

Abstract
Cell-penetrating peptides (CPPs), such as the HIV TAT peptide, are able to translocate across cellular membranes efficiently. A number of mechanisms, from direct entry to various endocytotic mechanisms (both receptor independent and receptor dependent), have been observed but how these specific amino acid sequences accomplish these effects is unknown. We show how CPP sequences can multiplex interactions with the membrane, the actin cytoskeleton, and cell-surface receptors to facilitate different translocation pathways under different conditions. Using “nunchuck” CPPs, we demonstrate that CPPs permeabilize membranes by generating topologically active saddle-splay (“negative Gaussian”) membrane curvature through multidentate hydrogen bonding of lipid head groups. This requirement for negative Gaussian curvature constrains but underdetermines the amino acid content of CPPs. We observe that in most CPP sequences decreasing arginine content is offset by a simultaneous increase in lysine and hydrophobic content. Moreover, by densely organizing cationic residues while satisfying the above constraint, TAT peptide is able to combine cytoskeletal remodeling activity with membrane translocation activity. We show that the TAT peptide can induce structural changes reminiscent of macropinocytosis in actin-encapsulated giant vesicles without receptors.
Keywords: protein transduction domain, polyarginine, peptide-lipid interactions, pore-forming peptide, antimicrobial peptide
Cell-penetrating peptides (CPPs) are effective intracellular delivery systems (1–5). These peptides are usually short (< 20 amino acids) and cationic. Examples include the TAT peptide from HIV, antennapedia (ANTP) from Drosophila, and even simple polyarginines. Although unique molecular architectures incorporating CPPs have been designed for drug delivery (3, 6–8), the molecular mechanisms of cellular entry, and the relations between them, are not well understood. Different uptake mechanisms have been proposed for CPPs (9). Cell-based assays have indicated that multiple endocytotic pathways are involved (10–15). In addition to these, CPPs are also capable of direct entry mechanisms* (17–20). In general, cell-penetrating activity of CPPs has proven to be difficult to eliminate completely using a specific set of conditions (3, 12, 21), suggesting the existence of multiple mechanisms. A unified understanding of CPPs, which is currently lacking, must engage why the same sequence can readily activate the qualitatively distinct outcomes.
How do relatively simple molecules like HIV TAT peptide facilitate mechanisms as different as direct translocation, and multiple endocytotic processes? Rather than debate priority between mechanisms, we focus on the physical chemistry of what these different mechanisms and CPPs have in common. Here, we show how the TAT peptide can multiplex different interactions with the same sequence, thus interacting with the membrane, the actin cytoskeleton, and specific receptors to produce multiple pathways of translocation under different conditions. Although it has been previously shown that TAT peptide can generate negative Gaussian membrane curvature that is conducive to different types of membrane translocation activity (22), the microscopic mechanism for curvature generation is not known, and no direct experimental tests are available. Nor is it known how different amino acids in a heterogeneous sequence collectively induce this type of curvature. For example, why do hydrophobic amino acids occur in some CPPs but not others? With fluorescence microscopy, we show the sensitivity of CPPs to changes in hydrophobicity: The addition of a single hydrophobic residue to purely hydrophilic CPPs can drastically modify the translocation mechanism. To investigate explicitly the role of peptide-induced membrane curvature strain, synchrotron X-ray scattering studies on a series of synthetic “nunchuck” CPPs were performed. Results reveal that the induced negative Gaussian curvature necessary for translocation can be turned on or off by controlling the organization of lipid head groups. Further, we find that the topological requirement for negative Gaussian curvature places constraints on the amino acid composition of CPPs, and we identify a trend relating arginines, lysines, and hydrophobic residues that is consistent with most CPP sequences. Importantly, because the requirements for pore formation underdetermine the full sequences of CPPs, there is significant sequence flexibility for taking on additional functions. The dense packing of cationic residues in TAT peptide allows it to not only optimize electrostatic interactions with proteoglycan receptors but also with the actin cytoskeleton. We show why the TAT peptide does not need receptors to induce endocytosis. By incubating with the TAT peptide, giant unilamellar vesicles (GUVs) with active encapsulated cytoskeletons but no receptors for endocytosis can be restructured into morphologies reminiscent of those in blebbing and macropinocytosis. Thus, the TAT peptide is able to multiplex interactions with different cellular components that allow it to induce direct membrane translocation, and to mediate endocytosis with or without receptors.
Results and Discussion
CPPs with hydrophobic residues (or hydrophobic cargo) act differently on membranes than those without. R6 (unlabeled to eliminate contributions from hydrophobic dyes) mediated permeation of GUVs that was sufficiently fast to rupture the GUV (Fig. 1 A and B), because there is no supporting cytoskeleton that can corral and stabilize the membrane. Similar results were observed for unlabeled TAT peptide. In contrast, the addition of a single tryptophan hydrophobic residue in R6W (unlabeled) resulted in slow leakage from a full vesicle to an empty intact vesicle, reminiscent of behavior of many antimicrobial peptides (AMPs) (Fig. 1 C and D). The same was observed for R6 attached to hydrophobic cargo, in this case fluorescein (R6-FITC), a dye rich in aromatic rings (Fig. 1 E and F). After the encapsulated dye is leaked out, further addition of R6-FITC results in the peptide traversing the GUV membrane and the peptide concentration equilibrating across it. This behavior is similar to that observed for labeled TAT peptide and R6 added to GUVs with no encapsulated dye. These results suggest that inclusion of a single hydrophobe drastically impacts translocation in R6. R6 with no hydrophobicity is able to cross the membrane quickly through transient pore-like structures, leading to rapid lysis of vesicles. The addition of hydrophobicity, however, stabilizes pores that allow for the leakage of the encapsulated dye without destroying vesicles. These results illustrate the sensitivity of CPPs to small changes in hydrophobicity and help reconcile the diverse range of observations for CPP-induced vesicle leakage, even for apparently similar experimental conditions (23–25).
Fig. 1.

Mechanism of membrane permeation depends on peptide sequence. (A and B) R6 (unlabeled) added to 20/60/20 PS/PE/PC GUVs (red) with encapsulated Alexa Fluor 633 dye (blue) induced permeation fast enough to rupture vesicles. (C and D) Addition of a single tryptophan hydrophobic residue drastically changed the dominant mode of translocation/escape: R6W (unlabeled) induced slow leakage of intact vesicles, similar to antimicrobial peptides. (E and F) Labeling R6 with the hydrophobic aromatic rings of fluorescein isothiocyanate (FITC, green) also gave rise to slow leakage of encapsulated dye, followed by eventual equilibrium distribution of the R6-FITC peptide (G). White scale bars are 20 μm.
To understand how changes in the CPP sequence (such as the addition of hydrophobicity) impinge on the translocation mechanism, we use polyarginine (polyR), the simplest prototypical CPP. Rather than directly solving the structure of a CPP-induced pore, we map out the conditions under which CPPs induce topologically active membrane curvature using small-angle X-ray scattering (SAXS). When exposed to R6 (Fig. 2A), small unilamellar vesicles (SUVs) underwent a drastic structural transition and displayed characteristic correlation peaks with ratios 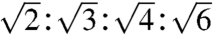 , which indicate the formation of a cubic Pn3m “double-diamond” lattice (Fig. 2A) with lattice parameter a = 13.3 nm. This is consistent with the behavior of the TAT peptide (22). The Pn3m is a bicontinuous cubic phase where two nonintersecting water channels are separated by a lipid bilayer (26). The bilayer traces out a minimal surface with saddle-splay curvature at every point, indicating that each constituent monolayer also has saddle-splay curvature at every point, the type of curvature necessary for “hole” formation in a membrane (Fig. 2D). Saddle-shaped surfaces are found inside “donuts,” which have a single hole each, but not on the surfaces of spheres, which lack holes. In addition to pore formation, negative Gaussian (or equivalently saddle-splay) curvature is a necessary condition for processes such as blebbing, which is observed in macropinocytosis.
, which indicate the formation of a cubic Pn3m “double-diamond” lattice (Fig. 2A) with lattice parameter a = 13.3 nm. This is consistent with the behavior of the TAT peptide (22). The Pn3m is a bicontinuous cubic phase where two nonintersecting water channels are separated by a lipid bilayer (26). The bilayer traces out a minimal surface with saddle-splay curvature at every point, indicating that each constituent monolayer also has saddle-splay curvature at every point, the type of curvature necessary for “hole” formation in a membrane (Fig. 2D). Saddle-shaped surfaces are found inside “donuts,” which have a single hole each, but not on the surfaces of spheres, which lack holes. In addition to pore formation, negative Gaussian (or equivalently saddle-splay) curvature is a necessary condition for processes such as blebbing, which is observed in macropinocytosis.
Fig. 2.

Membrane activity of CPPs controlled by lipid crowding effects and amino acid content. (A) SAXS data for R9, incubated with SUVs (DOPS∶DOPE = 20∶80) at peptide-to-lipid molar ratio (P/L) = 1/40 show diffraction peak positions well described by 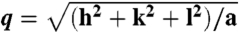 , characteristic of a bicontinuous cubic Pn3m phase (h, k, l are Miller indices; a is the lattice parameter). Tetraarginine blocks connected with a short PEG spacer (R4-(PEG)5-R4) induces a Pn3m phase (P/L = 1/40) with a larger lattice constant. SAXS data for R4-(PEG)27-R4, with a longer PEG spacer, show that the Pn3m phase is suppressed, replaced by HII and Lα phases with no saddle-splay curvature (P/L = 1/40). (B) Multidentate coordination of arginine’s guanidinium side chain induces positive curvature strain along the peptide. (C) Monodentate coordination of lysine’s amino side chain does not induce positive curvature. (D) The cubic Pn3m phase (the zero-mean-curvature surface at the midplane between the two membrane leaflets) (E) The average induced Gaussian curvature
, characteristic of a bicontinuous cubic Pn3m phase (h, k, l are Miller indices; a is the lattice parameter). Tetraarginine blocks connected with a short PEG spacer (R4-(PEG)5-R4) induces a Pn3m phase (P/L = 1/40) with a larger lattice constant. SAXS data for R4-(PEG)27-R4, with a longer PEG spacer, show that the Pn3m phase is suppressed, replaced by HII and Lα phases with no saddle-splay curvature (P/L = 1/40). (B) Multidentate coordination of arginine’s guanidinium side chain induces positive curvature strain along the peptide. (C) Monodentate coordination of lysine’s amino side chain does not induce positive curvature. (D) The cubic Pn3m phase (the zero-mean-curvature surface at the midplane between the two membrane leaflets) (E) The average induced Gaussian curvature 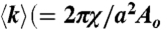 ; χ = -2 and Ao = 1.919 for Pn3m) is maximized near NR = 9, where observed membrane transduction activity is empirically highest. (F) ANTP, TAT peptide, R9 all induce the cubic Pn3m phase in membranes (DOPS∶DOPE = 20∶80 at P/L = 1/40). (G) The ratio of the number of arginines to the number of arginines + number of lysines (NR/(NR + NK)) plotted against hydrophobicity (Eisenberg hydrophobicity scale) using the amino acid sequences of 39 cell-penetrating peptides and 1080 cationic antimicrobial peptides shows that a reduction in arginine content can be compensated for by an increase in both lysine and hydrophobic content.
; χ = -2 and Ao = 1.919 for Pn3m) is maximized near NR = 9, where observed membrane transduction activity is empirically highest. (F) ANTP, TAT peptide, R9 all induce the cubic Pn3m phase in membranes (DOPS∶DOPE = 20∶80 at P/L = 1/40). (G) The ratio of the number of arginines to the number of arginines + number of lysines (NR/(NR + NK)) plotted against hydrophobicity (Eisenberg hydrophobicity scale) using the amino acid sequences of 39 cell-penetrating peptides and 1080 cationic antimicrobial peptides shows that a reduction in arginine content can be compensated for by an increase in both lysine and hydrophobic content.
At present, it is not clear how negative Gaussian curvature derives from membrane interactions with specific peptide sequences. It is known that charged polymers can induce electrostatic wrapping by oppositely charged membranes (27–29), so cationic peptides (e.g., polyarginine and polylysine) naturally generate negative mean curvature via induced electrostatic wrapping of the peptide by anionic cell membranes. The guanidinium side group in arginines can form multidentate hydrogen bonds with the phosphates of lipid head groups and associate with multiple lipid molecules (22, 30, 31). We hypothesize that this multidentate hydrogen bonding creates positive curvature buckling strain, which in combination with negative curvature strain along an orthogonal direction from electrostatic interactions, generates saddle-splay curvature (Fig. 2B). In contrast, the amino group in lysine forms monodentate hydrogen bonds and therefore generates only negative curvature from electrostatic interactions (Fig. 2C). To test this hypothesis, we investigated a series of artificial nunchuck CPPs based on two tetraarginine (R4) blocks connected by flexible PEG spacer chains of different lengths, so that the amount of positive curvature strain can be continuously varied. For lipid–peptide complexes made using CPPs with short PEG spacers (RPEG-5), the Pn3m phase was preserved, albeit at a larger lattice constant (a = 17.5 nm), consistent with decreased saddle-splay curvature: The average induced saddle-splay curvature of RPEG-5 is  , which is less than that from R9
, which is less than that from R9  . As the spacer length was increased (RPEG-27), the saddle-splay rich Pn3m structure was completely lost, replaced by a coexistence of a lamellar (Lα) phase with d = 8.7 nm (which corresponds to the sum of the bilayer thickness and diameter of the PEG spacer), and an inverted hexagonal (HII) phase, with a lattice constant of 8.0 nm, which is similar to the HII lattice constant for R4 alone (7.7 nm). These results show that the amount of saddle-splay curvature generated and the resultant propensity for inducing pore-like structures are controlled by the multidentate H-bonding induced strain. Interestingly, if we track how the average induced saddle-splay curvature varies with the number of arginine repeats (NR) in polyR, we see that it is maximized at NR = 9 (Fig. 2E), where cell-penetrating activity of polyR is highest from empirical observation (1, 32), with lower activity at significantly higher or lower peptide lengths.
. As the spacer length was increased (RPEG-27), the saddle-splay rich Pn3m structure was completely lost, replaced by a coexistence of a lamellar (Lα) phase with d = 8.7 nm (which corresponds to the sum of the bilayer thickness and diameter of the PEG spacer), and an inverted hexagonal (HII) phase, with a lattice constant of 8.0 nm, which is similar to the HII lattice constant for R4 alone (7.7 nm). These results show that the amount of saddle-splay curvature generated and the resultant propensity for inducing pore-like structures are controlled by the multidentate H-bonding induced strain. Interestingly, if we track how the average induced saddle-splay curvature varies with the number of arginine repeats (NR) in polyR, we see that it is maximized at NR = 9 (Fig. 2E), where cell-penetrating activity of polyR is highest from empirical observation (1, 32), with lower activity at significantly higher or lower peptide lengths.
Saddle-splay curvature was induced in similar CPPs, including polyR (a = 13.3 nm for NR = 9), HIV TAT (a = 10.7 nm), and ANTP (a = 15.9 nm) (Fig. 2F), consistent with the topological requirement for pore formation. In fact, generation of saddle-splay membrane curvature necessitates specific amino acid compositions in pore-forming peptides. As previously demonstrated, lipid head-group organization by arginine and lysine generates distinct types of membrane curvature deformations. Arginine simultaneously creates positive and negative curvatures along the two perpendicular principal directions (negative Gaussian curvature), whereas lysine creates negative curvature along one direction only. Hydrophobic amino acids create positive curvature by inserting into the membrane (33–35). This implies a compensatory relation between arginines and lysines/hydrophobes, as in AMPs (36), and illustrated in a comparison between the amino acid content of CPPs and AMPs. Fig. 2G shows a plot of NR/(NR + NK) (the ratio of the number of arginines to the sum of the number of arginines and lysines) vs. hydrophobicity as measured by the Eisenberg hydrophobicity scale. A strong trend consistent with this saddle-splay curvature selection rule can be clearly discerned in CPPs and AMPs, as both are capable of membrane permeation. The TAT peptide, which has two lysine residues, illustrates this rule. According to the selection rule, TAT peptide needs some hydrophobicity to balance the lysines. TAT peptide, in fact, has two hydrophobic amino acids. This allows the TAT peptide to generate more Gaussian curvature compared to R9, even though it has six arginines rather than nine, as shown by the lattice constants (10.7 nm vs. 13.3 nm, Fig. 2F).
Why is the hydrophobic content of the TAT peptide relatively low if hydrophobicity can help generate negative Gaussian curvature? A key difference between CPPs and AMPs is that the former systematically use fewer hydrophobic residues (and concomitantly more arginines) to generate saddle-splay curvature (Fig. 2G). This difference in sequences can potentially decrease the extent of membrane insertion and lead to shorter pore lifetimes for CPPs. This hypothesis is consistent with the behavior of R6 variants in Fig. 1. A comparison between TAT peptide and R6 is instructive. For R6, the addition of a single strong hydrophobe (one strong hydrophobe per seven amino acids) is able to change the observed translocation mechanism from the fast to the slow mode. TAT peptide is almost twice as long as R6. Moreover, TAT peptide has one strong hydrophobe and one that is quite weak. We have seen that unlabeled TAT peptide is capable of fast translocation, although not quite as fast as the R6. By adding one strong hydrophobic fluorescent dye to its sequence, TAT peptide has two strong hydrophobes in 12 residues (or one strong hydrophobe per six amino acids). The proportion of hydrophobic residues is now actually comparable to that of R6W and a slow dye leakage is observed, consistent with published experiments (25, 37, 38).
We hypothesize that the difference between peptides that kill and those that do not may be a time scale of the induced membrane deformation. CPPs only induce transient pore-like translocation structures in the membrane, perhaps reminiscent of those observed in recent simulations (39, 40), rather than the stable pores of AMPs.
The ability to generate saddle-splay membrane curvature is inherent in the sequences of CPPs. Such curvature is broadly enabling and is also evident in the dimples formed in caveolae-based endocytosis and in the cytoskeleton-driven protrusions in macropinocytosis (9). Moreover, CPPs are able to generate saddle-splay curvature in membranes even when incubated at 4 °C, consistent with direct translocation observed at low temperatures. Although induction of saddle-splay curvature accounts for the existence of direct translocation across cell membranes, it is not a sufficient condition for full CPP activity. We observed that carboxytetramethylrhodamine (TAMRA)-labeled TAT peptide (TAMRA-TAT) readily crossed the membranes of GUVs and the peptide concentration equilibrated across the membrane (Fig. 3A). However, the same was not true for TAT peptide conjugated to nanoparticles. We attached polylactide (PLA) nanoparticles (approximately 30 nm in diameter) stained with Cy5 dye to the TAT peptide (TAT-NP). TAT-NPs were unable to enter vesicles (Fig. 3B). The enhanced fluorescence intensity at the lipid bilayer indicated aggregation of TAT-NPs around the periphery of GUVs. The TAT peptide, although conjugated to the nanoparticles, is still able to induce negative Gaussian membrane curvature and permeabilize the membrane. TAT-NPs added to GUVs with encapsulated G-actin allowed Mg2+ ions to cross the membrane and polymerize actin (Fig. 3 C and D). These results suggest that large TAT-NPs are anchored to a perforated membrane but are not directly translocated, and require an endocytotic process for cellular uptake. This indicates that the debate of direct entry vs. endocytosis is not a simple “either-or” question with distinct and exclusive outcomes and is influenced by factors such as size of attached cargo. The above result shows that it is possible for the TAT peptide to perforate the membrane and anchor a large payload to it, but not translocate across the membrane. This leads to the question of how TAT peptide promotes endocytotic uptake of large cargos, as observed in cell-based studies (5, 11, 41).
Fig. 3.

The induction of saddle-splay curvature alone is not a sufficient condition for full strength CPP activity. (A) TAMRA-labeled TAT peptide (TAMRA-TAT; red) readily crossed the membrane of PS∶PC∶PE = 20∶40∶40 GUVs. (B) TAT peptide covalently coupled to Cy5-tagged PLA nanoparticles (TAT-NP, approximately 30-nm diameter, blue) did not cross the membrane but were localized at the periphery of GUVs. (C) PS∶PC∶PE = 20∶40∶40 GUVs loaded with G-actin and rhodamine phalloidin (red). (D) TAT-NPs (blue), added to these GUVs and localized at the periphery, were able to induce negative Gaussian membrane curvature and membrane permeation, enabling Mg2+ ions to cross GUV membrane and polymerize the encapsulated G-actin into F-actin bundles (red). White scale bars are 10 μm.
There is significant sequence flexibility in TAT peptide to develop additional functions, such as binding to receptors to the cytoskeleton, because the requirement for generating negative Gaussian curvature (Fig. 2) underdetermines the full CPP sequence, as stated above. The charge density of TAT peptide (linear charge density λT = +1e/4.5 Å) closely matches the charge density of cell-surface heparan sulfate proteoglycans (HSPG, λH ∼ -1e/5 Å), which are known receptors for endocytosis (42, 43). Electrostatic attractions in water are especially strong when the charged surfaces have comparable charge densities, due to the resultant maximal entropy gain of counterion release (44, 45). Indeed, thermodynamic studies show that the TAT peptide has significant affinity toward heparin sulfate (46, 47); studies using cells treated with chemicals that eliminate HSPGs or using mutant cells that are unable to synthesize proteoglycans have shown a significant reduction in TAT peptide internalization (13, 15, 48). However, recent studies have demonstrated transduction of TAT peptide even in mutant cells deficient in glycosaminoglycans (49, 50). These results suggest that even though cell-surface proteoglycans constitute an internalization pathway other independent pathways also exist.
Macropinocytosis has been suggested as a receptor-independent endocytosis mechanism for cellular uptake of TAT peptide (11, 49, 51), although the details are not completely clear. Macropinocytosis is a receptor-independent pathway for endocytosis that begins with outwardly directed growth of actin filaments that ultimately results in internalization of objects within large endocytotic vesicles (macropinosomes). Because the CPP internalization pathway depends on ambient conditions (3, 52, 53), we have performed experiments (see SI Text) to confirm that actin polymerization inhibitors significantly reduce CPP uptake levels under our experimental conditions, to compare with existing trafficking experiments (10, 11, 51): These results demonstrate the importance of actin polymerization in TAT activity, consistent with the existence of a parallel endocytotic pathway, but they do not indicate how endocytosis is achieved in the absence of receptors. Moreover, it is not clear how TAT peptide induces the observed actin accumulation and remodeling at the cell periphery (10, 54, 55).
At present, it is not known how the TAT peptide can nonspecifically induce actin remodeling. We note that CPPs are highly cationic with a charge density beyond the Manning limit (56, 57), which allows them to interact directly and strongly with the actin cytoskeleton. In fact, the cationic charge of the TAT peptide can efficiently interact with both heparan sulfate proteoglycans and filamentous actin (F-actin) (λA = -1e/2.5 Å), leading to strong attractive interactions. The former allows for receptor-mediated endocytosis as noted above, but the latter allows something previously undescribed. In order to eliminate the influence of all potential receptors for endocytosis, we investigated TAT peptide’s interactions with an active actin cytoskeleton encapsulated inside a GUV with divalent ion channels. Such channels provide a steady supply of Mg2+ ions to maintain a high level of dynamic actin polymerization inside the GUVs. In the absence of TAT peptide, no significant membrane deformations were observed (Fig. 4A). When these actin-encapsulated GUVs were exposed to TAT peptide (unlabeled), they underwent drastic shape transitions, reminiscent of “ruffling” and “fingering” deformations in cells (Fig. 4 B and C). In some cases, the actin network even broke symmetry and deformed the spherical vesicle into elongated structures (Fig. 4D). Collectively, these induced cytoskeletal changes are similar to those that occur in macropinocytosis. Moreover, in vitro results show that in addition to membrane effects, TAT peptide can also directly induce actin polymerization and bundle formation (SI Text). We hypothesize that the bundling of actin filaments reduces the number of contact points and concentrates the forces between the polymerizing actin and the membrane leading to observed membrane distortions (Fig. 4 B–D), which is similar to those observed for encapsulated microtubules (58). These results indicate that the TAT peptide can enhance membrane deformation and cytoskeleton reorganization necessary for endocytotic processes, even in a minimal model. The striking morphological changes in the vesicle observed above may also explain the puzzling observation that addition of free TAT peptide enhances the transfection efficiencies of TAT-DNA complexes (59). The free TAT peptide can cross the membrane and induce membrane ruffling and blebbing that promotes internalization of objects bound to the membrane surface.
Fig. 4.

TAT peptide can penetrate membranes and actively induce cytoskeletal actin response. (A) Confocal image of a GUV comprising 40/40/20 PE/PC/PS (labeled green with DiO) with 5% calcium ionophore (Calcimycin) and encapsulating 7 μM globular actin (G-actin). Exposure to 8 mM Mg2+, diffusing into the GUV via the ionophores, induced polymerization into filamentous actin (F-actin, labeled red with rhodamine phallodin) network without any accompanying deformation of vesicle. (B and C) Exposure to approximately 4 μM TAT peptide-induced dimple instabilities on the membrane, and promoted growth of F-actin bundles encapsulated within the GUV. (D) In certain cases, the F-actin bundles distorted originally spherical vesicles to form sharp filopodium-like protusions, reminiscent of membrane ruffling and macropinocytosis. (E) Schematic showing a proposed autonomous pathway for TAT cellular transduction. TAT peptide can generate saddle-splay membrane curvature and enter through an induced pore, but large conjugated cargos cannot. The TAT peptide interacts strongly with cytoplasmic actin to promote cellular uptake of anchored cargo via endocytotic pathways. White scale bars are 10 μm.
That CPPs can interact with the actin cytoskeleton directly suggests an interesting comparison with AMPs. Many AMPs have biological functions besides membrane permeation. For example, indolicidin (60), buforin (61), and tachyplesin (62) are also known to bind internal targets in bacteria such as DNA. In analogy to our arguments on CPPs, the amino acid composition criterion for generation of negative Gaussian membrane curvature underdetermines AMP sequences (36), so they are also able to optimize their sequences to bind to highly cationic biopolymers, in this case DNA rather than F-actin. When viewed in this way, CPPs are a special case of AMPs that specialize in short lifetime pores and cytoskeletal interactions.
Conclusion
Because the amino acid composition requirements for strong CPP-cytoskeleton and CPP-membrane interactions can be simultaneously satisfied, the TAT peptide can multiplex interactions to effect either direct translocation through the membrane or mediate endocytosis with or without receptors. We propose a general model for TAT peptide-mediated cellular transduction (Fig. 4E). The TAT peptide can bind to HSPG and get internalized via normal endocytosis pathways. It can also interact with the membrane to generate saddle-splay curvature and induce pore-like structures. If the cargo attached to the TAT peptide is small, it can be translocated directly. Cargos larger than a few nanometers do not get directly translocated but are anchored to the membrane by the TAT peptide. The existence of strong TAT-actin interactions on the cytoplasmic side can remodel the cytoskeleton, thus promoting alternate cellular uptake mechanisms like macropinocytosis.
Materials and Methods
Peptide Synthesis.
TAT protein transduction domain (Tyr-Gly-Arg-Lys-Lys-Arg-Arg-Gln-Arg-Arg-Arg), R4 [(Arg)4], RPEG-5 [(Arg)4-(PEG)5-(Arg)4], and RPEG-27 [(Arg)4-(PEG)27-(Arg)4] were synthesized using automated solid-state synthesis. R6 [(Arg)9] and TAMRA-labeled TAT were purchased from Anaspec, Inc. The R34, R46, and R60 polypeptides were synthesized as poly(ϵ-CBZ-L-lysine) chains of the desired lengths using transition metal-initiated α-amino acid N-carboxyanhydride polymerization using (PMe3)4Co. The primary amines on lysine were converted to guanidinium groups using an excess of the reagent 3,5-dimethyl-1-pyrazolylformaminidium nitrate as described elsewhere (6).
SUV Preparation.
The lipids 1,2-Dioleoyl-sn-glycero-3-[phospho-L-serine] (sodium salt) (DOPS), 1,2-dioleoyl-sn-glycero-3-phosphocholine, and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) were purchased from Avanti Polar Lipids, Inc. and were used without further preparation. Lipid solutions in chloroform were dried under N2 and desiccated under vacuum overnight. The dried lipids were rehydrated with Millipore water to a final concentration of 20 mg/mL at 37 °C overnight, sonicated, and extruded through a 0.2-μm nucleopore (Whatman, Inc.) filter. Peptides dissolved in Millipore water at 5 mg/mL were mixed with SUVs at specific peptide-to-lipid molar ratios and salt conditions and sealed in quartz capillaries.
Synchrotron X-Ray Scattering.
SAXS data were collected at Stanford Synchrotron Radiation Laboratory (BL4-2), Advanced Light Source (BL 7.3.3), and Advanced Photon Source (BESSRCAT BL-12ID) using 9 keV, 10 keV, and 12 keV X-rays, respectively. The scattered intensity was collected using a MAR-Research charge-coupled device detector (pixel size 79 μm). All experiments were conducted at room temperature. Representative samples were remeasured after several months to ensure that they were fully equilibrated.
Arginine/(Arginine + Lysine) Ratio vs. 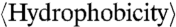 .
.
The sequences of 39 CPPs were compared against those of 1,080 cationic AMPs. For a given peptide, j, its average hydrophobicity is defined by
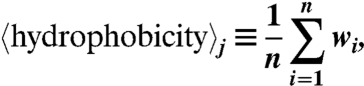 |
where n = number of amino acids in the peptide and wi = the hydrophobicity of the ith amino acid in the peptide as set by the Eisenberg hydrophobicity scale. The range of 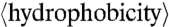 was divided into 20 equal bins in the case of CPPs and into 100 equal bins in the case of AMPs. For the M peptides in a given bin,
was divided into 20 equal bins in the case of CPPs and into 100 equal bins in the case of AMPs. For the M peptides in a given bin,
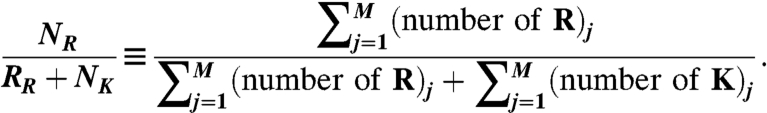 |
NR/(NR + NK) was plotted vs. 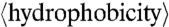 for each of the bins to see the trend.
for each of the bins to see the trend.
Confocal Microscopy.
GUVs labeled with DiO/DiI (Invitrogen) were prepared using electroformation. Lipid mixtures in chloroform were deposited and desiccated on indium tin oxide-coated glass slides, before swelling with sucrose (100 mM) solution containing Alexa Fluor 546/633 succinimidyl ester dyes (approximately 1 kDa at approximately 5 μM, pretreated with Tris) under 10-Hz ac electric field. After GUV detachment via 4-Hz square ac, the suspension was diluted into glucose (100 mM) and NaCl (100 mM) solution and imaged in a 200-μL chamber with a Leica TCS SP2 laser scanning confocal microscope (63×, 1.4 N.A. immersion objective).
Supplementary Material
Acknowledgments.
We thank S. Köhler and A. Bausch for their electroformation protocol. X-ray work was performed at the Stanford Synchrotron Radiation Lab, the Advanced Photon Source, the Advanced Light Source, and California NanoSystems Institute (CNSI). We also acknowledge microscopy resources at the Beckman Institute and CNSI. This work is supported by National Institutes of Health Grant 1UO1 AI082192-01 and National Science Foundation Grant DMR08-04363.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1108795108/-/DCSupplemental.
*Although initial observations of direct translocation were artifacts of cell fixation and inefficient removal of surface bound peptides (16) live-cell confocal microscopy studies have shown translocation both at 4 °C and in ATP-depleted conditions.
References
- 1.Futaki S, et al. Arginine-rich peptides: An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 2.El-Sayed A, Futaki S, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: Ways to overcome endosomal entrapment. AAPS J. 2009;11:13–22. doi: 10.1208/s12248-008-9071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wender PA, Galliher WC, Goun EA, Jones LR, Pillow TH. The design of guanidinium-rich transporters and their internalization mechanisms. Adv Drug Deliv Rev. 2008;60:452–472. doi: 10.1016/j.addr.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torchilin V, Rammohan R, Weissig V, Levchenko T. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitores. Proc Natl Acad Sci USA. 2001;98:8786–8791. doi: 10.1073/pnas.151247498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewin M, et al. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat Biotechnol. 2000;18:410–414. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]
- 6.Holowka EP, Sun VZ, Kamei DT, Deming TJ. Polyarginine segments in block copolypeptides drive both vesicular assembly and intracellular delivery. Nat Mater. 2007;6:52–57. doi: 10.1038/nmat1794. [DOI] [PubMed] [Google Scholar]
- 7.Wender P, Rothbard J, Jessop T, Kreider E, Wylie B. Oligocarbamate molecular transporters: Design, synthesis, and biological evaluation of a new class of transporters for drug delivery. J Am Chem Soc. 2002;124:13382–13383. doi: 10.1021/ja0275109. [DOI] [PubMed] [Google Scholar]
- 8.Wender P, Kreider E, Pelkey E, Rothbard J, VanDeusen C. Dendrimeric molecular transporters: Synthesis and evaluation of tunable polyguanidino dendrimers that facilitate cellular uptake. Org Lett. 2005;7:4815–4818. doi: 10.1021/ol051496y. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt NW, Mishra A, Lai GH, Wong GCL. Arginine-rich cell-penetrating peptides. FEBS Lett. 2010;584:1806–1813. doi: 10.1016/j.febslet.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 10.Nakase I, et al. Cellular uptake of arginine-rich peptides: Roles for macropinocytosis and actin rearrangement. Mol Ther. 2004;10:1011–1022. doi: 10.1016/j.ymthe.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 11.Wadia J, Stan R, Dowdy S. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 12.Richard JP, et al. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem. 2005;280:15300–15306. doi: 10.1074/jbc.M401604200. [DOI] [PubMed] [Google Scholar]
- 13.Ferrari A, et al. Caveolae-mediated internalization of extracellular HIV-1 Tat fusion proteins visualized in real time. Mol Ther. 2003;8:284–294. doi: 10.1016/s1525-0016(03)00122-9. [DOI] [PubMed] [Google Scholar]
- 14.Fittipaldi A, et al. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J Biol Chem. 2003;278:34141–34149. doi: 10.1074/jbc.M303045200. [DOI] [PubMed] [Google Scholar]
- 15.Tyagi M, Rusnati M, Presta M, Giacca M. Internalization of HIV-1 Tat requires cell surface heparan sulfate proteoglycans. J Biol Chem. 2001;276:3254–3261. doi: 10.1074/jbc.M006701200. [DOI] [PubMed] [Google Scholar]
- 16.Richard JP, et al. Cell-penetrating peptides: A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 17.Maiolo J, Ferrer M, Ottinger E. Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim Biophys Acta. 2005;1712:161–172. doi: 10.1016/j.bbamem.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 18.Ter-Avetisyan G, et al. Cell entry of arginine-rich peptides is independent of endocytosis. J Biol Chem. 2009;284:3370–3378. doi: 10.1074/jbc.M805550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwasa A, et al. Cellular uptake and subsequent intracellular trafficking of R8-liposomes introduced at low temperature. Biochim Biophys Acta. 2006;1758:713–720. doi: 10.1016/j.bbamem.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 20.Fretz MM, et al. Temperature-, concentration- and cholesterol-dependent translocation of L- and D-octa-arginine across the plasma and nuclear membrane of CD34+ leukaemia cells. Biochem J. 2007;403:335–342. doi: 10.1042/BJ20061808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Säälik P, et al. Protein cargo delivery properties of cell-penetrating peptides. A comparative study. Bioconjug Chem. 2004;15:1246–1253. doi: 10.1021/bc049938y. [DOI] [PubMed] [Google Scholar]
- 22.Mishra A, Gordon V, Yang L, Coridan R, Wong G. HIV TAT forms pores in membranes by inducing saddle-splay curvature: Potential role of bidentate hydrogen bonding. Angew Chem Int Ed Engl. 2008;47:2986–2989. doi: 10.1002/anie.200704444. [DOI] [PubMed] [Google Scholar]
- 23.Ziegler A. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv Drug Deliv Rev. 2008;60:580–597. doi: 10.1016/j.addr.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 24.Drin G, Demene H, Temsamani J, Brasseur R. Translocation of the pAntp peptide and its amphipathic analogue AP-2AL. Biochemistry. 2001;40:1824–1834. doi: 10.1021/bi002019k. [DOI] [PubMed] [Google Scholar]
- 25.Zhu WL, Shin SY. Effects of dimerization of the cell-penetrating peptide Tat analog on antimicrobial activity and mechanism of bactericidal action. J Pept Sci. 2009;15:345–352. doi: 10.1002/psc.1120. [DOI] [PubMed] [Google Scholar]
- 26.Schwarz US, Gompper G. Stability of inverse bicontinuous cubic phases in lipid-water mixtures. Phys Rev Lett. 2000;85:1472–1475. doi: 10.1103/PhysRevLett.85.1472. [DOI] [PubMed] [Google Scholar]
- 27.Koltover I, Salditt T, Raedler JO, Safinya CR. An inverted hexagonal phase of cationic liposome-DNA complexes related to DNA release and delivery. Science. 1998;281:78–81. doi: 10.1126/science.281.5373.78. [DOI] [PubMed] [Google Scholar]
- 28.Liang H, Harries D, Wong G. Polymorphism of DNA-anionic liposome complexes reveals hierarchy of ion-mediated interactions. Proc Natl Acad Sci USA. 2005;102:11173–11178. doi: 10.1073/pnas.0502416102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harries D, Ben-Shaul A, Szleifer I. Enveloping of charged proteins by lipid bilayers. J Phys Chem B. 2004;108:1491–1496. [Google Scholar]
- 30.Rothbard JB, Jessop TC, Wender PA. Adaptive translocation: The role of hydrogen bonding and membrane potential in the uptake of guanidinium-rich transporters into cells. Adv Drug Deliv Rev. 2005;57:495–504. doi: 10.1016/j.addr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 31.Li S, Su Y, Luo W, Hong M. Water-protein interactions of an arginine-rich membrane peptide in lipid bilayers investigated by solid-state nuclear magnetic resonance spectroscopy. J Phys Chem B. 2010;114:4063–4069. doi: 10.1021/jp912283r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. Polyarginine enters cells more efficiently than other polycationic homopolymers. J Pept Res. 2000;56:318–325. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 33.Huang HW. Action of antimicrobial peptides: Two-state model. Biochemistry. 2000;39:8347–8352. doi: 10.1021/bi000946l. [DOI] [PubMed] [Google Scholar]
- 34.Matsuzaki K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim Biophys Acta. 1999;1462:1–10. doi: 10.1016/s0005-2736(99)00197-2. [DOI] [PubMed] [Google Scholar]
- 35.Matsuzaki K, et al. Relationship of membrane curvature to the formation of pores by magainin 2. Biochemistry. 1998;37:11856–11863. doi: 10.1021/bi980539y. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt NW, et al. Criterion for amino acid composition of defensins and antimicrobial peptides based on geometry of membrane destabilization. J Am Chem Soc. 2011;133:6720–6727. doi: 10.1021/ja200079a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuchs SM, Raines RT. Pathway for polyarginine entry into mammalian cells. Biochemistry. 2004;43:2438–2444. doi: 10.1021/bi035933x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bourre L, et al. Effective photoinactivation of Gram-positive and Gram-negative bacterial strains using an HIV-1 Tat peptide-porphyrin conjugate. Photochem Photobiol Sci. 2010;9:1613–1620. doi: 10.1039/c0pp00146e. [DOI] [PubMed] [Google Scholar]
- 39.Herce HD, Garcia AE. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc Natl Acad Sci USA. 2007;104:20805–20810. doi: 10.1073/pnas.0706574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herce HD, et al. Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys J. 2009;97:1917–1925. doi: 10.1016/j.bpj.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Torchilin VP, et al. Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome-DNA complexes. Proc Natl Acad Sci USA. 2003;100:1972–1977. doi: 10.1073/pnas.0435906100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 43.Fuki IV, Meyer ME, Williams KJ. Transmembrane and cytoplasmic domains of syndecan mediate a multi-step endocytic pathway involving detergent-insoluble membrane rafts. Biochem J. 2000;351:607–612. [PMC free article] [PubMed] [Google Scholar]
- 44.Sanders L, et al. Structure and stability of self-assembled actin-lysozyme complexes in salty water. Phys Rev Lett. 2005;95:108302. doi: 10.1103/PhysRevLett.95.108302. [DOI] [PubMed] [Google Scholar]
- 45.Sanders L, et al. Control of electrostatic interactions between F-actin and genetically modified lysozyme in aqueous media. Proc Natl Acad Sci USA. 2007;104:15994–15999. doi: 10.1073/pnas.0705898104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goncalves E, Kitas E, Seelig J. Binding of oligoarginine to membrane lipids and heparan sulfate: Structural and thermodynamic characterization of a cell-penetrating peptide. Biochemistry. 2005;44:2692–2702. doi: 10.1021/bi048046i. [DOI] [PubMed] [Google Scholar]
- 47.Ziegler A, Blatter X, Seelig A, Seelig J. Protein transduction domains of HIV-1 and SIV TAT interact with charged lipid vesicles. Binding mechanism and thermodynamic analysis. Biochemistry. 2003;42:9185–9194. doi: 10.1021/bi0346805. [DOI] [PubMed] [Google Scholar]
- 48.Console S, Marty C, Garcia-Echeverria C, Schwendener R, Ballmer-Hofer K. Antennapedia and HIV transactivator of transcription (TAT) “protein transduction domains” promote endocytosis of high molecular weight cargo upon binding to cell surface glycosaminoglycans. J Biol Chem. 2003;278:35109–35114. doi: 10.1074/jbc.M301726200. [DOI] [PubMed] [Google Scholar]
- 49.Gump JM, June RK, Dowdy SF. Revised role of glycosaminoglycans in TAT protein transduction domain-mediated cellular transduction. J Biol Chem. 2010;285:1500–1507. doi: 10.1074/jbc.M109.021964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elson-Schwab L, et al. Guanidinylated neomycin delivers large, bioactive cargo into cells through a heparan sulfate-dependent pathway. J Biol Chem. 2007;282:13585–13591. doi: 10.1074/jbc.M700463200. [DOI] [PubMed] [Google Scholar]
- 51.Kaplan IM, Wadia JS, Dowdy SF. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J Control Release. 2005;102:247–253. doi: 10.1016/j.jconrel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 52.Brooks H, Lebleu B, Vives E. Tat peptide-mediated cellular delivery: Back to basics. Adv Drug Deliv Rev. 2005;57:559–577. doi: 10.1016/j.addr.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 53.Duchardt F, Fotin-Mleczek M, Schwarz H, Fischer R, Brock R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic. 2007;8:848–866. doi: 10.1111/j.1600-0854.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- 54.Gerbal-chaloin S, et al. First step of the cell-penetrating peptide mechanism involves Rac1 GTPase-dependent actin-network remodelling. Biol Cell. 2007;099:223–238. doi: 10.1042/BC20060123. [DOI] [PubMed] [Google Scholar]
- 55.Nakase I, et al. Interaction of arginine-rich peptides with membrane-associated proteoglycans is crucial for induction of actin organization and macropinocytosis. Biochemistry. 2006;46:492–501. doi: 10.1021/bi0612824. [DOI] [PubMed] [Google Scholar]
- 56.Wong G. Electrostatics of rigid polyelectrolytes. Curr Opin Coll Int Sci. 2006;11(6):310–315. [Google Scholar]
- 57.Holm C, Podgornik R. Electrostatic Effects in Soft Matter and Biophysics. Dordrecht, The Netherlands: Kluwer; 2001. [Google Scholar]
- 58.Fygenson DK, Marko JF, Libchaber A. Mechanics of microtubule-based membrane extension. Phys Rev Lett. 1997;79:4497–4500. [Google Scholar]
- 59.Ignatovich IA, et al. Complexes of plasmid DNA with basic domain 47–57 of the HIV-1 Tat protein are transferred to mammalian cells by endocytosis-mediated pathways. J Biol Chem. 2003;278:42625–42636. doi: 10.1074/jbc.M301431200. [DOI] [PubMed] [Google Scholar]
- 60.Hsu C-H, et al. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005;33:4053–4064. doi: 10.1093/nar/gki725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park CB, Kim HS, Kim SC. Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem Biophys Res Commun. 1998;244:253–257. doi: 10.1006/bbrc.1998.8159. [DOI] [PubMed] [Google Scholar]
- 62.Yonezawa A, Kuwahara J, Fujii N, Sugiura Y. Binding of tachyplesin I to DNA revealed by footprinting analysis: Significant contribution of secondary structure to DNA binding and implication for biological action. Biochemistry. 1992;31:2998–3004. doi: 10.1021/bi00126a022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.