

Abstract
The diheme enzyme MauG catalyzes the posttranslational modification of the precursor protein of methylamine dehydrogenase (preMADH) to complete biosynthesis of its protein-derived tryptophan tryptophylquinone (TTQ) cofactor. Catalysis proceeds through a high valent bis-Fe(IV) redox state and requires long-range electron transfer (ET), as the distance between the modified residues of preMADH and the nearest heme iron of MauG is 19.4 Å. Trp199 of MauG resides at the MauG-preMADH interface, positioned midway between the residues that are modified and the nearest heme. W199F and W199K mutations did not affect the spectroscopic and redox properties of MauG, or its ability to stabilize the bis-Fe(IV) state. Crystal structures of complexes of W199F/K MauG with preMADH showed no significant perturbation of the MauG-preMADH structure or protein interface. However, neither MauG variant was able to synthesize TTQ from preMADH. In contrast, an ET reaction from diferrous MauG to quinone MADH, which does not require the bis-Fe(IV) intermediate, was minimally affected by the W199F/K mutations. W199F/K MauGs were able to oxidize quinol MADH to form TTQ, the putative final two-electron oxidation of the biosynthetic process, but with kcat/Km values approximately 10% that of wild-type MauG. The differential effects of the W199F/K mutations on these three different reactions are explained by a critical role for Trp199 in mediating multistep hopping from preMADH to bis-Fe(IV) MauG during the long-range ET that is required for TTQ biosynthesis.
Keywords: cytochrome, electron hopping, peroxidase, protein oxidation, protein radical
Long-range electron transfer (ET) through proteins is required for biological processes including respiration, photosynthesis, and metabolism. Mechanisms by which ET occurs over large distances to specific sites within a protein have been extensively studied (1–4). For interprotein ET, kinetic mechanisms are more complex, as the overall redox reaction requires additional steps such as protein–protein association and reorientation of the protein complex to optimize the system for ET (5, 6). “Long-range catalysis” is a related process in which the redox center that provides the oxidizing or reducing power is physically distinct from the site of chemical reaction of the substrate, so that long-range ET is required for catalysis. Thus far two enzymes have been postulated to employ long-range catalysis. Ribonucleotide reductase (RNR) catalyzes the formation of deoxyribonucleotides from ribonucleotides by long-range ET via multiple tyrosyl residues (7, 8). DNA photolyase is a flavoprotein that catalyzes DNA repair of pyrimidine–pyrimidine dimers via multiple tryptophan residues (9). In these enzymes it is believed that the long-range ET proceeds by hopping (10) through residues that can stabilize a radical state, rather than via a single long-range electron tunneling event.
MauG catalyzes the final steps in the biosynthesis of the protein-derived cofactor (11), tryptophan tryptophylquinone (TTQ) in methylamine dehydrogenase (MADH) (12). MADH exhibits an α2β2 structure with TTQ in each β-subunit. The biosynthetic process is a six-electron oxidation of the precursor protein (preMADH) containing monohydroxylated βTrp57 (Fig. 1). During this process, a second oxygen atom is inserted into the indole ring of βTrp57, βTrp57 is covalently cross-linked to βTrp108, and the quinol species is oxidized to the quinone (13). These reactions require a high valent bis-Fe(IV) redox state of MauG that is generated by reaction of diferric MauG with H2O2 or diferrous MauG with O2 (14, 15).
Fig. 1.

Reactions of MauG with different forms of MADH and preMADH. The six-electron oxidation of preMADH to form TTQ (Top), the two-electron oxidation of quinol MADH to form TTQ (Middle), the nonbiosynthetic thermodynamically favorable ET reaction from diferrous MauG to TTQ (Bottom).
The crystal structure of the MauG-preMADH complex (16) revealed that βTrp57 and βTrp108 do not make direct contact with either heme of MauG (Fig. 2). The distance between the side chain of βTrp108 of preMADH and the iron of the oxygen-binding five-coordinate heme is 40.1 Å, and the closest distance to the iron of the six-coordinate heme is 19.4 Å. Nonetheless, addition of H2O2 to MauG-preMADH crystals causes synthesis of the mature TTQ cofactor, demonstrating that the crystallized complex is catalytically active. Trp199 of MauG resides on the protein surface at the site of interaction with preMADH and is positioned midway between βTrp108 and the nearest heme of MauG. Tryptophan side chains have been implicated in electron/radical transfer in DNA photolyase (9) as well as in heme proteins (17–22). However, the role of tryptophan in mediating hopping during long-range ET requires further elucidation. Trp199 of MauG was converted to Phe and Lys by site-directed mutagenesis to determine its role in MauG-dependent catalysis. The structures of the W199F/K-preMADH complexes were determined and the effect of each mutation was assessed on three different ET reactions (Fig. 1): preMADH to bis-Fe(IV) MauG, quinol MADH to bis-Fe(IV) MauG, and diferrous MauG to quinone MADH. The results are explained by a critical role for Trp199 in mediating hopping during ET from preMADH to bis-Fe(IV) MauG, which is required for TTQ biosynthesis.
Fig. 2.

Orientation of hemes and residues of interest within the MauG-preMADH complex. A portion of the crystal structure (PDB ID code 3L4M) is shown. Hemes and residues of interest are displayed as sticks colored by atom, with the remaining protein as cartoon (MauG, pink; α-subunit preMADH, blue; β-subunit preMADH, green). Irons are represented by orange spheres. The closest distances between the aromatic side chains of residues βTrp108 and Trp199 of MauG, and between Trp199 and the porphyrin ring of the six-coordinate heme, are indicated.
Results
Expression of MauG Mutant Proteins.
W199F/K MauGs were expressed with a yield of protein of 0.8–1.2 mg/L of culture. Mass spectrometry confirmed the expected mass differences relative to WT MauG [observed (calculated) atomic mass unit] of -38.8 (-39.0) for W199F MauG and -57.2 (-58.0) for W199K MauG.
Spectroscopic and Redox Properties of W199F/K MauGs.
The absorption spectra of the diferric and diferrous forms of each W199F/K MauG were essentially identical to those of WT MauG (Fig. S1). The diferric forms exhibited Soret peak maxima at 406 nm. In the spectrum of each diferrous protein, the Soret peak maximum shifts to 418 nm and α- and β-bands appear at 552 and 524 nm, respectively. A shoulder in the Soret peak of each diferrous W199F/K MauG is present at 427 nm, which was previously noted for WT MauG (23). EPR spectra of diferric W199F/K MauGs exhibit high-spin and low-spin heme signals that are very similar to those of WT MauG (11) (Fig. S2). MauG exhibits two redox potential (Em) values for the interconversion between diferric and diferrous states, which correspond to the sequential addition or removal of one electron from the diheme system. The Em values of W199F/K MauGs are essentially the same as for WT MauG (Table 1).
Table 1.
Effects of W199F/K mutations on the reactivity of MauG
| WT MauG* | W199F MauG | W199K MauG | |
| Redox potentials | |||
| Em1, mV | −158 ± 9 | −145 ± 3 | −144 ± 5 |
| Em2, mV | −246 ± 3 | −237 ± 5 | −253 ± 5 |
| Steady-state TTQ formation from preMADH | |||
| kcat, s-1 | 0.20 ± 0.01 | NR† | NR |
| Km, μM | 6.6 ± 0.6 | NR | NR |
| Oxidation of preMADH by bis-Fe(IV) MauG | |||
| k, s-1 | 0.8 ± 0.1 | NR | NR |
| Kd, μM | < 1.5 | NR | NR |
| Steady-state TTQ formation from quinol MADH | |||
| kcat, s-1 | 4.2 ± 0.2 | 0.55 ± 0.04 | 0.37 ± 0.02 |
| Km, μM | 11.1 ± 1.3 | 25.6 ± 3.1 | 12.5 ± 1.3 |
| kcat/Km × 105, M-1 s-1 | 3.7 ± 0.5 | 0.22 ± 0.03 | 0.29 ± 0.03 |
| Oxidation of quinol MADH by bis-Fe(IV) MauG‡ | |||
| k, s-1 | 20 ± 1.3 | — | — |
| Kd, μM | 11.2 ± 2.3 | — | — |
| k1 × 105, M-1s-1 | — | 0.17 ± 0.01 | 0.23 ± 0.01 |
| Oxidation of diferrous MauG by quinone MADH | |||
| k, s-1 | 0.07 ± 0.01 | 0.03 ± 0.01 | 0.03 ± 0.01 |
| Kd, μM | 10.6 ± 1.6 | 6.4 ± 2.4 | 6.0 ± 2.6 |
X-Ray Crystal Structures of W199F/K MauG-preMADH Complexes.
W199F/K MauGs were each cocrystallized with preMADH and the structures solved to resolutions of 1.74 and 2.08 Å, respectively. For the W199K MauG/preMADH complex, data to 1.93 Å were used in refinement, but due to the lower completeness in the higher resolution shell (46.6%), the structure is reported to 2.08-Å resolution (completeness in 2.12 to 2.08 Å, 84.4%). Data collection and refinement statistics are given in Table S1. Each W199F/K MauG-preMADH complex has the same overall structure as that of WT MauG/preMADH (PDB ID code 3L4M), with two crystallographically independent copies of W199F/K MauG in the asymmetric unit. Each structure is superimposable with WT MauG-preMADH within coordinate error (rmsd values on Cα positions in the range of 0.20–0.32 Å) (Fig. S3). The positions of the nascent TTQ sites, Ca2+ sites (24), both hemes, and the intervening Trp93 residue are preserved in the W199F/K MauGs relative to the WT MauG structure.
The positions of Phe199 and Lys199 in the MauG mutants at the interface with preMADH are essentially the same as that of the native Trp199 (Fig. 3). βGlu101 of the preMADH is the only residue that shows a change between the two mutant structures (Fig. 4 and Fig. S4). In the WT and W199K structures, the βGlu101 side chain is directed toward MauG residue 199 and forms a long hydrogen bond with the indole nitrogen of Trp199 in the WT MauG-preMADH structure (Fig. 4A). In the W199K MauG-preMADH structure, the terminal N of Lys199 is hydrogen bonded to a water that has the same position in the WT MauG/preMADH (Fig. 4C). In the W199F MauG-preMADH structure the βGlu101 side chain is rotated so that the carboxylate head group points away from Phe199 and hydrogen bonds to the preMADH side chains of αArg197 and βArg99 (Fig. 4B). This appears to be due to steric reasons, as one of the Phe199 aromatic carbons is almost coincident with the hydrogen bonding indole N of Trp199 in WT MauG. The rotation of βGlu101 places the closest preMADH β-subunit atom over 6 Å from any atom of Phe199. The protein interface has an unusually large number of solvent molecules that mediate interactions between MauG and preMADH, including around the Trp199 side chain, which only directly contacts preMADH through the side chain of βGlu101. This explains the minimal perturbations to the MauG-preMADH interface, as it is the water structure that primarily changes to accommodate the different residue 199 side chains.
Fig. 3.

Overlay of the interface regions of complexes of W199F MauG-preMADH (A) and W199K MauG-preMADH (B) with WT MauG-preMADH. Coordinates of the W/K199 side chains are shown as sticks colored by atom (carbon: gold), with 2Fo - Fc electron density in blue mesh [contour levels 1.5σ (W199F) and 1.0σ (W199K)]. The surrounding protein is shown as gold cartoon and the WT MauG-preMADH coordinates are overlaid as cartoon (MauG: pink; β-preMADH: green; and α-preMADH: blue).
Fig. 4.

Interface residues and water structure near residue 199. (A) Hydrogen bonding network in the WT MauG-preMADH structure involving Trp199 and βGlu101 of preMADH (PDB ID code 3L4M) colored by atom (carbon: WT MauG, pink; β-preMADH, green; α-preMADH, blue). (B) Overlay of 3L4M and the W199F MauG-preMADH coordinates. (C) Overlay of 3L4M and the W199K MauG-preMADH coordinates. B and C are colored by atom (carbon: W199F/K MauG, yellow; β-preMADH, orange; α-preMADH, blue; all WT MauG-preMADH carbon and water atoms, gray).
Effects of Trp199 Mutations on the Formation of bis-Fe(IV) MauG and Reactivity Toward preMADH.
EPR and Mössbauer spectroscopy studies previously showed that reaction of diferric MauG with H2O2 resulted in formation of a bis-Fe(IV) redox state (14). The formation of the bis-Fe(IV) species is accompanied by a decrease in intensity and shift of the Soret peak from 406 to 408 nm in the absorption spectrum of MauG, and a decrease in intensity of the diferric heme signals in the EPR spectrum (25). These spectral changes were also observed upon reaction of each diferric W199F/K MauG with H2O2 (Figs. S1 and S2). WT and W199F/K bis-Fe(IV) MauGs spontaneously decay to the diferric state over several minutes as gauged by the return to the resting state absorption spectra. Whereas addition of preMADH to WT bis-Fe(IV) MauG increased the rate of return to the diferric state to 0.8 s-1 with a Kd < 1.5 μM (25), addition of up to 20 μM preMADH to the W199F/K MauGs did not increase the rate of return to the diferric state indicating that bis-Fe(IV) W199F/K MauGs were unable to oxidize preMADH.
The reactivity of W199F/K MauGs was also examined using a steady-state assay for TTQ biosynthesis using preMADH as the substrate (25). Whereas WT MauG exhibited a kcat value of 0.2 s-1 and Km value of 6.6 μM for preMADH, no detectable TTQ biosynthesis was observed in this assay with W199F/K MauGs, using up to 20 μM preMADH as the substrate (Table 1). Mass spectrometry of the reaction mixture confirmed that no chemical alterations of preMADH had occurred (Fig. S5). The reactivity of W199F MauG was also examined in crystallo. Whereas incubation of WT MauG-preMADH crystals with 2.0 mM H2O2 for 2 min leads to generation of TTQ (16), similar treatment of a W199F MauG-preMADH crystal caused no changes at the preTTQ site (Fig. S6). These results demonstrate that although W199F/K MauGs can attain and stabilize the bis-Fe(IV) state, they cannot catalyze the initial two-electron oxidation of preMADH either in solution or in crystallo.
6-Hydroxyindole reacts with bis-Fe(IV) MauG (26) with a rate of 5 s-1 and a Kd of 2.5 mM. As this rate is faster than that with preMADH (0.8 s-1) and the Kd much greater than for preMADH (< 1.5 μM), it was concluded that 6-hydoxyindole was chemically reactive toward bis-Fe(IV) MauG, but reacted directly with the ferryl heme rather than bind to the same site as preMADH. Although W199F/K bis-Fe(IV) MauGs did not react with preMADH, addition of 3 mM 6-hydroxyindole did cause rapid return to the diferric state. These results establish that the lack of reactivity of W199F/K MauGs with preMADH is not due to a change in heme reactivity but to specific disruption of long-range catalysis.
Effects of Trp199 Mutations on the Reactivity of bis-Fe(IV) MauG Toward Quinol MADH.
It was postulated that the final step of MauG-dependent TTQ biosynthesis is the oxidation of quinol MADH to the TTQ quinone. Transient formation of the quinol was observed early in the steady-state reaction, and bis-Fe(IV) WT MauG can oxidize quinol MADH (15). The steady-state kinetic parameters using WT MauG are Km of 11.1 μM and kcat of 4.2 s-1 (27). W199F/K MauGs were able to catalyze this reaction (Fig. 5A and Table 1), but much less efficiently than WT MauG. The kcat/Km values for W199F/K MauGs decreased by approximately 90%. These data indicate that, whereas Trp199 is essential for the initial two-electron oxidation of preMADH, it is important, but not essential, for the two-electron oxidation of the quinol to the quinone.
Fig. 5.

(A) Steady-state kinetic analysis of oxidation of quinol MADH by W199F (O) and W199K (Δ) MauG. The lines are fits of the data by Eq. 3. (B) Single-turnover kinetics of the reaction of quinol MADH with bis-Fe(IV) W199F (O) and W199K (Δ) MauG. The lines are fits of the data by Eq. 6.
Under single-turnover conditions, the reaction of bis-Fe(IV) WT MauG with quinol MADH exhibits a hyperbolic dependence of kobs on [quinol MADH] with a limiting first-order rate constant of 20 s-1 and a Kd of 11.2 μM (27). In contrast, the reactions of the bis-Fe(IV) W199F/K MauGs exhibited a linear dependence of kobs on [quinol MADH] (Fig. 5B). Thus, these mutations have caused the binding to quinol MADH rather than the catalytic step to be rate-limiting for the overall reaction (i.e., (k1[S] + k2) < k3). As such it was possible to determine only the bimolecular rate constant (k1) for these reactions: 0.17 ± 0.01 (M-1 s-1) × 105 for W199F MauG and 0.23 ± 0.01 (M-1 s-1) × 105 for W199K MauG.
Analysis of the ET Reaction from Diferrous MauG to Quinone MADH.
Although not involved in TTQ biosynthesis, a thermodynamically favorable ET reaction from diferrous MauG to quinone MADH, which does not require formation of the bis-Fe(IV) state, has been characterized (27). In this reaction WT MauG exhibited a limiting first-order rate constant of 0.07 s-1 and Kd value of 10.1 μM. The results obtained for W199F/K MauGs (Fig. 6 and Table 1) in this reaction were similar to those for WT MauG.
Fig. 6.

Single-turnover kinetics of the reaction quinone MADH with diferrous W199F (•) and W199K (O) MauG. The lines are fits of the data by Eq. 5.
Electron Tunneling Rate Predictions.
When ET is rate-determining for an observed reaction, the rate will depend upon the activation energy for the reaction (ΔG°+λ) and the electronic coupling (HAB) according to Eq. 1 (28) where λ is the reorganization energy and ΔG° is determined from the ΔEm for the redox reaction. HAB is related to the ET distance and nature of the intervening medium between ET donor and acceptor. The structures of WT and W199F/K MauG-preMADH complexes were analyzed with the HARLEM computer program (29) to calculate relative values of HAB for electron tunneling segments between residues β57 and β108 of preMADH, and residue 199, and the hemes of MauG (Table 2). The effects of the W199F/K mutations on HAB for direct tunneling between the residues of preMADH and the nearest heme of MauG were minimal and cannot explain the lack of reactivity of W199F/K MauG toward preMADH. However, if Trp199 mediates hopping during ET, then the observed rate will be that of the slowest hopping segment (i.e., the segment with the smallest HAB). In WT MauG this is the segment from preMADH β57/β108 to Trp199, and hopping versus single-step tunneling leads to a 62-fold increase in the effective HAB. As 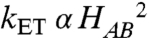 , in the absence of any other changes, the observed kET will be approximately 3,800-fold greater if the reaction occurs via hopping rather than direct long-range electron tunneling. If hopping could occur via Phe199 or Lys199, then a similar enhancement would be observed (Table 2) and the rate of reaction with preMADH would be similar to that of WT MauG. The undetectable reaction of W199K/F with preMADH is explained by the fact that only Trp, and not Phe or Lys, can mediate hopping.
, in the absence of any other changes, the observed kET will be approximately 3,800-fold greater if the reaction occurs via hopping rather than direct long-range electron tunneling. If hopping could occur via Phe199 or Lys199, then a similar enhancement would be observed (Table 2) and the rate of reaction with preMADH would be similar to that of WT MauG. The undetectable reaction of W199K/F with preMADH is explained by the fact that only Trp, and not Phe or Lys, can mediate hopping.
| [1] |
Table 2.
Effects of W199F/K mutations on the electronic coupling for possible ET reactions with the MauG-preMADH complex
| Tunneling segment | Relative HAB* | |||
| Donor | Acceptor | WT MauG | W199F MauG | W199K MauG |
| Direct single-step tunneling | ||||
| preMADH β57/β108 | MauG 6-coordinate heme | 1.0 | 0.92 | 0.98 |
| Steps in residue 199-mediated hopping | ||||
| preMADH β57/β108 | MauG residue 199 | 62 | 49 | 100 |
| MauG residue 199 | MauG 5-coordinate heme | 470 | 509 | 647 |
Discussion
The differential effects of the W199F/K MauG mutations on the three different reactions are best explained by a hopping mechanism in which Trp199 is reversibly oxidized and reduced during the long-range ET that is required for TTQ biosynthesis. Two alternative mechanisms of hopping may be considered. If preMADH reduces Trp199 which in turn reduces bis-Fe(IV) MauG, then the mechanism is electron hopping. If bis-Fe(IV) MauG oxidizes Trp199, which in turn oxidizes preMADH, then it is hole hopping. As discussed below, the latter is likely the mechanism that is used for MauG-dependent TTQ biosynthesis.
The initial oxidation reaction of bis-Fe(IV) WT MauG with preMADH yields a preMADH-based radical intermediate that has yet to be characterized (14). The Em value associated with the conversion of preMADH to this radical species is not known. However, the residues of preMADH that are modified are tryptophans and Em values for Trp•+/Trp redox couples have been determined to be in the range of 640–1080 mV (30–33). The Em value for the bis-Fe(IV)/diferric MauG redox couple is also unknown, but Em values for Fe(IV)/Fe(III) couples in many heme-dependent peroxidases have been determined. These values range from 724–1160 mV (18) and are similar to the values for Trp•+/Trp couples. This suggests that the ΔEm for the reaction may be close to zero. Given the low driving force and long distance for this reaction, the loss of activity of W199F/K MauGs is likely due to disruption of Trp199-mediated hopping during the long-range ET. The bis-Fe(IV) species is a sufficiently strong oxidant to oxidize Trp199, which in turn oxidizes preMADH. Conversely, Phe199 and Lys199 cannot be oxidized by the bis-Fe(IV). Thus, hole hopping via residue 199 cannot occur in the W199K and W199F MauGs, and no reaction with preMADH is observed.
In contrast to the biosynthetic reaction with preMADH, the ET reactions of diferrous W199F/K MauGs with quinone MADH are relatively unaffected by the mutations. This is consistent with a common mechanism for WT and W199F/K MauG in this reaction. As Phe and Lys cannot mediate hopping, this common mechanism is likely one of electron tunneling via sigma bonds, which is consistent with the crystal structures that show minimal structural impact. The Em value of +102 mV for the quinone/quinol redox couple is not enough for quinone MADH to oxidize Trp199, as does the more potent bis-Fe(IV) oxidant. However, as the diferric/diferrous MauG couple is -201 mV, the overall reaction is thermodynamically favorable with a ΔEm of 303 mV (23, 34), and this supports the long-range electron tunneling. This difference in mechanism is also consistent with the previous observation that, for WT MauG (27), this favorable reaction is slower (0.07 s-1) than the reaction of preMADH with bis-Fe(IV) MauG (0.8 s-1), for which we have estimated a ΔEm of about zero, but which can occur via hopping.
Quinol MADH does react with W199F/K MauGs. Again, the ΔEm value for this reaction is not known but can be estimated using the range of Em values for the Fe(IV)/Fe(III) couple of 724–1160 mV and the known value of +102 mV for quinone/quinol MADH. This yields a ΔEm value for the reaction of bis-Fe(IV) MauG with quinol MADH in the range of 622–1058 mV. This driving force is much greater than that for the reaction with preMADH (ΔEm ∼ 0) and allows the reaction of quinol MADH with W199F/K MauGs to proceed via long-range electron tunneling in the absence of Trp199-mediated hopping, albeit much less efficiently. Unfortunately it was not possible to obtain first-order rate constants for the reaction of bis-Fe(IV) W199F/K MauG with quinol MADH because binding was rate-limiting for the observed reaction (Fig. 5B). Therefore, it is not possible to directly compare the rate constant for this reaction with the rate constants for the other two.
It is noteworthy that a tryptophan is used by MauG as a hopping relay, as the residues on preMADH that are being oxidized are also tryptophans. Another tryptophan (Trp93 in Fig. 2) is believed to mediate ET between the two hemes of MauG (16). Using the same amino acid for different points in a multistep hopping mechanism may facilitate the reaction. The reported range of Em values for the heme Fe(IV)/Fe(III) and Trp•+/Trp redox couples are similar and suggest that the driving force for the reaction of bis-Fe(IV) MauG with preMADH is very low. Thus, a hopping mechanism is required to reduce the effective ET distance. It should be noted that the rate of the slowest hopping step in the overall reaction will depend not only on distance, but also on the ΔGo for that hop. By using the same amino acid as staging points, the ΔGo for each hop will be nearly zero. If a different amino acid residue that is more difficult to oxidize is present, then this would lessen the rate of hopping from that residue. Alternatively, if a residue that is more difficult to reduce is present, then this would lessen the rate of hopping into that residue. The use of hopping points that are nearly isopotential eliminates effects of ΔGo on the rates of the hopping steps. This notion is consistent with the observations that hopping through RNR proceeds primarily via a sequence of tyrosine residues, and that hopping through DNA photolyase proceeds primarily via a sequence of tryptophan residues. The results with MauG demonstrate that by employing a mechanism of hopping with appropriately spaced radical sites, distance can be removed as a limiting factor in enzyme-mediated long-range catalysis, as well as long-range interprotein ET reactions.
Materials and Methods
Protein Expression and Purification.
Recombinant MauG (11) and MADH (35) were purified from Pseudomonas denitrificans as described previously. PreMADH (13) was expressed in Rhodobacter sphaeroides and purified as described previously (36). Trp199 of MauG was converted to Lys and Phe by site-directed mutagenesis of double-stranded pMEG391 (11), which contains mauG, using the QuikChange kit (Strategene). W199F/K MauGs were expressed in P. denitrificans and isolated from the periplasmic fraction as described for recombinant WT MauG (11).
Mass Spectrometry.
WT and W199F/K MauGs, and samples from reaction mixtures of these proteins with preMADH and H2O2, were subjected to mass spectrometry. The latter reactions were performed in 10 mM potassium phosphate containing 100 μM preMADH and 10 μM WT or W199F/K MauG, initiated by addition of 1.0 mM H2O2, and incubated for 10 min. More details are provided in SI Text.
Redox Titrations.
Em values of W199F/K MauGs were determined by anaerobic spectrochemical titrations, as described previously for WT MauG (23). Data were fit using Eq. 2, which describes the redox behavior of a system with two redox active centers where a is the fraction of the total absorbance change attributable to one center and (1 - a) is the fraction of the total absorbance change attributable to the other.
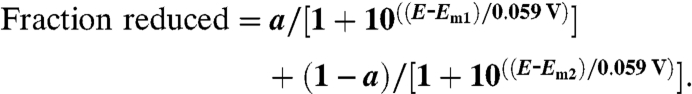 |
[2] |
Steady-State Spectrophotometric Kinetic Assays.
W199F/K MauGs were tested in previously described steady-state assays of TTQ biosynthesis from preMADH (37) and TTQ biosynthesis from quinol MADH (15). Each assay was performed in 0.01 M potassium phosphate buffer, pH 7.5 at 25 °C. The rate of appearance of TTQ (quinone) MADH was monitored by the increase in ΔA440 and the data were fit by Eq. 3.
| [3] |
Single-Turnover Kinetic Studies.
Two different reactions were studied. The dependence of the observed rate constant (kobs) on the concentration of the varied reactant was analyzed using a two-step kinetic model (formula 4) fit by either Eq. 5 for a hyperbolic concentration dependence or Eq. 6 for a linear concentration dependence. Each reaction was performed in 0.01 M potassium phosphate buffer, pH 7.5 at 25 °C. In one reaction varied concentrations of quinol MADH were mixed with 2 μM bis-Fe(IV) MauG that had been generated by stoichiometric addition of H2O2 (27). Reactions were monitored from 366–446 nm to observe the conversion of bis-Fe(IV) MauG (E) to diferric MauG (E′). In the other reaction varied concentrations of quinone MADH were mixed with 1.25 μM diferrous MauG under anaerobic conditions (27). The reaction was monitored by the decrease in A550, which corresponds to the conversion of diferrous MauG (E) to diferric MauG (E′) (11).
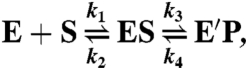 |
[4] |
| [5] |
| [6] |
EPR Spectroscopy.
EPR samples were prepared in 50 mM potassium phosphate buffer, pH 7.4, with 150 μM WT or W199F/K MauG. For the H2O2-treated samples, stoichiometric H2O2 was added to the enzyme in a stirred reaction vial at room temperature, and the reaction mixtures were immediately transferred to quartz EPR tubes and frozen in liquid nitrogen. The total reaction time was 30–40 s. Continuous wave X-band EPR spectra were taken by a Bruker E200 spectrometer at 100-kHz modulation frequency using a dual mode resonator. Temperature was maintained at 10 K with an ESR910 liquid helium cryostat and an ITC503 temperature controller.
Crystallization and X-Ray Structure Determinations of the W199F/K MauG-preMADH Complexes.
W199F/K MauGs were each cocrystallized with preMADH from a solution containing 100 μM W199F/K MauG and 50 μM preMADH. The details of crystallization conditions, data collection, and structure solution are in SI Text.
Electron Tunneling Rate Predictions.
The HARLEM computer program (29) was used to calculate relative values of HAB from the crystal structures of WT and W199F/K MauG-preMADH complexes using the approach of Dutton and coworkers (1).
Supplementary Material
Acknowledgments.
We thank Elizabeth Graichen and Yu Tang for technical assistance. This work was supported by National Institute of Health Grants GM-41574 (to V.L.D.), GM-66569 (to C.M.W.), Y1-CO-1020 and Y1-GM-1104 [National Institute of General Medical Science/National Cancer Institute Collaborative Access Team, Advanced Photon Source (APS)], National Science Foundation Grant MCB-0843537 (to A.L.), and Minnesota Partnership for Biotechnology and Medical Genomics Grant SPAP-05-0013-P-FY06 (to C.M.W.). Use of the APS was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, Contract DE-AC02-06CH11357. Computing was provided by the Minnesota Supercomputing Institute for Advanced Computational Research.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3RMZ (W199F-MauG/preMADH), 3RN0 (W199K-MauG/preMADH), and 3RLM (W199F-MauG/preMADH treated with H2O2).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1109423108/-/DCSupplemental.
References
- 1.Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 2.Davidson VL. Control of true, gated, and coupled electron transfer reactions. Acc Chem Res. 2008;41:730–738. doi: 10.1021/ar700252c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beratan DN, et al. Steering electrons on moving pathways. Acc Chem Res. 2009;42:1669–1678. doi: 10.1021/ar900123t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gray HB, Winkler JR. Electron tunneling through proteins. Q Rev Biophys. 2003;36:341–372. doi: 10.1017/s0033583503003913. [DOI] [PubMed] [Google Scholar]
- 5.Davidson VL. Unraveling the kinetic complexity of interprotein electron transfer reactions. Biochemistry. 1996;35:14035–14039. doi: 10.1021/bi961577p. [DOI] [PubMed] [Google Scholar]
- 6.Davidson VL. What controls the rates of interprotein electron-transfer reactions. Acc Chem Res . 2000;33:87–93. doi: 10.1021/ar9900616. [DOI] [PubMed] [Google Scholar]
- 7.Baldwin J, et al. Mechanism of rapid electron transfer during oxygen activation in the R2 subunit of Escherichia coli ribonucleotide reductase. 1. Evidence for a transient tryptophan radical. J Am Chem Soc. 2000;122:12195–12206. [Google Scholar]
- 8.Minnihan EC, Seyedsayamdost MR, Stubbe J. Use of 3-aminotyrosine to examine the pathway dependence of radical propagation in Escherichia coli ribonucleotide reductase. Biochemistry. 2009;48:12125–12132. doi: 10.1021/bi901439w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lukacs A, Eker AP, Byrdin M, Brettel K, Vos MH. Electron hopping through the 15 A triple tryptophan molecular wire in DNA photolyase occurs within 30 ps. J Am Chem Soc. 2008;130:14394–14395. doi: 10.1021/ja805261m. [DOI] [PubMed] [Google Scholar]
- 10.Giese B, Graber M, Cordes M. Electron transfer in peptides and proteins. Curr Opin Chem Biol. 2008;12:755–759. doi: 10.1016/j.cbpa.2008.08.026. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, et al. MauG, a novel diheme protein required for tryptophan tryptophylquinone biogenesis. Biochemistry. 2003;42:7318–7325. doi: 10.1021/bi034243q. [DOI] [PubMed] [Google Scholar]
- 12.Davidson VL. Pyrroloquinoline quinone (PQQ) from methanol dehydrogenase and tryptophan tryptophylquinone (TTQ) from methylamine dehydrogenase. Adv Protein Chem. 2001;58:95–140. doi: 10.1016/s0065-3233(01)58003-1. [DOI] [PubMed] [Google Scholar]
- 13.Pearson AR, et al. Further insights into quinone cofactor biogenesis: Probing the role of MauG in methylamine dehydrogenase TTQ formation. Biochemistry. 2004;43:5494–5502. doi: 10.1021/bi049863l. [DOI] [PubMed] [Google Scholar]
- 14.Li X, et al. A catalytic di-heme bis-Fe(IV) intermediate, alternative to an Fe(IV) = O porphyrin radical. Proc Natl Acad Sci USA. 2008;105:8597–8600. doi: 10.1073/pnas.0801643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Jones LH, Pearson AR, Wilmot CM, Davidson VL. Mechanistic possibilities in MauG-dependent tryptophan tryptophylquinone biosynthesis. Biochemistry. 2006;45:13276–13283. doi: 10.1021/bi061497d. [DOI] [PubMed] [Google Scholar]
- 16.Jensen LM, Sanishvili R, Davidson VL, Wilmot CM. In crystallo posttranslational modification within a MauG/pre-methylamine dehydrogenase complex. Science. 2010;327:1392–1394. doi: 10.1126/science.1182492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stayton PS, Sligar SG. Structural microheterogeneity of a tryptophan residue required for efficient biological electron transfer between putidaredoxin and cytochrome P-450cam. Biochemistry. 1991;30:1845–1851. doi: 10.1021/bi00221a017. [DOI] [PubMed] [Google Scholar]
- 18.Battistuzzi G, Bellei M, Bortolotti CA, Sola M. Redox properties of heme peroxidases. Arch Biochem Biophys. 2010;500:21–36. doi: 10.1016/j.abb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Goodin DB, McRee DE. The Asp-His-Fe triad of cytochrome c peroxidase controls the reduction potential, electronic structure, and coupling of the tryptophan free radical to the heme. Biochemistry. 1993;32:3313–3324. [PubMed] [Google Scholar]
- 20.Henry ER, Hochstrasser RM. Molecular dynamics simulations of fluorescence polarization of tryptophans in myoglobin. Proc Natl Acad Sci USA. 1987;84:6142–6146. doi: 10.1073/pnas.84.17.6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colin J, Wiseman B, Switala J, Loewen PC, Ivancich A. Distinct role of specific tryptophans in facilitating electron transfer or as [Fe(IV) = O Trp( ∗ )] intermediates in the peroxidase reaction of Bulkholderia pseudomallei catalase-peroxidase: A multifrequency EPR spectroscopy investigation. J Am Chem Soc. 2009;131:8557–8563. doi: 10.1021/ja901402v. [DOI] [PubMed] [Google Scholar]
- 22.Pogni R, et al. A tryptophan neutral radical in the oxidized state of versatile peroxidase from Pleurotus eryngii: A combined multifrequency EPR and density functional theory study. J Biol Chem. 2006;281:9517–9526. doi: 10.1074/jbc.M510424200. [DOI] [PubMed] [Google Scholar]
- 23.Li X, Feng M, Wang Y, Tachikawa H, Davidson VL. Evidence for redox cooperativity between c-type hemes of MauG which is likely coupled to oxygen activation during tryptophan tryptophylquinone biosynthesis. Biochemistry. 2006;45:821–828. doi: 10.1021/bi052000n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shin S, et al. The tightly bound calcium of MauG is required for tryptophan tryptophylquinone cofactor biosynthesis. Biochemistry. 2011;50:144–150. doi: 10.1021/bi101819m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee S, Shin S, Li X, Davidson VL. Kinetic mechanism for the initial steps in MauG-dependent tryptophan tryptophylquinone biosynthesis. Biochemistry. 2009;48:2442–2447. doi: 10.1021/bi802166c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin S, Lee S, Davidson VL. Suicide inactivation of MauG during reaction with O2 or H2O2 in the absence of its natural protein substrate. Biochemistry. 2009;48:10106–10112. doi: 10.1021/bi901284e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shin S, Abu Tarboush N, Davidson VL. Long range electron transfer reactions between hemes of MauG and different forms of tryptophan tryptophylquinone of methylamine dehydrogenase. Biochemistry. 2010;49:5810–5816. doi: 10.1021/bi1004969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marcus RA, Sutin N. Electron transfers in chemistry and biology. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 29.Kurnikov IV. HARLEM Computer Program. Pittsburgh: University of Pittsburgh; 2000. [Google Scholar]
- 30.Navarathnam S, Parsons BJ. Reduction potential of histidine free radicals: A pulse radiolysis study. J Chem Soc Faraday Trans. 1998;94:2577–2581. [Google Scholar]
- 31.Tommos C, Skalicky JJ, Pilloud DL, Wand AJ, Dutton PL. De novo proteins as models of radical enzymes. Biochemistry. 1999;38:9495–9507. doi: 10.1021/bi990609g. [DOI] [PubMed] [Google Scholar]
- 32.Milligan JR, et al. Repair of oxidative DNA damage by amino acids. Nucleic Acids Res. 2003;31:6258–6263. doi: 10.1093/nar/gkg816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeFelippis MR, Murthy CP, Faraggi M, Klapper MH. Pulse radiolytic measurement of redox potentials: The tyrosine and tryptophan radicals. Biochemistry. 1989;28:4847–4853. doi: 10.1021/bi00437a049. [DOI] [PubMed] [Google Scholar]
- 34.Brooks HB, Davidson VL. Free energy dependence of the electron transfer reaction between methylamine dehydrogenase and amicyanin. J Am Chem Soc. 1994;116:11201–11202. [Google Scholar]
- 35.Davidson VL. Methylamine dehydrogenases from methylotrophic bacteria. Methods Enzymol. 1990;188:241–246. doi: 10.1016/0076-6879(90)88040-h. [DOI] [PubMed] [Google Scholar]
- 36.Graichen ME, et al. Heterologous expression of correctly assembled methylamine dehydrogenase in Rhodobacter sphaeroides. J Bacteriol. 1999;181:4216–4222. doi: 10.1128/jb.181.14.4216-4222.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, Fu R, Liu A, Davidson VL. Kinetic and physical evidence that the diheme enzyme MauG tightly binds to a biosynthetic precursor of methylamine dehydrogenase with incompletely formed tryptophan tryptophylquinone. Biochemistry. 2008;47:2908–2912. doi: 10.1021/bi702259w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.