

In PNAS, Hu et al. (1) present strong evidence that pancellular expression of an important chromatin plasticity gene may serve as a biomarker for the state of Huntington disease (HD) “activity,” which, in turn, presumably implies the injurious events caused by polyglutamine cytotoxicity and/or the cellular response to that toxicity.
Biomarkers of brain disease are especially valuable because the tissue of interest cannot be conveniently subjected to biopsy for histological analysis. The “gold standard” brain disease biomarker effort to date is represented by the National Institute on Aging Alzheimer's Disease Neuroimaging Initiative (ADNI) (2). The main focus of ADNI is the annual, serial neuropsychological examination and neuroradiological examination of an age-matched cohort of 400 subjects: 100 cognitively intact elderly subjects, 200 subjects with mild cognitive impairment, and 100 subjects with mild Alzheimer's disease (AD). One of the most important outcomes of ADNI is the recent realization that approximately one-third of cognitively intact elders have cerebral amyloidosis but no detectable cognitive impairment (3). These biomarker data have revolutionized the approach to AD, leading to an emphasis on prevention trials (4).
The ideal disease biomarker (5) has been described as possessing as many of the following characteristics as possible. (i) Visible early, before histopathological changes, and should then be of a magnitude that is proportional to disease progression. (ii) Sensitive, but it should also correlate with the severity of damage. (iii) Accessible in the peripheral tissue. (iv) Analytically stable in tissue, so it can be measured from biopsy and autopsy material. (v) Translational; that is, it should bridge across species. (vi) Associated with a known mechanism. Many current biomarkers are identified through statistical analyses of gene expression, but one should be able to understand the pathophysiologic link between the biomarker and the disease. (vii) Able to localize damage. For example, it should pinpoint the particular area of the organ that has been damaged rather than just indicating toxicity in general.
Imaging has also been widely used to develop biomarkers for presymptomatic HD (6–8), but a peripheral marker is most desirable both for convenience of serial measurement and for low-cost screening to determine which subjects should be referred for more costly neuroimaging. Hu et al. (1) propose that the chromatin modulator H2AFY (Fig. 1) may be a useful biomarker for the active phase of HD. Does H2AFY meet the aforementioned criteria for the ideal biomarker? First, independent replication by other laboratories is crucial. Assuming that the observation is readily confirmed, we can move on to consideration of the qualification of the molecule as a biomarker and to consideration of how to link H2AFY to HD pathogenesis.
Fig. 1.
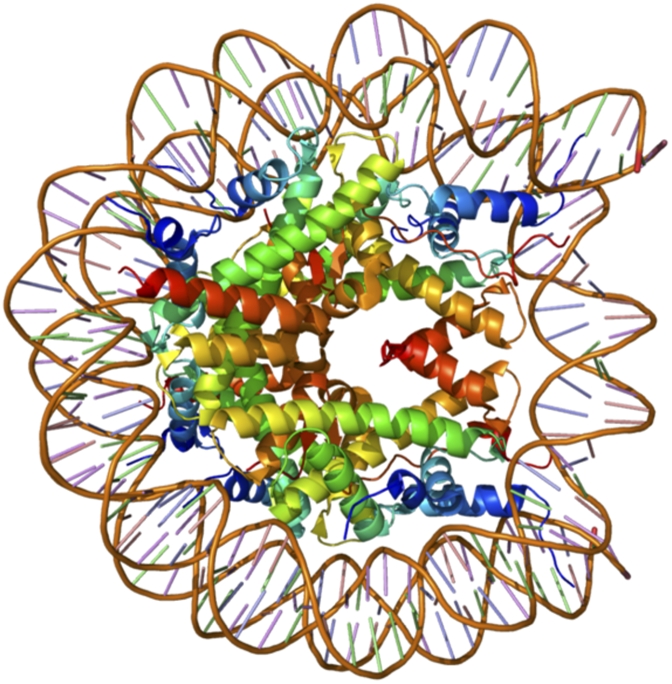
X-ray crystal structure of a nucleosome assembled with macroH2A (Protein Data Bank 1U35; α-satellite DNA + Mus musculus H3, H4, H2B + Homo sapiens macroH2A11). [Reprinted from ref. 12 with the permission of the authors.]
Hu et al. present evidence that H2AFY levels climb in both human and mutant huntingtin (htt) transgenic mouse blood and striatum before the onset of histological damage and, at least early in the disease, H2AFY levels are reported to change in proportion to disease severity. It is unclear whether H2AFY will be useful in later stages of HD. Still, because therapeutics are typically aimed at presymptomatic or very early clinical disease, it is hoped that H2AFY may be a useful addition to the armamentarium of assays used to follow response to experimental therapeutics, regardless of the targeted pathway (e.g., glutamate receptor antagonists, histone deacetylase inhibitors).
This brings us to the one of the most interesting and challenging questions about H2AFY: What is the role of H2AFY in the pathogenesis of symptoms in HD? Why should this molecule be both a central and peripheral marker of brain HD activity? In the case of the amyloid imaging agent Pittsburgh compound b (PiB) and AD, we understand (at least in familial forms of the disease) the role of Aβ as a key toxin involved in the initiation of the disease. Mutations in amyloid precursor protein (APP) and presenilins 1 and 2 alter APP processing so as to promote Aβ oligomerization and neurotoxicity (9). No molecule has been claimed to reflect AD activity, although much attention is devoted to discovering such a molecule. If H2AFY is replicated in other populations, perhaps it will be a template for the discovery of a central and peripheral marker of AD activity.
Regarding H2AFY as a biomarker for HD, as noted above, one of the criteria for an ideal biomarker is that it should be associated with a known mechanism. The studies of this gene began with the discovery of H2AFY in 1992. The protein products of the H2AFY gene, known as macroH2As, are histone variants that contain nonhistone regions. H2AFY is associated with, but not required for, X-chromosome inactivation (10). More relevant perhaps to HD, the macroH2As are associated with regulation of chromatin structure, particularly after DNA damage and activation of poly-ADP ribosylation enzymes, upon which the recruitment of the DNA repair molecules Ku70–Ku80 is inhibited. Evidence suggests that DNA repair is disrupted in the presence of mutant htt and that overexpression of Ku70 can correct abnormalities in an HD mouse model. The increase in the H2AFY protein product macroH2A may therefore contribute to this abnormality (11). Furthermore, macroH2A can directly inhibit binding of transcription factors and also repress transcription by recruitment of class I histone deacetylases, leading to a hypoacetylated chromatin state. In other contexts, macroH2A1 can induce transcription (Fig. 2) (12–14). Therefore, an increase of macroH2A1 may participate in the transcriptional dysregulation that is so prevalent in HD.
Fig. 2.

Schematic diagram contrasting the molecular changes in macroH2A occupancy that occur at genes activated or repressed by macroH2A. [Reprinted from ref. 12 with the permission of the authors.]
Changes in macroH2A1 biology are dependent on cell cycle, phosphorylation state, and senescence (12), but whether any of these is relevant to the HD activity-related phenomena described by Hu et al. remains to be explored. As noted by the authors, study of the role of macroH2A in HD, and in the brain under healthy conditions, will now be of intense interest, as will the mechanism by which H2AFY is regulated by mutant htt. H2AFY-null mice are viable and fertile (13), but no description of the brains of these mice has yet been reported. Interestingly, however, microarray analysis of gene expression in their liver shows abnormalities of expression of genes involved in lipid metabolism (13). Perhaps this will dovetail with the lipid-modulated events that have been identified in HD (14).
The explanation for the peripheral increase in H2AFY mRNA in HD, and its consequences, will also require further investigation. Because htt is expressed in all cells, the mechanism certainly may overlap with that which occurs in the striatum. Another simple speculation that should be excluded is the possibility that certain conformers of aggregated htt interact directly with certain macroH2A species. The fact that macroH2A species have apparently not been detected in any of the several published htt interactor discovery experiments (reviewed in refs. 15 and 16) argues against this proposal, but direct examination will have more sensitivity and should be evaluated. Additionally, given the role of thyroid hormones and cellular bioenergetics, another frequent theme in HD pathogenesis, a possible association between H2AFY and thyroid function warrants investigation (10).
In summary, genetics and genomics are at their most powerful when they point to molecules and pathways that would not have been contemplated relying upon logic alone. There is no obvious reason to have associated HD with pancellular changes in H2AFY; yet the data are tantalizing, and only deeper inquiry will reveal the robustness of the biomarker function, and only direct studies in cell and mouse model systems will elucidate the role of H2AFY in HD pathogenesis.
Acknowledgments
M.E.E. acknowledges the support of National Institutes of Health/National Institute of Neurological Diseases and Stroke Grant R01 NS059936.
Footnotes
The authors declare no conflict of interest.
See companion article on page 17141.
References
- 1.Hu Y, et al. Transcriptional modulator H2A histone family, member Y (H2AFY) marks Huntington disease activity in man and mouse. Proc Natl Acad Sci USA. 2011;108:17141–17146. doi: 10.1073/pnas.1104409108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mueller SG, et al. Ways toward an early diagnosis in Alzheimer's disease: The Alzheimer's Disease Neuroimaging Initiative (ADNI) Alzheimers Dement. 2005;1:55–66. doi: 10.1016/j.jalz.2005.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aizenstein HJ, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gandy S. Perspective: Prevention is better than cure. Nature. 2011;475:S15. doi: 10.1038/475S15a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robinson S, Pool R, Giffin R. Emerging Safety Science. Forum on Drug Discovery, Development, and Translation. Washington, DC: National Academies Press; 2008. [Google Scholar]
- 6.Kassubek J, et al. Fully automated atlas-based MR imaging volumetry in Huntington disease, compared with manual volumetry. AJNR Am J Neuroradiol. 2011;32:1328–1332. doi: 10.3174/ajnr.A2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esmaeilzadeh M, Kullingsjö J, Ullman H, Varrone A, Tedroff J. Regional cerebral glucose metabolism after pridopidine (ACR16) treatment in patients with Huntington disease. Clin Neuropharmacol. 2011;34:95–100. doi: 10.1097/WNF.0b013e31821c31d8. [DOI] [PubMed] [Google Scholar]
- 8.Squitieri F, Esmaeilzadeh M, Ciarmiello A, Jankovic J. Caudate glucose hypometabolism in a subject carrying an unstable allele of intermediate CAG(33) repeat length in the Huntington's disease gene. Mov Disord. 2011;26:925–927. doi: 10.1002/mds.23623. [DOI] [PubMed] [Google Scholar]
- 9.Gandy S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–1129. doi: 10.1172/JCI25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanasijevic B, Rasmussen TP. X chromosome inactivation and differentiation occur readily in ES cells doubly-deficient for macroH2A1 and macroH2A2. PLoS ONE. 2011;6:e21512. doi: 10.1371/journal.pone.0021512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Enokido Y, et al. Mutant huntingtin impairs Ku70-mediated DNA repair. J Cell Biol. 2010;189:425–443. doi: 10.1083/jcb.200905138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gamble MJ, Kraus WL. Multiple facets of the unique histone variant macroH2A: From genomics to cell biology. Cell Cycle. 2010;9:2568–2574. doi: 10.4161/cc.9.13.12144. [DOI] [PubMed] [Google Scholar]
- 13.Boulard M, et al. Histone variant macroH2A1 deletion in mice causes female-specific steatosis. Epigenetics Chromatin. 2010;3:8. doi: 10.1186/1756-8935-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valenza M, Cattaneo E. Emerging roles for cholesterol in Huntington's disease. Trends Neurosci. 2011;34:474–486. doi: 10.1016/j.tins.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 15.Bhattacharyya NP, Banerjee M, Majumder P. Huntington's disease: Roles of huntingtin-interacting protein 1 (HIP-1) and its molecular partner HIPPI in the regulation of apoptosis and transcription. FEBS J. 2008;275:4271–4279. doi: 10.1111/j.1742-4658.2008.06563.x. [DOI] [PubMed] [Google Scholar]
- 16.Wu LL, Zhou XF. Huntingtin associated protein 1 and its functions. Cell Adh Migr. 2009;3:71–76. doi: 10.4161/cam.3.1.7511. [DOI] [PMC free article] [PubMed] [Google Scholar]