

Abstract
Despite evidence for liquid water at the surface of Mars during the Noachian epoch, the temperature of early aqueous environments has been impossible to establish, raising questions of whether the surface of Mars was ever warmer than today. We address this problem by determining the precipitation temperature of secondary carbonate minerals preserved in the oldest known sample of Mars’ crust—the approximately 4.1 billion-year-old meteorite Allan Hills 84001 (ALH84001). The formation environment of these carbonates, which are constrained to be slightly younger than the crystallization age of the rock (i.e., 3.9 to 4.0 billion years), has been poorly understood, hindering insight into the hydrologic and carbon cycles of earliest Mars. Using “clumped” isotope thermometry we find that the carbonates in ALH84001 precipitated at a temperature of approximately 18 °C, with water and carbon dioxide derived from the ancient Martian atmosphere. Furthermore, covarying carbonate carbon and oxygen isotope ratios are constrained to have formed at constant, low temperatures, pointing to deposition from a gradually evaporating, subsurface water body—likely a shallow aquifer (meters to tens of meters below the surface). Despite the mild temperatures, the apparently ephemeral nature of water in this environment leaves open the question of its habitability.
The extreme difficulty in achieving mild surface temperatures in early Mars climate models (1) is in disagreement with widespread geomorphological evidence for surface water runoff during the Noachian epoch (2, 3). A robust determination of the temperature at or near the surface of Noachian Mars would provide insight into this apparent paradox, but has been challenging to establish. Carbonate minerals can record and preserve information regarding the temperature and chemistry of their formation environment through aspects of their oxygen and carbon isotopic composition (δ18O and δ13C, respectively). These minerals have been observed in a range of Martian geological materials, including dust, bedrock outcrops, and several meteorites (4–9). Collectively, these observations highlight a role for carbonate formation in the global Martian carbon cycle and the evolution of its atmosphere. The oldest known carbonate from Mars, or any other planetary body, occurs as a minor constituent (approximately 1% by weight) in the meteorite Allan Hills 84001 (ALH84001), which has a crystallization age of approximately 4.1 billion years (10). The carbonate in ALH84001, geochronologically constrained to be slightly younger in age [between 3.9 and 4.0 billion years old; (11)], provides a unique window into the hydrologic cycle, carbon cycle, and climate of Noachian Mars.
Texturally, the carbonates in ALH84001 occur as (i) chemically and isotopically zoned ovoid concretions, veins, and void fillings and (ii) regions of massive carbonate, variably intergrown with unweathered feldspathic glass and orthopyroxene, the latter of which makes up the bulk of the meteorite (12–14). The concretions display wide systematic variation in chemical and isotopic composition. They have Ca-Fe-rich cores with 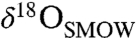 (O isotope ratios reported relative to Standard Mean Ocean Water) values as low as approximately 5‰ and
(O isotope ratios reported relative to Standard Mean Ocean Water) values as low as approximately 5‰ and 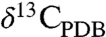 (C isotope ratios reported relative to the Pee Dee Belemnite standard) values of 25–35‰ (15–18). Their rims are Mg-rich, sometimes nearly pure MgCO3, and have
(C isotope ratios reported relative to the Pee Dee Belemnite standard) values of 25–35‰ (15–18). Their rims are Mg-rich, sometimes nearly pure MgCO3, and have 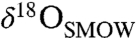 and
and 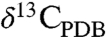 values as high as approximately 30‰ and 65‰, respectively. The veins and void fillings display covarying chemical and isotopic compositions similar to the concretions (12). The massive carbonates are usually internally homogeneous, though chemical and isotopic variability has been observed between isolated occurrences. Several studies have reported an ankeritic (Ca-rich) and isotopically light (
values as high as approximately 30‰ and 65‰, respectively. The veins and void fillings display covarying chemical and isotopic compositions similar to the concretions (12). The massive carbonates are usually internally homogeneous, though chemical and isotopic variability has been observed between isolated occurrences. Several studies have reported an ankeritic (Ca-rich) and isotopically light (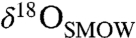 of 0–10‰) composition for the massive carbonates (e.g., ref. 16).
of 0–10‰) composition for the massive carbonates (e.g., ref. 16).
The carbonates in ALH84001 have been studied by a variety of geochemical and petrographic methods, but no agreement exists concerning the processes and environment of their precipitation. Hypotheses for their formation span a range of temperatures between subfreezing and more than 700 °C, a range of depths between the surface and several kilometers, and a range of processes including impact melting of preexisting carbonates, hydrothermal alteration of crustal rocks, precipitation from a rapidly evaporating water body, and biological activity (13–16, 19–28). Textural observations indicate that after carbonate growth, ALH84001 experienced brief, localized, shock-related heating, but natural remanent magnetization and argon thermochronometry place upper limits of approximately 40 °C on the short-duration (approximately 10 min) and approximately 20–30 °C on the long-duration (billions of years) thermal history of this rock (29, 30).
Previous methods for estimating the temperature of carbonate formation in ALH84001, though informative, have placed only loose constraints (e.g., refs. 29 and 30) or have depended on assumptions about the chemical and isotopic composition of the carbonate’s parent fluid (e.g., refs. 16 and 22). Multiply substituted, or “clumped,” isotope thermometry avoids this problem because it is based on homogeneous thermodynamic equilibrium within the carbonate mineral (31, 32). The abundance of rare isotopologues, measured as mass 47 CO2 (mostly 13C18O16O) evolved from acid digestion of carbonate minerals, is higher than predicted from a random distribution of the heavy isotopes. This excess is denoted Δ47 and varies systematically with carbonate precipitation temperature (32, 33). With temperature known, measurements of the δ18O and δ13C of the carbonates provide direct constraints on the isotopic compositions of their parent fluid (water and CO2).
Results
We performed stepped phosphoric acid digestions of three aliquots of ALH84001 and measured the bulk and clumped isotopic composition of the evolved CO2 (see Methods and SI Text). The amount of CO2 evolved from the digestion steps was about one-tenth to one-third the amount typically required for clumped isotope analysis. Additionally, the acid digestion duration was up to 36 times longer than typically applied, increasing the potential for contamination. This combination of factors necessitated special extraction apparatus, purification steps, and analytical protocols (SI Text, Table S1, and Figs. S1–S3). The δ18O and δ13C values of the carbonate extracted in the three digestion steps (Table 1 and Table S2) were reproducible among the three different meteorite aliquots and fall within the range of previous measurements (15–18, 21, 22). Combining these δ18O and δ13C measurements with prior constraints on the relationship between stable isotope and major element composition in ALH84001 carbonates (15–18), we estimated the chemical composition of carbonate sampled by the digestion steps (Fig. 1 and SI Text). The sequential extraction technique yielded increasing Mg content with reaction time, a relationship expected from the slower dissolution of the Mg-rich outer concretion layers (34).
Table 1.
Isotopic composition of ALH84001 carbonates extracted by stepped acid digestion
| 0–1 h | 1–4 h | 4–12 h | |||||||||||||
| N | δ18O‰ | δ13C‰ | Δ47‰ | T °C | N | δ18O‰ | δ13C‰ | Δ47‰ | T °C | N | δ18O‰ | δ13C‰ | Δ47‰ | T °C | |
| Aliquot A (320 mg) | — | 13.05 | 35.52 | — | — | — | 16.68 | 40.87 | — | — | — | 18.96 | 40.82 | — | — |
| Aliquot B (1,625 mg) | 5 | 13.38 (±0.28) | 34.60 (±0.07) | 0.642 (±0.039) | 27 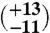
|
5 | 17.32 (±0.28) | 41.05 (±0.07) | 0.638 (±0.039) | 28 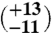
|
1 | 17.99 (±0.28) | 40.94 (±0.07) | 0.742 (±0.098) | 3 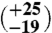
|
| Aliquot C (1,550 mg) | 8 | 14.11 (±0.16) | 35.14 (±0.02) | 0.693 (±0.027) | 14 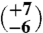
|
9 | 16.27 (±0.16) | 40.67 (±0.02) | 0.643 (±0.027) | 26 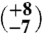
|
7 | 18.17 (±0.16) | 42.39 (±0.02) | 0.711 (±0.028) | 10 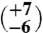
|
δ18O and δ13C are reported relative to the SMOW and PDB standards, respectively. N is the number of data acquisitions suitable for clumped isotope analysis (typical clumped isotope measurements comprise eight acquisitions), reflecting the gas yield from the digestion steps. Error estimates are based on identical treatment of carbonate standards of known bulk and clumped isotopic composition, which was only done for aliquots B and C (SI Text)
Fig. 1.

Major element composition of the ALH84001 carbonates digested in the three steps, estimated from their δ18O and δ13C values and correlations between major element and isotopic composition (see text).
The first aliquot of the meteorite (approximately 320 mg) yielded too little gas for reliable Δ47 measurements, but has δ18O and δ13C values that are consistent with subsequent extractions of two larger aliquots (1,625 and 1,550 mg, respectively). The Δ47 measurements of these two larger aliquots (Table 1) yield six temperatures between 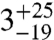 and
and 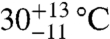 , all within uncertainty of an error-weighted average temperature of 18 ± 4 °C.
, all within uncertainty of an error-weighted average temperature of 18 ± 4 °C.
Discussion
Establishing the Robustness of the Temperature Determination.
We addressed potential sources of uncertainty (outside of analytical uncertainty) in interpreting the Δ47 data as a precipitation temperature for the carbonates in ALH84001. The crystal orientation distribution of the carbonate concretions (SI Text and Fig. S4) suggests that the carbonates have not been subject to recrystallization since their radial growth from aqueous solution. In the absence of recrystallization, resetting of the carbonates’ isotopic composition to yield the observed, strongly covarying relationship between δ18O and δ13C is unlikely. Together, these observations indicate that the measured Δ47 was acquired during initial carbonate precipitation.
Precipitation at high temperature and subsequent reordering of carbonate clumped isotopes upon cooling is unlikely because the temperature recorded under such a scenario (i.e., blocking temperature) would be approximately 200–300 °C (35, 36). Mixing between two physical or isotopic end members with bulk isotopic composition at either end of the observed δ13C and δ18O range in ALH84001 carbonate may lead to spuriously low apparent temperatures, but is also unlikely, as indicated both by previous observations of the distribution of ALH84001 carbonate chemical and isotopic composition (15–18) and by limits on the time scale for isotopic equilibrium (SI Text and Fig. S5).
Several additional hypotheses for interpreting the Δ47 data can be rejected on the basis of the relationships observed among the δ18O, δ13C, and Δ47 measured in the carbonate (Fig. 2). Carbonate precipitation over a range of temperatures (e.g., ref. 22) does not fit the observed δ18O and δ13C array (Fig. 2A). Because the stepped acid digestion could not perfectly separate the concretion cores from their rims, the range of 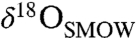 we observed was narrow (approximately 13–19‰) relative to the full range observed in microprobe studies (approximately 5–30‰). As a result, the uncertainty in a linear fit to the reliable δ18O-Δ47 data (aliquots B and C) is large at the ends of the full δ18O range (Fig. 2B, dashed curves). Thus, in a formal hypothesis testing sense, the 95% confidence bounds marginally include an explanation of the data by kinetic isotope effects (KIEs) of the sort observed in some cave and cryogenic carbonates (37–40). These KIEs, which are thought to arise in part from rapid, disequilibrium CO2 degassing, have been observed to lead to spurious apparent values of Δ47 that are lower than the true values by as much as 0.12‰. Applied to our measured Δ47, this would translate into an error-weighted average temperature of carbonate formation in ALH84001 of -7 °C (instead of approximately 18 °C if the Δ47 we measure is correct). Temperatures below 0 °C can be rejected because they would cause the water from which the carbonates formed to freeze and are thus inconsistent with the increase in δ18O from the concretion cores to their rims. The remaining temperature range between 0 and 18 °C is possible, but the large uncertainty in the fit and the better correspondence to a hypothesis of constant temperature (Fig. 2B) lead us to conclude that KIEs of the type discussed above are unlikely in the case of ALH84001 carbonates. Altogether the data are best explained by carbonate precipitation at a constant temperature of approximately 18 °C. This result rules out a high-temperature origin of ALH84001 carbonates (e.g., refs. 13, 14, and 19) and imposes strict constraints on the remaining low-temperature hypotheses.
we observed was narrow (approximately 13–19‰) relative to the full range observed in microprobe studies (approximately 5–30‰). As a result, the uncertainty in a linear fit to the reliable δ18O-Δ47 data (aliquots B and C) is large at the ends of the full δ18O range (Fig. 2B, dashed curves). Thus, in a formal hypothesis testing sense, the 95% confidence bounds marginally include an explanation of the data by kinetic isotope effects (KIEs) of the sort observed in some cave and cryogenic carbonates (37–40). These KIEs, which are thought to arise in part from rapid, disequilibrium CO2 degassing, have been observed to lead to spurious apparent values of Δ47 that are lower than the true values by as much as 0.12‰. Applied to our measured Δ47, this would translate into an error-weighted average temperature of carbonate formation in ALH84001 of -7 °C (instead of approximately 18 °C if the Δ47 we measure is correct). Temperatures below 0 °C can be rejected because they would cause the water from which the carbonates formed to freeze and are thus inconsistent with the increase in δ18O from the concretion cores to their rims. The remaining temperature range between 0 and 18 °C is possible, but the large uncertainty in the fit and the better correspondence to a hypothesis of constant temperature (Fig. 2B) lead us to conclude that KIEs of the type discussed above are unlikely in the case of ALH84001 carbonates. Altogether the data are best explained by carbonate precipitation at a constant temperature of approximately 18 °C. This result rules out a high-temperature origin of ALH84001 carbonates (e.g., refs. 13, 14, and 19) and imposes strict constraints on the remaining low-temperature hypotheses.
Fig. 2.

Comparison of Δ47, δ18O, and δ13C values (see SI Text) with models for the isotopic variation in ALH84001 carbonates. (A) Relationship between δ13C and δ18O. The solid black line and the broken black curves are a weighted total least squares linear fit to the data and a 95% confidence bound on the fit, respectively. The diagonal gray line shows the equilibrium relationship between δ13C and δ18O over the range of temperatures required to explain the range of 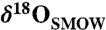 observed in microprobe studies (approximately 5–30‰). A hypothesis of evolving temperature can be rejected on the basis of the misfit between these lines. (B) Relationship between Δ47 and δ18O. The solid black line and the broken black curves are a weighted total least squares linear fit to the data and a 95% confidence bound on the fit, respectively. The diagonal gray line shows the equilibrium relationship between Δ47 and δ18O over the range of temperatures required to explain the observed
observed in microprobe studies (approximately 5–30‰). A hypothesis of evolving temperature can be rejected on the basis of the misfit between these lines. (B) Relationship between Δ47 and δ18O. The solid black line and the broken black curves are a weighted total least squares linear fit to the data and a 95% confidence bound on the fit, respectively. The diagonal gray line shows the equilibrium relationship between Δ47 and δ18O over the range of temperatures required to explain the observed 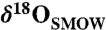 range (approximately 5–30‰). The horizontal gray line shows the isotopic evolution due to evaporation at a constant temperature. The temperature of 21.2 °C is the average temperature of the two leftmost data points. The light and dark orange wedges are model predictions for the relationship between Δ47 and δ18O that is expected from rapid degassing and associated kinetic isotope effects (38). The confidence bounds on the fit to the data are most consistent with constant temperature, less consistent with an evolving temperature (which can be ruled out by the relationship between δ13C and δ18O) and only marginally include kinetic isotope effects due to rapid degassing (see text).
range (approximately 5–30‰). The horizontal gray line shows the isotopic evolution due to evaporation at a constant temperature. The temperature of 21.2 °C is the average temperature of the two leftmost data points. The light and dark orange wedges are model predictions for the relationship between Δ47 and δ18O that is expected from rapid degassing and associated kinetic isotope effects (38). The confidence bounds on the fit to the data are most consistent with constant temperature, less consistent with an evolving temperature (which can be ruled out by the relationship between δ13C and δ18O) and only marginally include kinetic isotope effects due to rapid degassing (see text).
Formation Environment of ALH84001 Carbonates.
Previously only loose constraints on the temperature of carbonate growth in ALH84001 were available, and past models for their formation required one or more assumptions about temperature, the open/closed nature of the system, the ratio of CO2 to H2O, or the isotopic composition of the parent fluid. Here, by independently constraining temperature, we are able to use the isotopic variation in the carbonates to develop a model for their formation environment and mechanism.
From the Δ47 constraints, we find that the striking core to rim coevolution of 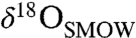 (approximately 5 → 30‰) and
(approximately 5 → 30‰) and 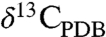 (approximately 25 → 65‰), observed in previous ion-microprobe studies (15–18) is best explained by gradual evaporation of a shallow subsurface aqueous solution at a constant, low temperature. The evaporative water loss drives carbonate precipitation and CO2 degassing under equilibrium conditions. The observed isotopic variation in the carbonates suggests a system in poor short-term communication with the atmosphere—otherwise the δ18O and δ13C would be buffered by exchange with the atmospheric CO2 reservoir. This rules out, for example, formation of the ALH84001 carbonates in a long-lasting, open lake. Instead we suggest that distillative loss of the isotopically light vapor phase (gaseous CO2 and water vapor) from a semiisolated environment (Fig. 3A) gradually enriched the residual water and dissolved inorganic carbon in the heavy isotopes, resulting in the coupled increase in δ18O and δ13C values from the concretion cores to their rims. A system open to the atmosphere, where evaporation and carbonate precipitation occurred too rapidly to allow equilibrium with the gas phase, may also be consistent with the coevolution of δ18O and δ13C values, but is not supported by the observed δ18O-Δ47 relationship (Fig. 2B). Finally, although the observed δ13C increase can also be explained by organic matter synthesis or oxidation, the existence of extraterrestrial organic matter in ALH84001 remains controversial, due mainly to the difficulty in ruling out contamination by terrestrial organic matter (27). Furthermore, the covariation in carbon and oxygen isotope ratios suggests a common process affected both reservoirs.
(approximately 25 → 65‰), observed in previous ion-microprobe studies (15–18) is best explained by gradual evaporation of a shallow subsurface aqueous solution at a constant, low temperature. The evaporative water loss drives carbonate precipitation and CO2 degassing under equilibrium conditions. The observed isotopic variation in the carbonates suggests a system in poor short-term communication with the atmosphere—otherwise the δ18O and δ13C would be buffered by exchange with the atmospheric CO2 reservoir. This rules out, for example, formation of the ALH84001 carbonates in a long-lasting, open lake. Instead we suggest that distillative loss of the isotopically light vapor phase (gaseous CO2 and water vapor) from a semiisolated environment (Fig. 3A) gradually enriched the residual water and dissolved inorganic carbon in the heavy isotopes, resulting in the coupled increase in δ18O and δ13C values from the concretion cores to their rims. A system open to the atmosphere, where evaporation and carbonate precipitation occurred too rapidly to allow equilibrium with the gas phase, may also be consistent with the coevolution of δ18O and δ13C values, but is not supported by the observed δ18O-Δ47 relationship (Fig. 2B). Finally, although the observed δ13C increase can also be explained by organic matter synthesis or oxidation, the existence of extraterrestrial organic matter in ALH84001 remains controversial, due mainly to the difficulty in ruling out contamination by terrestrial organic matter (27). Furthermore, the covariation in carbon and oxygen isotope ratios suggests a common process affected both reservoirs.
Fig. 3.

Model for carbonate formation in ALH84001. (A) Physical model of a shallow subsurface aquifer. The depth of carbonate formation is constrained by a combination of the cosmic ray exposure pattern of ALH84001 and Martian meteorite ejection models (48, 49). (B) Reduction of the physical model to a geochemical model of carbonate precipitation and CO2 degassing driven by gradual evaporation of water in a confined volume, coupled to loss of the vapor phase.
Support for the hypothesis of carbonate formation from an evaporating subsurface aqueous reservoir comes from observed covariation between δ18O and δ13C in terrestrial carbonates from cold or arid regions. These analogs are imperfect because even in cold, dry, and relatively organic-poor environments, organic matter fixation or respiration contributes to the preserved isotopic signal to variable degree. Even so, the isotopic composition of Mg-bearing carbonates formed on ultramafic mine tailings in northwestern Canada (latitude ≥58 °N) form arrays strikingly similar to those observed in ALH84001 carbonates in δ18O-δ13C space (41), as do endostromatolites in an impact structure in the Canadian high Arctic (42), soil carbonates from the Mojave and Atacama deserts (43, 44), and caliche formed on basalts in young volcanic fields in Arizona (45). All of these studies discuss hypotheses that explain the observed isotopic trends with evaporation and CO2 degassing.
To further examine this hypothesis we developed a model of carbonate precipitation (Fig. 3B) in which carbonate mineral saturation and charge balance are maintained, as is chemical and isotopic equilibrium among the aqueous solution, the instantaneous precipitate, and the gas phase in the regolith during gradual evaporation (Methods and SI Text). The range of δ18O values in the concretions requires distillative loss of 42–92% of the water, depending on whether the transport of water toward the surface is rate limited by vapor-phase diffusion. The observed covariation in δ18O and δ13C can be reproduced (Fig. 4) if approximately 87–98% of the noncarbonate carbon loss from solution is transported to the surface. The remaining 2–13% causes a pore pCO2 increase of 5–15%, consistent with the modest pressure gradient that would be required to cause distillative CO2 loss from the subsurface in the scenario considered here. These results are relatively insensitive to the choice of initial pCO2, the nonprecipitating ion concentration, and the particular carbonate mineralogy (SI Text and Fig. S6).
Fig. 4.

Comparison of observed and modeled isotopic composition. Gray crosses are combinations of δ18O and δ13C microprobe data correlated by Mg content, including uncertainty in the correlation (SI Text). The black curves show a weighted total least squares linear fit through these data (solid) and 95% confidence bounds on the fit (broken). The black triangles are measurements from this study. The gray rectangle brackets the highest values of 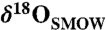 achievable by water–rock oxygen isotope exchange, given the uncertainty on temperature and on the measured δ18O of the ALH84001 silicates (SI Text). The light orange field shows the coevolution of δ18O and δ13C during 42% evaporation and diffusion-limited water loss from a subsurface reservoir, accompanied by carbonate precipitation and CO2 degassing and diffusion, at a temperature of 17.5 ± 5 °C (SI Text). The darker orange field is for 92% evaporative water loss and accompanying carbonate precipitation if transport to the surface is not rate-limited by diffusion. The gray line with a negative slope shows the δ18O and δ13C coevolution if the water slowly freezes instead of evaporating. The initial pCO2 in the calculations was 600 millibar and the nonprecipitating solute concentrations were 0 mol liter-1. The results are relatively insensitive to this choice (SI Text and Fig. S6).
achievable by water–rock oxygen isotope exchange, given the uncertainty on temperature and on the measured δ18O of the ALH84001 silicates (SI Text). The light orange field shows the coevolution of δ18O and δ13C during 42% evaporation and diffusion-limited water loss from a subsurface reservoir, accompanied by carbonate precipitation and CO2 degassing and diffusion, at a temperature of 17.5 ± 5 °C (SI Text). The darker orange field is for 92% evaporative water loss and accompanying carbonate precipitation if transport to the surface is not rate-limited by diffusion. The gray line with a negative slope shows the δ18O and δ13C coevolution if the water slowly freezes instead of evaporating. The initial pCO2 in the calculations was 600 millibar and the nonprecipitating solute concentrations were 0 mol liter-1. The results are relatively insensitive to this choice (SI Text and Fig. S6).
Although the fluid in an environment such as the one we describe could, in principle, come from depth (magmatic or hydrothermal fluid), there is evidence that the parent fluid of the ALH84001 carbonates came from the surface (atmosphere–hydrosphere). The carbonates have an oxygen isotopic composition that is anomalous with respect to Martian igneous rocks [Δ17O > 0.3; (46)]. Similarly, sulfides in this sample have a “mass-independent” sulfur isotopic composition [Δ33S ≠ 0; (47)]. Both signatures were likely generated by atmospheric reactions and support a surface source of the fluid. Furthermore, water in oxygen isotope equilibrium with the Ca-rich concretion cores (determined by microprobe studies) would, at the temperatures we measured, have δ18O more than 10‰ lower than equilibrium with coexisting silicate minerals (Fig. S7) and CO2 in equilibrium with the concretion cores is tens of permil too high in δ13C to have come directly from the Martian mantle (though the δ13C of the Martian mantle is poorly constrained, SI Text).
At the measured growth temperature, the Mg-rich carbonates in the concretion rims have δ18O 10–15‰ heavier than equilibrium with the silicates (Fig. S7). This, together with the persistence of the mass-anomalous oxygen isotope composition, implies that the combination of time and temperature was insufficient to allow appreciable oxygen isotope exchange between water and rock, supporting a low-temperature, transient aqueous environment. This is consistent also with the absence of alteration minerals (e.g., hydrous phyllosilicates) in ALH84001 other than the carbonates and the associated sulfides and magnetite (13, 14).
Conclusions
In sum, the evidence points to a short-lived, low-temperature, aqueous environment, initially containing surface-derived CO2 and H2O but subsequently characterized by poor communication with the atmosphere, such that isotopic buffering did not occur. Distillative loss of CO2 and H2O can explain the observed δ18O and δ13C trends. This combination of characteristics is consistent with partial to complete evaporation of a subsurface aqueous reservoir, coupled to carbonate precipitation and CO2 degassing under equilibrium conditions. We imagine that an ephemeral regolith aquifer at a depth of a few to tens of meters would be a plausible setting for this process (Fig. 3). The existence of a near-surface aqueous environment during the Noachian epoch, recorded by the carbonates in ALH84001, is consistent with, though need not require, a warmer early Mars—a hydrothermal or impact-related source for the required heat is equally possible. Though the mild temperatures point to an environment that might be considered habitable, the presence of water was also ephemeral, suggesting a time frame probably too short for life to have evolved de novo.
Methods
Three aliquots of ALH84001 were subjected to stepped phosphoric acid digestion. Because of the slower dissolution kinetics of magnesian carbonates, the first step in each of the digestions (0–1 h) sampled the faster-reacting, Ca-rich carbonates, whereas the second and third steps (1–4 and 4–12 h, respectively) sampled more Mg-rich compositions (34). Finer separation (e.g., by lower reaction temperature or shorter reaction intervals) was not attempted due to the relatively large amount of CO2 required for clumped isotope analysis, the susceptibility of small samples to contamination, and the precious nature of the sample. The bulk and clumped isotopic composition of evolved CO2 was measured on a Thermo Finnigan MAT 253 isotope ratio gas source mass spectrometer. The small sample size necessitated measurement using a microvolume. Measurements were standardized to carbonates of known bulk and clumped isotopic composition, treated identically to the meteorite aliquots and measured using a microvolume. See SI Text for full analytical methods.
With knowledge of the precipitation temperature, we constructed an isotopic evolution model and examined hypotheses for carbonate precipitation that could also quantitatively explain the observed covariation of carbon and oxygen isotope ratios in the carbonate. Prescribing values for the model-free parameters—the initial CO2 pressure (pCO2) and the concentration of nonprecipitating ions—we solve for the pH, solute concentrations, and pCO2 as the water evaporates. The oxygen isotopic evolution depends on the cumulative fraction of water lost to evaporation and vapor-phase diffusion. The carbon removed by carbonate precipitation is equal to the loss of the precipitating cation, which is solved by the model. The remaining carbon lost from solution is divided between transport to the surface and an increase in pore pCO2. The carbon isotopic evolution, therefore, depends on the relative proportions of these removal processes (carbonate precipitation, diffusive transport, and pore overpressurization). See SI Text for a full description of the model and parametric sensitivity analysis results.
Supplementary Material
Acknowledgments.
We thank N. Kitchen for assistance with the isotopic analyses. We thank two anonymous reviewers for valuable comments that have improved this work. I.H. was funded by a Texaco Postdoctoral Fellowship (Division of Geological and Planetary Sciences, Caltech). J.M.E. acknowledges funding from National Aeronautics and Space Administration and National Science Foundation-Division of Earth Sciences.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1109444108/-/DCSupplemental.
References
- 1.Haberle RM. Early Mars climate models. J Geophys Res. 1998;103:28467–28479. [Google Scholar]
- 2.Carr MH. Water on Mars. New York: Oxford Univ Press; 1996. p. vii. [Google Scholar]
- 3.Fassett CI, Head JW. Valley network-fed, open-basin lakes on Mars: Distribution and implications for Noachian surface and subsurface hydrology. Icarus. 2008;198:37–56. [Google Scholar]
- 4.Bandfield JL, Glotch TD, Christensen PR. Spectroscopic identification of carbonate minerals in the martian dust. Science. 2003;301(5636):1084–1087. doi: 10.1126/science.1088054. [DOI] [PubMed] [Google Scholar]
- 5.Boynton WV, et al. Evidence for calcium carbonate at the Mars Phoenix Landing Site. Science. 2009;325:61–64. doi: 10.1126/science.1172768. [DOI] [PubMed] [Google Scholar]
- 6.Ehlmann BL, et al. Orbital identification of carbonate-bearing rocks on Mars. Science. 2008;322:1828–1832. doi: 10.1126/science.1164759. [DOI] [PubMed] [Google Scholar]
- 7.Michalski JR, Niles PB. Deep crustal carbonate rocks exposed by meteor impact on Mars. Nat Geosci. 2010;3:751–755. [Google Scholar]
- 8.Morris RV, et al. Identification of carbonate-rich outcrops on Mars by the Spirit Rover. Science. 2010;329:421–424. doi: 10.1126/science.1189667. [DOI] [PubMed] [Google Scholar]
- 9.Grady MM, Wright I. The carbon cycle on early Earth—and on Mars? Philos Trans R Soc Lond B Biol Sci. 2006;361:1703–1713. doi: 10.1098/rstb.2006.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lapen TJ, et al. A younger age for ALH84001 and its geochemical link to shergottite sources in Mars. Science. 2010;328(5976):347–351. doi: 10.1126/science.1185395. [DOI] [PubMed] [Google Scholar]
- 11.Borg LE, et al. The age of the carbonates in martian meteorite ALH84001. Science. 1999;286:90–94. doi: 10.1126/science.286.5437.90. [DOI] [PubMed] [Google Scholar]
- 12.Corrigan CM, Harvey RP. Multi-generational carbonate assemblages in martian meteorite Allan Hills 84001: Implications for nucleation, growth, and alteration. Meteorit Planet Sci. 2004;39:17–30. [Google Scholar]
- 13.Mittlefehldt DW. ALH84001, a cumulate orthopyroxenite member of the Martian meteorite clan. Meteoritics. 1994;29:214–221. [Google Scholar]
- 14.Treiman AH. A petrographic history of Martian meteorite ALH84001: 2 shocks and an ancient age. Meteoritics. 1995;30:294–302. [Google Scholar]
- 15.Leshin LA, McKeegan KD, Carpenter PK, Harvey RP. Oxygen isotopic constraints on the genesis of carbonates from Martian meteorite ALH84001. Geochim Cosmochim Acta. 1998;62:3–13. [Google Scholar]
- 16.Eiler JM, Valley JW, Graham CM, Fournelle J. Two populations of carbonate in ALH84001: Geochemical evidence for discrimination and genesis. Geochim Cosmochim Acta. 2002;66:1285–1303. [Google Scholar]
- 17.Saxton JM, Lyon IC, Turner G. Correlated chemical and isotopic zoning in carbonates in the Martian meteorite ALH84001. Earth Planet Sci Lett. 1998;160:811–822. [Google Scholar]
- 18.Niles PB, Leshin LA, Guan Y. Microscale carbon isotope variability in ALH84001 carbonates and a discussion of possible formation environments. Geochim Cosmochim Acta. 2005;69:2931–2944. [Google Scholar]
- 19.Scott ERD, Yamaguchi A, Krot AN. Petrological evidence for shock melting of carbonates in the Martian meteorite ALH84001. Nature. 1997;387:377–379. doi: 10.1038/387377a0. [DOI] [PubMed] [Google Scholar]
- 20.Harvey RP, McSween HY. A possible high-temperature origin for the carbonates in the Martian meteorite ALH84001. Nature. 1996;382:49–51. doi: 10.1038/382049a0. [DOI] [PubMed] [Google Scholar]
- 21.Valley JW, et al. Low-temperature carbonate concretions in the Martian meteorite ALH84001: Evidence from stable isotopes and mineralogy. Science. 1997;275:1633–1638. doi: 10.1126/science.275.5306.1633. [DOI] [PubMed] [Google Scholar]
- 22.Romanek CS, et al. Record of fluid-rock interactions on Mars from the meteorite ALH84001. Nature. 1994;372:655–657. doi: 10.1038/372655a0. [DOI] [PubMed] [Google Scholar]
- 23.Warren PH. Petrologic evidence for low-temperature, possibly flood evaporitic origin of carbonates in the ALH84001 meteorite. J Geophys Res. 1998;103:16759–16773. doi: 10.1029/98je01544. [DOI] [PubMed] [Google Scholar]
- 24.McSween HY, Harvey RP. An evaporation model for formation of carbonates in the ALH84001 Martian meteorite. Int Geol Rev. 1998;40:774–783. [Google Scholar]
- 25.Eiler JM, Kitchen N, Leshin L, Strausberg M. Hosts of hydrogen in Allan Hills 84001: Evidence for hydrous martian salts in the oldest martian meteorite? Meteorit Planet Sci. 2002;37:395–405. [Google Scholar]
- 26.McKay DS, et al. Search for past life on Mars: Possible relic biogenic activity in Martian meteorite ALH84001. Science. 1996;273:924–930. doi: 10.1126/science.273.5277.924. [DOI] [PubMed] [Google Scholar]
- 27.Jull AJT, Courtney C, Jeffrey DA, Beck JW. Isotopic evidence for a terrestrial source of organic compounds found in martian meteorites Allan Hills 84001 and Elephant Moraine 79001. Science. 1998;279:366–369. doi: 10.1126/science.279.5349.366. [DOI] [PubMed] [Google Scholar]
- 28.Golden DC, et al. A simple inorganic process for formation of carbonates, magnetite, and sulfides in Martian meteorite ALH84001. Am Mineral. 2001;86:370–375. [Google Scholar]
- 29.Weiss BP, et al. A low temperature transfer of ALH84001 from Mars to Earth. Science. 2000;290:791–795. doi: 10.1126/science.290.5492.791. [DOI] [PubMed] [Google Scholar]
- 30.Cassata WS, Shuster DL, Renne PR, Weiss BP. Evidence for shock heating and constraints on Martian surface temperatures revealed by 40Ar/39Ar thermochronometry of Martian meteorites. Geochim Cosmochim Acta. 2010;74:6900–6920. [Google Scholar]
- 31.Eiler JM, Schauble E. 18O13C16O in Earth’s atmosphere. Geochim Cosmochim Acta. 2004;68:4767–4777. [Google Scholar]
- 32.Ghosh P, et al. 13C-18O bonds in carbonate minerals: A new kind of paleothermometer. Geochim Cosmochim Acta. 2006;70:1439–1456. [Google Scholar]
- 33.Huntington KW, et al. Methods and limitations of ‘clumped’ CO2 isotope (Δ47) analysis by gas-source isotope ratio mass spectrometry. J Mass Spectrom. 2009;44:1318–1329. doi: 10.1002/jms.1614. [DOI] [PubMed] [Google Scholar]
- 34.Al-Aasm IS, Taylor BE, South B. Stable isotope analysis of multiple carbonate samples using selective acid extraction. Chem Geol. 1990;80:119–125. [Google Scholar]
- 35.Eiler JM, Bonifacie M, Daeron M. ‘Clumped isotope’ thermometry for high-temperature problems. Geochim Cosmochim Acta. 2009;73:A322. [Google Scholar]
- 36.Dennis KJ, Schrag DP. Clumped isotope thermometry of carbonatites as an indicator of diagenetic alteration. Geochim Cosmochim Acta. 2010;74:4110–4122. [Google Scholar]
- 37.Daeron M, et al. 13C18O Clumping in speleothems: Observations from natural caves and precipitation experiments. Geochim Cosmochim Acta. 2011;75:3303–3317. [Google Scholar]
- 38.Guo W. Pasadena, CA: California Inst of Technology; 2009. Carbonate clumped isotope thermometry: Application to carbonaceous chondrites and effects of kinetic isotope fractionation. PhD dissertation. [Google Scholar]
- 39.Affek HP, Bar-Matthews M, Ayalon A, Matthews A, Eiler JM. Glacial/interglacial temperature variations in Soreq cave speleothems as recorded by ‘clumped isotope’ thermometry. Geochim Cosmochim Acta. 2008;72:5351–5360. [Google Scholar]
- 40.Affek HP, Guo W, Daeron M, Eiler JM. ‘Clumped isotopes’ in speleothem carbonate and atmospheric CO2—Is there a kinetic isotope effect? Geochim Cosmochim Acta. 2008;72:A6. [Google Scholar]
- 41.Wilson SA, et al. Carbon dioxide fixation within mine wastes of ultramafic-hosted ore deposits: Examples from the Clinton Creek and Cassiar Chrysotile Deposits, Canada. Econ Geol. 2009;104:95–112. [Google Scholar]
- 42.Lacelle D, Pellerin A, Clark ID, Lauriol B, Fortin D. (Micro) morphological, inorganic-organic isotope geochemisty and microbial populations in endostromatolites (cf. fissure calcretes), Haughton impact structure, Devon Island, Canada: The influence of geochemical pathways on the preservation of isotope biomarkers. Earth Planet Sci Lett. 2009;281:202–214. [Google Scholar]
- 43.Quade J, Cerling TE, Bowman JR. Systematic variations in the carbon and oxygen isotopic composition of pedogenic carbonate along elevation transects in the Southern Great-Basin, United-States. Geol Soc Am Bull. 1989;101:464–475. [Google Scholar]
- 44.Quade J, et al. Soils at the hyperarid margin: The isotopic composition of soil carbonate from the Atacama Desert, Northern Chile. Geochim Cosmochim Acta. 2007;71:3772–3795. [Google Scholar]
- 45.Knauth LP, Brilli M, Klonowski S. Isotope geochemistry of caliche developed on basalt. Geochim Cosmochim Acta. 2003;67:185–195. [Google Scholar]
- 46.Farquhar J, Thiemens MH, Jackson T. Atmosphere-surface interactions on Mars: Δ17O measurements of carbonate from ALH 84001. Science. 1998;280:1580–1582. doi: 10.1126/science.280.5369.1580. [DOI] [PubMed] [Google Scholar]
- 47.Farquhar J, Savarino J, Jackson TL, Thiemens MH. Evidence of atmospheric sulphur in the martian regolith from sulphur isotopes in meteorites. Nature. 2000;404:50–52. doi: 10.1038/35003517. [DOI] [PubMed] [Google Scholar]
- 48.Fritz J, Artemieva N, Greshake A. Ejection of Martian meteorites. Meteorit Planet Sci. 2005;40:1393–1411. [Google Scholar]
- 49.Warren PH. Lunar and Martian meteorite delivery services. Icarus. 1994;111:338–363. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.