increased iNOS and COX-2 expression in SS hearts but very weakly in BN hearts (Fig. 4).
Fig. 4. The expression of iNOS and COX-2 in the hearts.
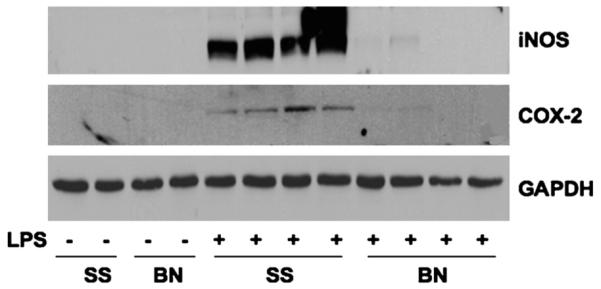
Six hours after LPS injection, hearts from SS and BN rats were homogenated and analyzed by Western blot analysis. The blots were probed with antibodies against iNOS and COX-2 and reprobed with GAPDH antibody (n = 4).
Expression of TLR-4 and MyD88 in hearts from BN and SS rats
To explore the possibility that the differences in cytokines and proinflammatory cytokines are a result of the differences in the expression of TLR-4 and MyD88, we probed heart homogenates from SS and BN rats with specific antibodies. No significant differences in basal levels of TLR-4 and MyD88 expression were observed in the hearts from SS and BN rats. In addition, LPS did not induce TLR-4 or MyD88 expression in the hearts of either strain of rat (Fig. 5, A–C; n = 5).
Fig. 5. The expression of TLR-4 and MyD88 in the hearts.

Six hours after LPS injection, hearts from SS and BN rats were homogenated and analyzed for Western blot analysis. A, Representative blots of 5 repeats for TLR-4, MyD88, and GAPDH. B, Relative expression of TLR-4. C, MyD88 after normalization with GAPDH. NS indicates no significant difference versus SS rats with or without LPS (n = 5).
The effects of LPS on the activation of NF-κB pathway
In 30 min, LPS induced IκB phosphorylation and NF-κB subunit p65 phosphorylation (at Ser536) as shown in Figure 6, but both IκB and p65 phosphorylation were much weaker in BN hearts compared with the level of transcription factor activation in SS hearts (Fig. 6).
Fig. 6. The NF-κB activation in the hearts.
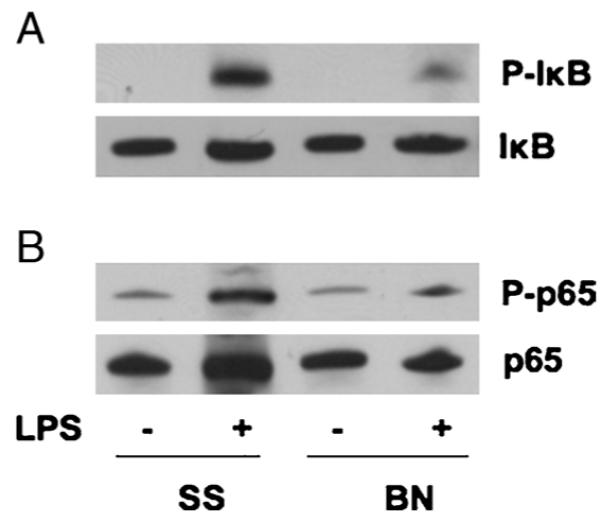
Thirty minutes after LPS injection (20 mg/kg), hearts from BN and SS rats were homogenated and analyzed for Western blot analysis. The blots were probed with antibodies against P-IκB (A) and P-p65 (B) and reprobed with antibodies against IκB and p65 after stripping. The blots were representative of 3 repeats.
DISCUSSION
In this study, we compared the effects of endotoxin-induced myocardial dysfunction in BN and SS rats. We reported here for the first time that hearts from BN rats are more resistant to LPS-induced injury than hearts from SS rats. Furthermore, LPS-induced inflammatory cytokine production in both plasma and hearts was much lower in BN rats than in SS rats. Activation of NF-κB was weaker in the hearts of BN rats compared with SS rats. However, LPS did not affect TLR-4 or MyD88 expression in the hearts from either strain. Our data indicate that reduced production of proinflammatory cytokines and diminished activation of NF-κB are the major mechanisms by which BN hearts are more resistant to LPS-induced myocardial dysfunction than SS hearts.
Our results demonstrate that LPS depresses cardiac function in both SS and BN rats as shown by decreases in LVDP and ±dP/dt. These data are consistent with observations in septic patients and different animal models of sepsis (1). Furthermore, LPS directly inhibits cardiac contractility in vitro in isolated hearts and cardiomyocytes (13). In a similar study in LPS-treated Sprague-Dawley male rats (14), LVDP decreased to approximately 50% of the LVDP of the controls, which is close to values observed in the SS rats in our study. Interestingly, LPS reduced coronary flow to a similar extent in both BN and SS rats, indicating that global ischemia was likely not responsible for the differences in LPS-induced myocardial dysfunction in these animals.
Many myocardial depressant factors have been proposed to play a causal role in inducing myocardial dysfunction in septic shock (13, 15). TNF-α, IL-1β, IL-6, ·NO, and prostanoids are the major factors mediating cardiac depression. TNF-α is mainly derived from activated macrophages, cardiomyocytes (16), and cardiac fibroblasts after LPS stimulation (17). Our results showed that the TNF-α response to LPS stimulation was blunted in BN hearts and plasma. When we isolated activated macrophages from LPS-treated BN and SS rats, we also found that TNF-α production by macrophages isolated from BN rats was only approximately 20% of the level that was produced by macrophages isolated from SS rats (data not shown). On the basis of these data, it is possible that the high levels of TNF-α in SS rat plasma were secreted by tissues and cells other than the heart. Although LPS significantly increased IL-1β and IL-6 levels in plasma and hearts from both BN and SS rats, BN rats had much lower levels of induction, suggesting that the blunted cytokine response may have been responsible for a better outcome after LPS. It is known that all 3 isoforms of NOS exist in the heart, and during sepsis, iNOS is dramatically up-regulated, resulting in increased ·NO production. Overproduction of ·NO is well recognized as a myocardial depressant (18). In addition, ·NO could also react with superoxide to form cytotoxic peroxynitrite. In our study, iNOS was strongly up-regulated by LPS in SS rats but weakly induced in BN rats, which is consistent with the changes in cardiac function. Furthermore, the induction of COX-2, the enzyme responsible for producing prostanoids from arachidonic acid, was also blunted in BN rats. It should be noted that in BN rats, the induction of TNF-α and iNOS in the heart by LPS was not increased appreciably, although myocardial depression was reduced by approximately 20% by LPS. These data suggest that IL-1β and IL-6 may be more important in LPS-induced cardiomyopathy than TNF-α. Alternatively, LPS may not act directly on the myocardium. However, these questions remain to be defined in future studies.
LPS-induced myocardial dysfunction depends on the presence of TLR-4. Toll-like receptor 4–deficient mice are protected from LPS-induced cardiac dysfunction (19, 20). Eighteen hours after treatment with LPS, TLR-4 expression is induced in myocardium of mice (21). However, in macrophages, TLR-4 expression decreases within 1 h after LPS treatment and remains suppressed for more than 24 h (11). Interestingly, in monocytes repeatedly treated with LPS, TLR-4 expression remains unchanged (22). In our study, LPS (6 h treatment) did not affect TLR-4 expression in either BN or SS rats, demonstrating that LPS-induced myocardial dysfunction is not dependent on alteration of the expression of TLR-4 in our experimental model. The difference between our results and those of others may be a result of differences in time points of analysis or severity of shock or even differences in animal species/strains.
Myeloid differentiation factor 88 is a critical regulator of TLR-4 signaling. Down-regulation of MyD88 results in impaired TLR-4 signaling in response to LPS (23). Although we did not observe differences in MyD88 expression before and after LPS treatments of BN and SS rats, it is still possible that the formation of TLR-4 and MyD88 complex is impaired in BN rats, which needs further investigation. For example, others have shown in monocytes that a decrease in TLR-4–MyD88 complex formation contributes to an LPS-tolerant phenotype (22). In addition to MyD88, TLR-4 also interacts with other adaptor proteins known to activate NF-κB. After LPS stimulation, IκB is phosphorylated by IKK, which triggers IκBα degradation and activation of the NF-κB p65 subunit (24). Our results demonstrated that 30 min after injection, LPS induced significant increases in IκB and p65 phosphorylation in the hearts of SS rats but not in the hearts of BN rats (Fig. 5), confirming that LPS does not activate NF-κB downstream signaling pathway in BN rats to the same extent as it does in SS rats. After NF-κB translocates to the nucleus, it increases the expression of a multitude proinflammatory genes (24), such as the genes mentioned here: iNOS, COX-2, TNF-α, IL-6, and IL-1β. Our results indicate that BN rats may be more resistant to LPS because of impaired NF-κB activation, which would logically reduce subsequent proinflammatory cytokine and enzyme production. However, our data also indicate that cytokines produced at sites other than the myocardium may be more important to myocardial dysfunction in that LPS-induced cytokines increased in the plasma of BN and SS rats more than in the hearts (Figs. 2 and 3) and that LPS did not change cardiac TLR-4 or MyD88 expression. Accordingly, additional research is required to determine whether BN hearts are more resistant to direct LPS stimulation than SS hearts or whether an overall blunting of cytokine production from other sites in BN rats increases resistance.
In conclusion, BN rats are more resistant to LPS-induced myocardial dysfunction than SS rats. Additional data reveal that LPS-induced NF-κB activation, proinflammatory cytokines, and enzymes are reduced in BN rats. The importance of this study is that the phenotypes we described here are very relevant to sepsis research. For example, detailed identification of the defect that leads to the weaker activation of NF-κB may open new avenues in the search for new therapeutic interventions for sepsis. Therefore, determining the difference in the response to sepsis in these animals makes it easier for us to dissect the mechanisms that are responsible for increasing resistance to sepsis caused cardiomyopathy.
ACKNOWLEDGMENTS
The authors appreciate the administrative assistance of Meghann Sytsma (BS) and Anne Laulederkind (BSN, BA).
This study was supported by the National Institutes of Health, Bethesda, Maryland (grant no. HL080468 to Y. S.).
REFERENCES
- 1.Fernandes CJ, Jr, Akamine N, Knobel E. Myocardial depression in sepsis. Shock. 2008;30(Suppl 1):14–17. doi: 10.1097/SHK.0b013e3181818617. [DOI] [PubMed] [Google Scholar]
- 2.Ichinose F. Impact of nitric oxide synthase 3 on myocardial dysfunction in sepsis. Masui. 2008;57:294–301. [PubMed] [Google Scholar]
- 3.Rudiger A, Singer M. Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med. 2007;35:1599–1608. doi: 10.1097/01.CCM.0000266683.64081.02. [DOI] [PubMed] [Google Scholar]
- 4.Ao L, Song Y, Fullerton DA, Dinarello CA, Meng X. The interaction between myocardial depressant factors in endotoxemic cardiac dysfunction: role of TNF-alpha in TLR4-mediated ICAM-1 expression. Cytokine. 2007;38:124–129. doi: 10.1016/j.cyto.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muller-Werdan U, Engelmann H, Werdan K. Cardiodepression by tumor necrosis factor-alpha. Eur Cytokine Netw. 1998;9:689–691. [PubMed] [Google Scholar]
- 6.Merx MW, Weber C. Sepsis and the heart. Circulation. 2007;116:793–802. doi: 10.1161/CIRCULATIONAHA.106.678359. [DOI] [PubMed] [Google Scholar]
- 7.Damas P, Ledoux D, Nys M, Vrindts Y, De Groote D, Franchimont P, Lamy M. Cytokine serum level during severe sepsis in human IL-6 as a marker of severity. Ann Surg. 1992;215:356–362. doi: 10.1097/00000658-199204000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salomao R, Martins PS, Brunialti MK, Fernandes Mda L, Martos LS, Mendes ME, Gomes NE, Rigato O. TLR signaling pathway in patients with sepsis. Shock. 2008;30(Suppl 1):73–77. doi: 10.1097/SHK.0b013e318181af2a. [DOI] [PubMed] [Google Scholar]
- 9.Matsuguchi T, Masuda A, Sugimoto K, Nagai Y, Yoshikai Y. JNK-interacting protein 3 associates with Toll-like receptor 4 and is involved in LPS-mediated JNK activation. EMBO J. 2003;22:4455–4464. doi: 10.1093/emboj/cdg438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi Y, Hutchins W, Ogawa H, Chang CC, Pritchard KA, Jr, Zhang C, Khampang P, Lazar J, Jacob HJ, Rafiee P, et al. Increased resistance to myocardial ischemia in the Brown Norway vs. Dahl S rat: role of nitric oxide synthase and Hsp90. J Mol Cell Cardiol. 2005;38:625–635. doi: 10.1016/j.yjmcc.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Nomura F, Akashi S, Sakao Y, Sato S, Kawai T, Matsumoto M, Nakanishi K, Kimoto M, Miyake K, Takeda K, et al. Cutting edge: endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J Immunol. 2000;164:3476–3479. doi: 10.4049/jimmunol.164.7.3476. [DOI] [PubMed] [Google Scholar]
- 12.Shi Y, Baker JE, Zhang C, Tweddell JS, Su J, Pritchard KA., Jr Chronic hypoxia increases endothelial nitric oxide synthase generation of nitric oxide by increasing heat shock protein 90 association and serine phosphorylation. Circ Res. 2002;91:300–306. doi: 10.1161/01.res.0000031799.12850.1e. [DOI] [PubMed] [Google Scholar]
- 13.Stein B, Frank P, Schmitz W, Scholz H, Thoenes M. Endotoxin and cytokines induce direct cardiodepressive effects in mammalian cardiomyocytes via induction of nitric oxide synthase. J Mol Cell Cardiol. 1996;28:1631–1639. doi: 10.1006/jmcc.1996.0153. [DOI] [PubMed] [Google Scholar]
- 14.Markel TA, Crisostomo PR, Wang M, Herrmann JL, Abarbanell AM, Meldrum DR. Right ventricular TNF resistance during endotoxemia: the differential effects on ventricular function. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1893–R1897. doi: 10.1152/ajpregu.00359.2007. [DOI] [PubMed] [Google Scholar]
- 15.Smahi A, Courtois G, Rabia SH, Doffinger R, Bodemer C, Munnich A, Casanova JL, Israel A. The NF-kappaB signalling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet. 2002;11:2371–2375. doi: 10.1093/hmg/11.20.2371. [DOI] [PubMed] [Google Scholar]
- 16.Wagner DR, Combes A, McTiernan C, Sanders VJ, Lemster B, Feldman AM. Adenosine inhibits lipopolysaccharide-induced cardiac expression of tumor necrosis factor-alpha. Circ Res. 1998;82:47–56. doi: 10.1161/01.res.82.1.47. [DOI] [PubMed] [Google Scholar]
- 17.Yokoyama T, Sekiguchi K, Tanaka T, Tomaru K, Arai M, Suzuki T, Nagai R. Angiotensin II and mechanical stretch induce production of tumor necrosis factor in cardiac fibroblasts. Am J Physiol. 1999;276:H1968–H1976. doi: 10.1152/ajpheart.1999.276.6.H1968. [DOI] [PubMed] [Google Scholar]
- 18.Fernandes D, Assreuy J. Nitric oxide and vascular reactivity in sepsis. Shock. 2008;30(Suppl 1):10–13. doi: 10.1097/SHK.0b013e3181818518. [DOI] [PubMed] [Google Scholar]
- 19.Nemoto S, Vallejo JG, Knuefermann P, Misra A, Defreitas G, Carabello BA, Mann DL. Escherichia coli LPS-induced LV dysfunction: role of toll-like receptor-4 in the adult heart. Am J Physiol Heart Circ Physiol. 2002;282:H2316–H2323. doi: 10.1152/ajpheart.00763.2001. [DOI] [PubMed] [Google Scholar]
- 20.Tavener SA, Long EM, Robbins SM, McRae KM, Van Remmen H, Kubes P. Immune cell Toll-like receptor 4 is required for cardiac myocyte impairment during endotoxemia. Circ Res. 2004;95:700–707. doi: 10.1161/01.RES.0000144175.70140.8c. [DOI] [PubMed] [Google Scholar]
- 21.Niu J, Azfer A, Kolattukudy PE. Protection against lipopolysaccharide-induced myocardial dysfunction in mice by cardiac-specific expression of soluble Fas. J Mol Cell Cardiol. 2008;44:160–169. doi: 10.1016/j.yjmcc.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 22.Medvedev AE, Lentschat A, Wahl LM, Golenbock DT, Vogel SN. Dysregulation of LPS-induced Toll-like receptor 4-MyD88 complex formation and IL-1 receptor-associated kinase 1 activation in endotoxin-tolerant cells. J Immunol. 2002;169:5209–5216. doi: 10.4049/jimmunol.169.9.5209. [DOI] [PubMed] [Google Scholar]
- 23.Noman AS, Koide N, Hassan F, I-E-Khuda I, Dagvadorj J, Tumurkhuu G, Islam S, Naiki Y, Yoshida T, Yokochi T. Thalidomide inhibits lipopolysac-charide-induced tumor necrosis factor-alpha production via down-regulation of MyD88 expression. Innate Immun. 2009;15:33–41. doi: 10.1177/1753425908099317. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Lin A. Wiring the cell signaling circuitry by the NF-kappa B and JNK1 crosstalk and its applications in human diseases. Oncogene. 2007;26:3267–3278. doi: 10.1038/sj.onc.1210417. [DOI] [PubMed] [Google Scholar]