

Abstract
Long interspersed nuclear elements-1 (L1s) are highly repetitive DNA elements that are capable of altering the human genome through retrotransposition. To protect against L1 retroposition, the cell downregulates the expression of L1 proteins by various mechanisms, including high-density cytosine methylation of L1 promoters and DICER-dependent destruction of L1 mRNAs. In this report, a large number of p53 responsive elements, or p53 DNA binding sites, were detected in L1 elements within the human genome. At least some of these p53 responsive elements are functional and can act to increase the levels of L1 mRNA expression. The p53 protein can directly bind to a short 15-nucleotide sequence within the L1 promoter. This p53 responsive element within L1 is a recent addition to evolution, appearing ~20 million years ago. This suggests an interplay between L1 elements, which have a rich history of causing changes in the genome, and the p53 protein, the function of which is to protect against genomic changes. To understand these observations, a model is proposed in which the increased expression of L1 mRNAs by p53 actually increases, rather than decreases, the genomic stability through amplification of p53-dependent processes for genomic protection.
Keywords: p53, LINE-1, genome stability
Introduction
p53 is one of the most important tumor-suppressor genes, as indicated by the fact that it is mutant in about half of the solid tumors (Hollstein et al., 1991). The p53 gene product functions as a sequence-specific transcription factor (Kern et al., 1991) with many target genes (Zhao et al., 2000; Kannan et al., 2001). Two 10-mer (10 nucleotides (nt)) half-sites, linked by a short spacer region of 0–13 nt, comprise a p53-binding site. The two half-sites have a degenerate, palindromic sequence: 5′-RRRCWWGYYY-3′, where R represents purines, W represents adenine or thymine and Y represents pyrimidines (Funk et al., 1992; el-Deiry et al., 1992). On the basis of X-ray crystallography of the p53–DNA complex, the most critical bases in → ← are the 6-nt 5′-RCWWGY-3′, which come into close contact with the core DNA-binding domain of the p53 protein (Cho et al., 1994).
Normally a short-lived protein, the activity of p53 is triggered by an increased half-life under conditions of cellular stress (Maltzman and Czyzyk, 1984; Lavin and Gueven, 2006). Typical stresses that activate p53 include insults that lead to mutation, including exposure to chemicals, exposure to radiation and reactive oxygen species created by increased metabolism (Harris and Levine, 2005). Upon activation, the p53 protein can trigger various responses. For instance, p53 can arrest cell cycle, or initiate apoptosis or cellular senescence. All of these pathways have one common theme: the prevention of alterations in the genome and the propagation of these events. For this reason, p53 has been known as the guardian of the genome (Lane, 1992).
A known agent of genomic change is retroposition of repetitive DNA elements, such as L1 elements. L1, also known as long interspersed nuclear element-1 or LINE-1, is 6 kb in length and is present 850 000 times within the human genome (Lander et al., 2001). Although generally silenced by methylation in normal somatic tissues, L1 promoters become demethylated in many tumors, and the L1-ORF1p protein encoded by this element has been observed in tumor samples. L1 elements can amplify their copy number by producing a reverse transcriptase/endonuclease protein (L1-ORF2p), which copies L1 mRNA and inserts it into a new position within the genome (Babushok and Kazazian, 2007). Insertion of an L1 into an exon can decrease gene expression (Han et al., 2004), and insertion of L1s into tumor-suppressor loci would probably hasten tumorigenesis by the inactivation of gene function.
In human tissue culture cells, L1 activity in p53-competent cells can trigger a DNA damage response leading to apoptosis (Haoudi et al., 2004). Therefore, another method by which p53 may function to protect the genome may be to prevent such insertions. Given the number of pathways that could be activated by p53, the prevention of L1 insertion could occur by various mechanisms, including apoptosis (Haoudi et al., 2004). A survey of p53 DNA binding sites in the genome uncovered such sites in the L1 element. The hypothesis that p53, which is a DNA-binding protein capable of activating or repressing transcription (Ori et al., 1998; Budhram-Mahadeo et al., 1999; Lee et al., 1999), might also prevent L1 retroposition by directly repressing the L1 promoter was tested. The experiments presented in this study show that the p53 protein does indeed bind to L1 promoters, but interestingly does not repress L1 transcription; rather, L1 transcription becomes activated by the p53 protein. The binding site for p53 within the L1 DNA is a slightly different motif from that previously described for most p53 responsive elements, in that it is shortened by 5 nt.
Results
Predicted versus observed distributions of p53REs
As noted in the ‘Introduction’ section, p53 responsive elements generally occur as a pair of 10-mer ‘half-site’ sequences separated by a spacer of variable length, usually <14 nt. We denote standard half-sites as → ←. p53 responsive elements can also have reversed quarter sites; that is, instead of 5′-RRRCWWGYYY-3′, the reversed sequence is 5′-WGYYYRRRCW-3′. Such a reversed half-site is found in the promoter of the strongly p53-responsive p21 gene (Resnick-Silverman et al., 1998). We denote reversed half-sites as ← →.
Using a Poisson probability model, we set out to determine spacing between half-sites within the human genome. A total of 732 422 palindromic → ← sites were predicted to reside in the human genome, whereas 750 231 were found. Figure 1 shows the observed and predicted frequencies of spacer lengths separating two → ←s (Figure 1a) and ← →s (Figure 1b). Six frequency peaks were found at spacers of 0, 14, 24, 129, 1895 and 3056 bp, respectively (Figure 1a), occurring on each of the chromosomes but with substantial variability in frequency (http://variation.yale.edu/genome-p53/). The significance of the spacer frequencies was further confirmed by a wavelet shrinkage analysis, from which random noise was removed (SOM). A non-random pattern was also observed between ← → sites with spacers having significantly high relative frequencies at 15, 26, 46, 155, 156 and 157 bp, respectively (Figure 1b). In contrast, ⇒⇐ was randomly distributed in the human genome, the frequency of which was exactly as predicted by the Poisson distribution. A Poisson distribution was also observed for → ← in a randomly shuffled human genome.
Figure 1.
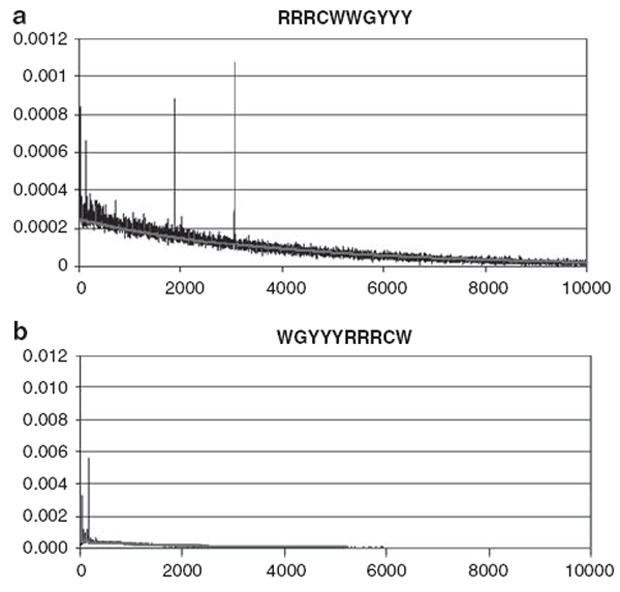
Distributions of p53REs in the human genome. The distributions of the p53REs (a) → ← and (b) ← → in the human genome are shown. Spacer lengths from 0 to 10 kb are on the x axis, and relative frequencies on the y axis. The (red) line represents the exponential density function. Both observed and expected frequencies of spacers > 10 kb are close to 0 and hence are omitted. A full colour version of this figure is available at the Oncogene journal online.
When we compared distributions in different species, it became apparent that each species has its own unique frequency peaks in terms of spacer distributions between → ← sites. The six peaks in humans stand in sharp contrast to those in mouse, worm and fly genomes. Fly and worm genomes have more peaks than does the human genome. In contrast, the mouse genome is more similar to that of the human in number of peaks, but the peak spacers are of shorter lengths, for example, all are <100 bp (results not presented).
p53REs in retrotransposable elements
On the basis of Repbase Update (RepBase7.40–RepBase14.04), a database for consensus sequences of the transposable elements (Jurka, 2000), clusters of → ← with 14- and 24-bp spacers were found in the long-terminal repeat-related sequences of MER41A and MER4D/1, respectively (Table 1). These two long-terminal repeats, ~900 bp in length, are homologous to each other except for a 35-bp sequence at their 3′-ends. More than half of the 129-bp spacers between → ← were found in the pol domain of HERV-H (Table 1). Full-length HERV-H is 7713 bp in length, containing the typical components for a retrovirus: two long-terminal repeats and the gag, pol and env genes. However, in the human genome, all MERs and HERV-H are fragmented.
Table 1.
Peak spacer frequencies and numbers matching to transposable elements
| Spacer length (base pairs)
|
||||||
|---|---|---|---|---|---|---|
| 0 | 14 | 24 | 129 | 1895 | 3056 | |
| Expected | 184 | 115 | 92 | 69 | 46 | 23 |
| Total observed | 527 | 597 | 434 | 502 | 667 | 777 |
| MER41A (LTR of HERV17)a | — | 230 | — | — | — | — |
| MER4D/1(LTR)a | — | — | 143 | — | — | — |
| HERVH (pol region)b | — | — | — | 26 | — | — |
| LINE-1 (L1)c | — | — | — | — | 529 | 661 |
Abbreviation: LTR, long-terminal repeat.
Observed and expected numbers of copies of the primary p53 DNA-binding motif → ← identified in the human genome at the six peak spacer frequencies. The spacer sequences were compared with known transposable elements and the number of spacers within each spacer length with homology to the listed transposable elements is provided.
>80% homology.
>85% homology.
>90% homology.
The majority of → ← with spacers of 1895 and 3056 bp in the human genome were in LINE-1 non-long-terminal repeat retrotransposon elements (L1s). The great bulk of L1 elements are fragmented, but unlike MER and HERV-H elements, full-length actively transposing L1s do exist within the genome. L1 elements contain a 5′-untranslated region (UTR), two intact open reading frames (ORF1 and ORF2), as well as a 3′UTR that ends with a poly(A) tail (Figure 2). The ORF1 encodes an RNA-binding protein, whereas ORF2 encodes an endonuclease, a reverse transcriptase and a cysteine-rich domain of unknown function. The L1s contain → ← of both 1895- and 3056-bp spacers with one → ← on ORF2, stretching from 5′UTR to ORF1 and ORF2 (Figure 2).
Figure 2.
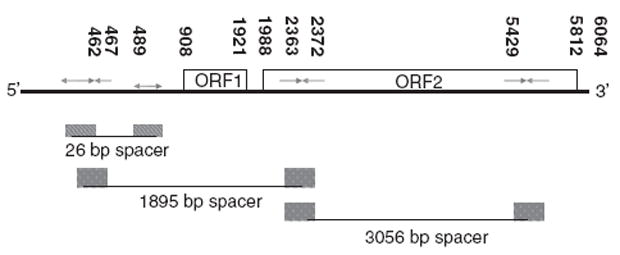
Genomic structure of L1 and its p53 responsive elements. Numbers in black represent gene coordinates in base pairs from the transcriptional start site.
For the p53 half-sites with ← → orientation and separated by 26 bp, we found that 26 bp spacers also occur within L1s and are found in 5′UTR, sharing a → with → ←1895 bp→ ← (Figure 2). The peak spacers of 15, 155, 156 and 157 bp between ← → occur mostly in Alu repeats.
It should be noted that another p53 responsive element, with the structure → ← (0-nt spacer) → ←, was recently discovered in repetitive DNA derived from certain human endogenous retroviruses (ERVs) (Wang et al., 2007).
p53 induces L1 transcription
To determine whether the presence of a p53 responsive element affects L1 transcription, the first 802 nt from the 5′-end of L1 were fused to luciferase, and this DNA was cotransfected into a p53-null cell line with DNA encoding either wild-type p53 or a point mutant of p53, which cannot bind to DNA. We had anticipated that p53 might downregulate the transcription of this promoter, given that L1 expression would have the potential to cause mutations in the genome through retroposition. Surprisingly, cotransfection of the L1 promoter fusion with wild-type p53 actually resulted in a three-fold induction of transcription from this L1 promoter when compared with cotransfection with a DNA-binding mutant form of p53, p53 (273H) (Figure 3a). A luciferase fusion to a known p53-responsive promoter, p21, was induced fivefold in the same experiment (not shown). Thus, the L1 promoter functions as a positively regulated p53 promoter, not as a negatively regulated p53 promoter.
Figure 3.
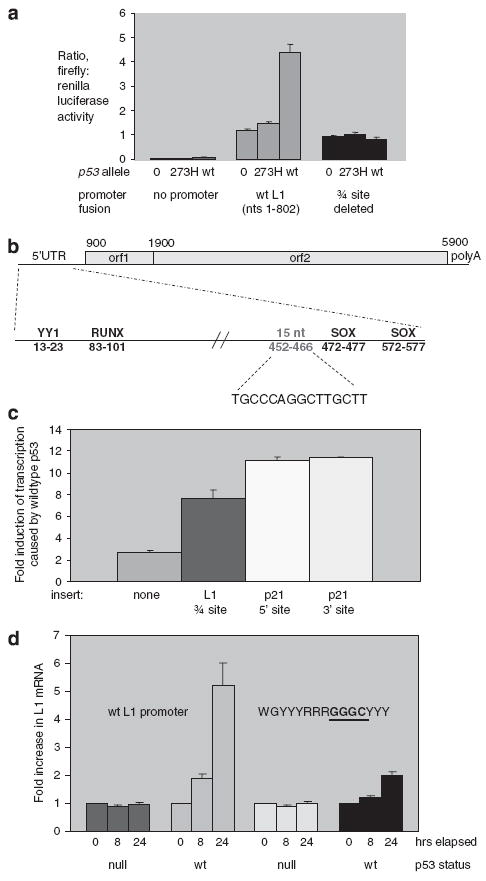
L1 (sense promoter) induction by p53, and mutational analysis. (a) Levels of transcription produced by an L1 (1–802)–firefly luciferase fusion plasmid upon cotransfection with plasmids encoding wild-type or mutant p53 were compared by luciferase assay. Twenty-four hours after cotransfection, cells were lysed and both firefly luciferase, which was expressed by the L1 promoter, and renilla luciferase, which acts as a transfection control, were assayed. Expression from L1–luciferase fusions containing mutations in the ← → ← or ← → sites were also tested. (b) Three-quarter site within the L1 promoter is shown, relative to binding site for the YY-1, RUNX and SOX transcription factor. (c) Effect of placing the three-quarter site from L1 promoter in front of a pdk4–luciferase promoter fusion. Response of the pdk4 promoter fusion to sequences added from p21 is included for comparison. (d) Levels of full-length L1 transcription produced by stable transfectants of HCT116, which has wild-type p53, in comparison with expression in HCT116 (p53–/–), which has both copies of p53 deleted. To activate p53, the cells were treated with 2 μM camptothecin, or mock-treated with dimethyl sulfoxide (DMSO) before harvesting RNA. RNA was collected after 8 and 24 h after camptothecin or DMSO treatments. The fold-increase in L1 mRNA upon camptothecin treatment is plotted on this graph. The plasmids contain either wild-type L1, or an L1 with a mutation in the site. The GGGG site is the mutant site
The L1–luciferase fusion that responded to p53 contains one of the sites identified by our informatics study: the two ← → half-sites, separated by 26 bp. Moreover, these sequences are very close to another sequence previously shown to cause response of the L1 promoter to SOX transcription factors (Tchenio et al., 2000), indicating that this region can participate in promoter activity. To test whether these sequences were responsible for the p53 response of the L1 promoter, we made point mutations in the promoter fusion plasmid and tested their effects. The first half-site was essential for p53 response (Table 2 and Figure 3a), whereas the second half-site was not (Table 2).
Table 2.
Effect of mutations on response to p53 by an L1–luciferase promoter fusion
| Three-quarter (5′) site | Fold response to p53 |
|---|---|
| 1. TGCCCAGGCTTGCTT (wild type) | 3.2±0.3 |
| 2. ΔΔΔΔΔΔΔΔΔΔΔΔΔΔ | 0.9±0.05 |
| 3. TTCCCAGGTTTTCTT | 0.9±0.2 |
| 4. TGCCCAGGGTTGCTT | 0.8±0.1 |
| 5. TTCCCAGGCTTGCTT | 1.0±0.2 |
| 6. TGCCCAGGCTTCTT | 0.9±0.05 |
| 7. gaactTGCCCAGGCT ΔΔΔΔΔ | 3.1±0.02 |
| 3′-site | Fold response to p53 |
|
| |
| 8. AGCTCGAACT (wild type) | 3.2±0.3 |
| 9. ΔΔΔΔΔΔΔΔΔΔΔΔΔΔ | 3.2±0.3 |
| 10. ATCTCGAATT | 3.2±0.3 |
Note: Δ indicates a deleted nucleotide, lower case letters are inserted nucleotides and bold underlined letters are point mutations.
Contiguous to the 5′-WGYYYRRRCW-3′ half-site sequence that drives p53 responsiveness of the L1 promoter is another quarter-site for p53 binding: 5′-RRRCW-3′ (Figure 3b). We denote this sequence as → ← →, which makes it a ‘three-quarter’ site. Three-quarter sites were recently found in other p53-responsive genes (Jordan et al., 2008), and we next determined whether all three of the quarter sites within this 15-nt sequence were important in L1 response to p53. Indeed, this seems to be the case. As shown in Table 2, replacing C nucleotides from each of the quarter sites individually blocked p53 response of the promoter. We wanted to test whether three-quarter sites function in either orientation; therefore, we replaced the 5′-WGYYYRRRCWWGYYY-3′ sequence with 5′-RRRCWWGYYYRRRCW-3′. As shown in Table 2, both orientations appeared to promote L1 transcription equivalently.
To show that the three-quarter site from L1 is sufficient for a p53 response, we placed this 15-nt sequence upstream of the promoter for the pyruvate dehydrogenase kinase-4 promoter in a pdk4–luciferase fusion plasmid. The pdk4 promoter is already responsive to p53 (Figure 3c), but the presence of the additional 15-nt three-quarter site from L1 greatly increases its activation by p53. For a comparison, we also inserted the two known p53 responsive elements from p21 into the pdk4–luciferase fusion plasmid, and observed a slightly higher increase in p53 response (Figure 3c).
Transient cotransfection experiments, such as those shown in Figures 3a and c, often overproduce proteins, and might not effectively mimic the effects of other important features of transcription, such as DNA methylation. To test for effects of p53 on the L1 promoter under more physiological conditions, we made a stable L1 transfectant in HCT116, a human colon cell line that has wild-type p53 alleles at the normal sites of the genome. We also made a stable transfectant of the HCT116 (p53–/–) cell line, which is isogenic to HCT116, except that both alleles of p53 have been knocked out genetically (Bunz et al., 1998). The transfectants contained a full-length L1 allele, with a sequence tag at its extreme 3′-end to allow its expression to be examined above the background of other L1 alleles expressed by these cell lines. This full-length L1 gene was not under the control of viral promoters, and instead was under the exclusive control of its endogenous promoter, and transcription termination signals immediately adjacent to the gene prevent adjacent sequences from influencing its expression. To activate p53, the transfected cell lines were treated for 24 h with the DNA-damaging reagent, camptothecin. In the cell line without p53, L1 mRNA was not induced by camptothecin (data not shown), whereas in the cell line with two copies of the p53 gene, L1 mRNA was induced fivefold (Figure 3d). The amount of induction of L1 is actually higher than that of GADD45a, which was induced 3.5-fold in the same experiment (data not shown). These data indicate that L1 expression can be induced by camptothecin in a p53-dependent manner.
p53 interacts with L1 in vitro
An electrophoretic mobility shift assay was carried out to determine whether the three-quarter site within the LINE-1 promoter could be bound by a purified p53 protein in vitro. We synthesized DNA corresponding to nt 427–523 from the 5′-end of L1. The ability of an L1 sequence to bind to p53 was compared with that of a sequence within the third intron of GADD45, a well-established, p53-responsive element (Hollander et al., 1993). The p53 responsive element from GADD45 has a more prototypical ‘four-quarter’-binding site, with no spacer between the two 10-mer half-sites. As seen in Figure 4, the presence of p53 causes the LINE-1 sequence to shift in apparent molecular weight, indicative of p53 binding. Specific binding is further shown by competition with non-radioactive self-probe and by a supershift on addition of a monoclonal antibody against p53. The apparent affinity of p53 for LINE-1 seems less than that of the GADD45 response element, yet these data show that p53 can bind to LINE-1 sequences.
Figure 4.
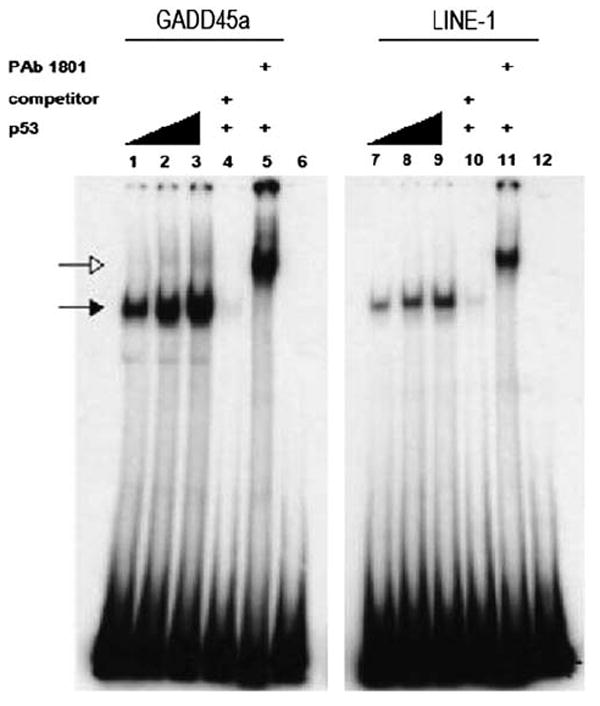
Electrophoretic mobility shift assay (EMSA) for p53 binding to L1 sequences. EMSA reaction mixtures containing 32P-labeled oligonucleotides with either the p53-binding sites from the GADD45 gene, or putative binding sites from L1. The LINE-1 oligonucleotide includes the ← → 26 bp ← → and the 15-nt three-quarter site (← → ←) found at the 5′-end of L1s (see Figure 2). Radiolabeled oligonucleotides were incubated with 40, 60 or 80 ng of bacterially expressed and purified His-tagged p53 protein (lanes 1–3 and 7–9)). Unlabeled DNA (50× molar excess) of the corresponding sequence was added as a specific competitor (lanes 4 and 10). The p53-specific antibody PAb1801 was added to supershift p53-DNA complexes (lanes 5 and 11). Closed arrows indicate p53–DNA complexes, whereas open arrows indicate p53-PAb 1801-DNA complexes.
p53 interacts with L1 promoters in a whole cell assay
We assayed p53 binding to L1s using chromatin immunoprecipitation. To make the assays robust, we focused on L1 elements that were immediately adjacent to unique, non-repetitive sequences. Using antisera specific for p53 on extracts of H1299/V138, a human lung cell line containing a temperature-inducible allele of p53 (Pochampally et al., 1999), we found that p53 co-immunoprecipitated the 5′-ends of the three L1s (Figures 5a–c). Other than the 5′-p53REs present within these L1s, there are no other p53-binding sequences within 500 nt. The binding of p53 to L1 elements may depend on the local chromatin structure. For instance, p53 did not appear to bind to another L1 element, as shown in Figure 5d. As a positive control, we showed that under the same conditions, p53 immunoprecipitated a segment of the p21 gene, which harbors p53REs in the promoter region (Figure 5e). A control antibody raised against green fluorescent protein (GFP) did not immunoprecipitate the same sequences (Figure 5). Furthermore, the p53 sera did not immunoprecipitate the positive control p21 sequence in the p53-null cell line H1299, nor did they immunoprecipitate negative control sequences that were known to contain no p53REs (Figures 5f and g).
Figure 5.
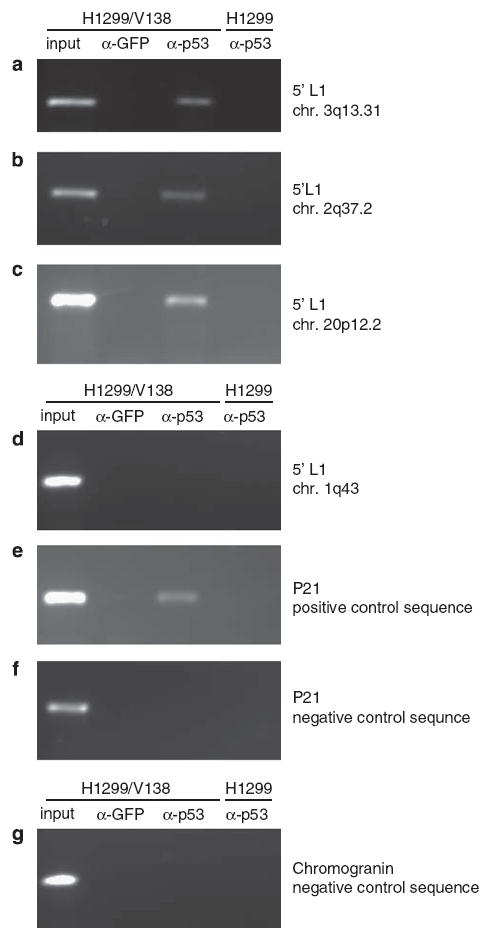
Binding of p53 to L1 sequences in whole cells. Cells from the H1299/V138 cancer cell line were incubated at 32 °C for 7 h to induce p53 activity then briefly treated with formaldehyde to crosslink chromatin to DNA-binding proteins. The isogenic H1299 cell line does not express p53 and is included as a negative control. A chromatin immunoprecipitation reaction using p53 or GFP antisera was then performed as described in the ‘Materials and methods’ section. The immunoprecipitated DNA was probed for specific L1 sequences, as well as for a p21waf1/cip1 positive control sequence and negative control sequences using PCR. Accession numbers for each are given in parentheses. (a) 5′-sequence from L1 at chromosomal position 3q13.31 (AC046136); (b) 5′-sequence from L1 at chromosomal position 2q37.2 (AC010148); (c) 5′-sequence from L1 at chromosomal position 20p12.2 (AL078623); (d) 5′-sequence from L1 at chromosomal position 1q43 (AC015901); (e) p21waf1/cip1 sequence containing a known p53 RE; (f) p21waf1/cip1 containing no p53 RE; (g) chromogranin A sequence containing no p53 RE.
Three-quarter sites in L1s are a recent acquisition
To determine the age of the p53-binding sites in L1, we compared consensus sequences of the six youngest L1 subfamilies (Khan et al., 2006), dating back to ~40 million years ago, to determine the presence or absence of p53REs. As shown in Figure 6, the upstream p53-binding site located in 5′UTR did not appear until ~20 million years ago, with the emergence of L1PA3. It resulted from several mutations: a two-base transversion of AG→CT to create the → ← half-site, and another G→C transversion to create the ← → half-site. However, 16 of the other 17 nt within these two half-sites, as well as the ← → half-site closer to the 3′-portion of this element, were present as earlier as L1PA6. To confirm the emergence of the upstream p53-binding site sequence before speciation of the African primates, we identified its presence in multiple full-length chimpanzee L1s available in GenBank (data not shown).
Figure 6.
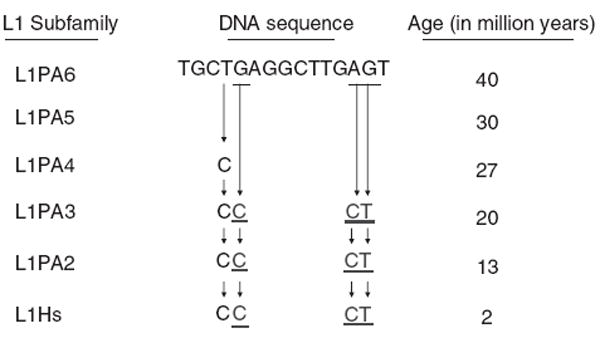
Lineage of p53 target DNA sites within the primate L1s. Consensus sequences in the region spanning the L1 site are shown for several L1 subfamilies (Khan et al., 2006). Three point mutations occurred in the L1PA3 subfamily to create the L1 site.
Discussion
We have previously designed and reported a computational method to identify p53 responsive elements of the structure → ← spacer → ←, with 0–14-nt spacers between the palindromic sequences, in both human and mouse genomes. This algorithm, p53MH (Hoh et al., 2002), showed ~4500 sequences, and identified several novel p53 responsive genes. To extend that analysis, we examined 2 kinds of 10-mer palindromes and found 6 spacers of 0, 14, 24, 129, 1895 3056 bp between → ← p53RE half-sites and 15, 26, 46, 155, 156 and 157 bp between ← → occurring with significantly high frequencies in the human genome. Nearly all of the spacer peaks appear because of the presence of half-sites within repetitive elements. For many of these sequences within repetitive elements, we detected no response to p53. For instance, the more 3′-half-site of the ← → pair separated by 26 bp had no effect on the p53 response of the L1 promoter (Table 2).
However, as we tested repetitive element promoters for response to p53, we discovered a transcriptional response by L1 that derived not from one of the pairs of widely spaced half-sites, but from a three-quarter site that fell within that pair of half-sites. Among the L1 subclasses, only the most recent L1PA1 subclass is currently active for retroposition. Three-quarter sites for p53 binding are thus part of potentially active L1 elements (so called ‘hot’ L1s) that belong to the youngest L1 retrotransposon family (Brouha et al., 2003). An oligonucleotide containing promoter sequence from L1, mimicking those in the full-length ‘hot’ L1s was constructed. Using a gel shift assay, the p53 protein was found to have a high affinity to this L1 sequence (Figure 4). p53-L1 binding was further confirmed in whole cells by chromatin immunoprecipitation (Figure 5). To explore the functional significance of these retroposon-associated three-quarter sites, the L1 transcript levels both in promoter fusions and in transcripts from the full-length L1 gene was measured. The L1 mRNA expression was elevated as much as fivefold on activation of p53 (Figure 3). Mutations that destroyed the three-quarter site reduced p53-dependent induction of transcription (Figure 3).
It was surprising that p53 would increase the transcription of L1 elements, because this might suggest that p53 increases the spread of these retroposons, whereas we generally consider p53 as a protein that prevents genomic changes. However, when considering ‘selfish’ DNA elements such as L1s, one should consider whether a given property is advantageous to the DNA element, to its host or to both. It seems important to note that most selfish elements prefer to function within the cells of the germ line, and that L1s appear to be no exception as their expression and movement is mainly detected within the germline tissue (Ostertag et al., 2002).
On the basis of the results presented in this study, we propose a model by which p53 might help to limit transposition in somatic cells but not in the germ line. In this model, p53 would have no effect on transposition in the germ line, as p53 fails to be functional in the germ line so as to allow recombination intermediates to occur without stimulating apoptotic responses. Similarly, intermediates in the retroposition of L1 could be produced at this time, without the risk of activating DNA damage response pathways. However, p53 is functional in somatic cells, and responds strongly in these cells to the presence of double-stranded breaks in the chromatin, such as those that would be created by endonuclease activity of the L1-ORF2 protein (ORF2p). As we have shown, p53 activates L1 transcription, which in turn would create even more ORF2p, more doublestranded breaks and more p53 activity. Thus, a positive feedback loop would be created, which would amplify not only the amount of DNA damage but would also amplify the p53-mediated DNA damage response. Eventually, sufficient DNA damage would be produced to result in apoptosis, which would destroy the cell. Such a mechanism would reduce L1 transposition in somatic cells.
Other mechanisms of L1 regulation, most notably DNA methylation, are important for the germ line-specific transposition of L1s (Woodcock et al., 1997). Methylation of cytosines within somatic cells induces a closed chromatin structure, upon which transcription factors such as p53 cannot access the DNA (Lorincz et al., 2004). Cytosine methylation can be lost at discrete sites within somatic cells in tumors, and if a given L1 becomes hypomethylated, transcribed and translated, p53 could provide a method by which to limit the DNA damage caused by such a ‘rogue’ L1 element. Interestingly, an enzyme required to re-establish cytosine methylation, DNMT1, can bind directly to p53 (Esteve et al., 2005). If double-stranded breaks created by L1s are increased by a small amount because of loss of methylation, it is possible that re-methylation of the responsible rogue L1 could occur with great specificity through the recruitment of p53, and co-recruitment of DNMT1, to p53REs. However, if levels of doublestranded breaks become too high, p53 levels would increase, more L1 expression and DNA damage would result, and apoptosis would ensue. Differing responses of repair or apoptosis, depending on the degree of DNA damage, is a theme that occurs many times within p53 biology (Jin and Levine, 2001; Bensaad and Vousden, 2005).
The ability of three-quarter sites to function in L1s and in other genes raises interesting questions about p53 biology. It has been shown previously that a p53 responsive element within another class of repetitive elements, derived from ERVs, can affect p53 response of the nearby genes (Wang et al., 2007). As these ERVderived sequences are ~40 million years old and are no longer transposing, the ERV-derived responses to p53 should be common for all humans. Conversely, L1 retroposition continues to take place. Although the rate of L1 retroposition is quite low, many new L1s have nevertheless been created over the past several 1000 years and could introduce diversity in p53 responses between humans, including p53-influenced processes, such as tumor formation, longevity and fertility (Donehower, 2005; Hu et al., 2007).
Materials and methods
The genome-wide results of p53 responsive elements are available at the website http://linkage.rockefeller.edu/p53-genome.
Electromobility shift assays
Bacterially expressed 6xHis-p53 protein was purified by Ni-NTA-agarose chromatography. After 30 min of incubation with a DNA probe at room temperature, mixtures were subjected to electrophoresis on a 4% polyacrylamide gel (29:1 acrylamide/bisacrylamide) containing 0.5× Tris-borate-EDTA (ethylenediaminetetraacetic acid) buffer at 165V for 1.25 h at room temperature. The gel was dried and exposed to the Kodax Biomax MR film (Kodak, Rochester, NY, USA). Sequences of DNA probes were GADD45: 5′-GTAGCTGA TATCGAATTCTCGAGCAGAACATGTCTAAGCATGCTGGGCTCGAGAATTCCTGCAGCC-3′ and LINE-1: 5′-GAGGCTGGGGGAGGGGCGCCCGCCATTGCCCAGGCTTGCTTAGGTAAACAAAGCAGCCGGGAAGCTCGAACTGGGTGGAGCCCACCACAGCTCAAGG-3′.
Plasmids
Plasmids encoding wild-type human p53 under the control of the CMV promoter, or the mutant p53-273H, were a gift from Dr Z Feng (University of Medicine and Dentistry of New Jersey). The 848-pdk4 plasmid, encoding a pdk4–luciferase promoter fusion, was provided by Dr R Harris (Indiana University School of Medicine) and has been described previously (Kwon et al., 2006). The plasmid 99PUR-RPS-eGFP was a gift from H Kazazian (University of Pennsylvania) and has been described previously (Ostertag et al., 2000). The CMV promoter was removed from the 99PUR-RPS-eGFP plasmid by eliminating the small BstBI fragment. To generate an L1–luciferase fusion plasmid, the first 802 nt of the L1 promoter were amplified from 99PUR-RPS-eGFP by PCR, using 5′-CCCCACGCGTGCTCCGGTCTACAGCTCCCAGCGTGAG-3′ and 5′-CCCCAAGCTTCTGGTTGTTAGTTTTCCTTCTAAC-3′ as primers. The PCR fragment was digested with MluI and HindIII, then ligated into the same sites of the plasmid pGL3-Basic (Promega, San Luis Obispo, CA, USA).
Point mutations were introduced into plasmids using a Quikchange II XL Kit (Stratagene, Torrey Pines, CA, USA). Sequences of mutagenic oligonucleotides are provided in Supplementary Table S1.
Gene expression assays
To assay an L1–luciferase fusion gene, cotransfection of HCT116 (p53–/–) cells was performed using the L1-luciferase plasmid along with plasmids encoding either wild-type p53 or p53-273H. The plasmid pRL-SV40 (Promega) was also used to normalize the transfection levels between experiments. A total of 4μg of DNA was combined with 10μ of Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). The ratio of p53 plasmid to L1-luciferase plasmid to pRL-SV40 (Promega) was 5:1:0.01. Twenty-four hours after cotransfection, the cells were washed, lysed and assayed using a Dual Luciferase Reporter Kit (Promega) as recommended by the manufacturer.
To assay the full-length L1 gene, isogenic cell lines HCT116, which has two wild-type copies of p53, and HCT116 (p53–/–) (Bunz et al., 1998), which has both copies deleted, were transfected with a plasmid containing full-length L1 gene with a 3′-sequence tag, or with the plasmid containing a mutant site. Transfections were performed using Lipofectamine 2000 (Invitrogen) as recommended by the manufacturer. Transfectants were selected for puromycin resistance pooled, and passed at least four times, but not more than ten times. Transfectants were exposed to 2 μM camptothecin (Sigma, St Louis, MO, USA) for up to 24 h, or mock-treated with dimethyl sulfoxide. RNA was prepared using an RNeasy kit (Qiagen, Valencia, CA, USA), and converted into cDNA using reverse transcriptase (Applied Biosystems, Foster City, CA, USA). cDNA quantitation was performed by real-time PCR using SYBR green reagent and a Prism 7500 instrument (Applied Biosystems). Primers used to assay the L1 mRNA were 5′-GGGGGTGTTCTGCTGGTAGTGGTC-3′ and 5′-AAGCAGAAGAACGGCATCAAGGTG-3′, which are specific to the sequence-tagged mRNA produced by the plasmid.
Chromatin immunoprecipitation assays
H1299/V138 and H1299 human lung cancer cells were sub-cultured and grown at 39 °C for 16 h until 50% confluent, then shifted to 32°C for 7 h to induce the temperature-sensitive allele of p53 within V138. Formaldehyde was added for 10 min at 32 °C, and chromatin immunoprecipitation was then performed with a kit purchased from Upstate Cell Signaling Solutions (New York, NY, USA), using the procedure recommended by the manufacturer. Chromatin was sonicated thrice for a total of 45 s using a model 50 Fisher Scientific Sonic Dismembrator at 30% power. Chromatin–protein complexes were immunoprecipitated using the DO-1 monoclonal antiserum raised against p53 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Monoclonal antisera raised against GFP (Santa Cruz Biotechnology) were used as a negative control. PCR of sequences from the p21waf1/cip1 were used as both positive and negative controls. The chromogranin A gene also served as a negative control. p53REs within L1 sequences throughout the genome were used, including 1q43 (AL015901), 2q37.2 (AC010148), 20p12.2 (AL078623; two p53REs were used, one at the 5′-prime end and the other at the 3′-end), 3q13.31 (AC046136) and 4q28.3 (AC018680). All PCR primer sequences can be found in Supplementary Table S2. For each of the L1-specific PCRs, one of the oligonucleotides anneals to a sequence internal to the L1, which is present in many copies within the genome, and the other oligonucleotide anneals to a site within unique, flanking DNA that is specific to a particular L1. PCRs were carried out for 34 cycles.
Supplementary Material
Acknowledgments
This work was primarily supported by the Verto Institute, with additional support provided by NIH Grant nos K25HG00060, R01EY015771, R01MH44292 and P01CA87497.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
Conflict of interest The authors declare no conflict of interest.
References
- Babushok DV, Kazazian HH., Jr Progress in understanding the biology of the human mutagen LINE-1. Hum Mutat. 2007;28:527–539. doi: 10.1002/humu.20486. [DOI] [PubMed] [Google Scholar]
- Bensaad K, Vousden KH. Savior and slayer: the two faces of p53. Nat Med. 2005;11:1278–1279. doi: 10.1038/nm1205-1278. [DOI] [PubMed] [Google Scholar]
- Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, et al. Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci USA. 2003;100:5280–5285. doi: 10.1073/pnas.0831042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budhram-Mahadeo V, Morris PJ, Smith MD, Midgley CA, Boxer LM, Latchman DS. p53 suppresses the activation of the Bcl-2 promoter by the Brn-3a POU family transcription factor. J Biol Chem. 1999;274:15237–15244. doi: 10.1074/jbc.274.21.15237. [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- Donehower LA. p53: guardian AND suppressor of longevity? Exp Gerontol. 2005;40:7–9. doi: 10.1016/j.exger.2004.10.007. [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- Esteve PO, Chin HG, Pradhan S. Human maintenance DNA (cytosine-5)-methyltransferase and p53 modulate expression of p53-repressed promoters. Proc Natl Acad Sci USA. 2005;102:1000–1005. doi: 10.1073/pnas.0407729102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk WD, Pak DT, Karas RH, Wright WE, Shay JW. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol Cell Biol. 1992;12:2866–2871. doi: 10.1128/mcb.12.6.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Szak ST, Boeke JD. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature. 2004;429:268–274. doi: 10.1038/nature02536. [DOI] [PubMed] [Google Scholar]
- Haoudi A, Semmes OJ, Mason JM, Cannon RE. Retrotransposition-competent human LINE-1 induces apoptosis in cancer cells with intact p53. J Biomed Biotechnol. 2004;2004:185–194. doi: 10.1155/S1110724304403131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- Hoh J, Jin S, Parrado T, Edington J, Levine AJ, Ott J. The p53MH algorithm and its application in detecting p53-responsive genes. Proc Natl Acad Sci USA. 2002;99:8467–8472. doi: 10.1073/pnas.132268899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander MC, Alamo I, Jackman J, Wang MG, McBride OW, Fornace AJ., Jr Analysis of the mammalian gadd45 gene and its response to DNA damage. J Biol Chem. 1993;268:24385–24393. [PubMed] [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- Hu W, Feng Z, Teresky AK, Levine AJ. p53 regulates maternal reproduction through LIF. Nature. 2007;450:721–724. doi: 10.1038/nature05993. [DOI] [PubMed] [Google Scholar]
- Jin S, Levine AJ. The p53 functional circuit. J Cell Sci. 2001;114:4139–4140. doi: 10.1242/jcs.114.23.4139. [DOI] [PubMed] [Google Scholar]
- Jordan JJ, Menendez D, Inga A, Nourredine M, Bell D, Resnick MA. Noncanonical DNA motifs as transactivation targets by wild type and mutant p53. PLoS Genet. 2008;4:e1000104. doi: 10.1371/journal.pgen.1000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–420. doi: 10.1016/s0168-9525(00)02093-x. [DOI] [PubMed] [Google Scholar]
- Kannan K, Kaminski N, Rechavi G, Jakob-Hirsch J, Amariglio N, Givol D. DNA microarray analysis of genes involved in p53 mediated apoptosis: activation of Apaf-1. Oncogene. 2001;20:3449–3455. doi: 10.1038/sj.onc.1204446. [DOI] [PubMed] [Google Scholar]
- Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, et al. Identification of p53 as a sequence-specific DNA-binding protein. Science. 1991;252:1708–1711. doi: 10.1126/science.2047879. [DOI] [PubMed] [Google Scholar]
- Khan H, Smit A, Boissinot S. Molecular evolution and tempo of amplification of human LINE-1 retrotransposons since the origin of primates. Genome Res. 2006;16:78–87. doi: 10.1101/gr.4001406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HS, Huang B, Ho Jeoung N, Wu P, Steussy CN, Harris RA. Retinoic acids and trichostatin A (TSA), a histone deacetylase inhibitor, induce human pyruvate dehydrogenase kinase 4 (PDK4) gene expression. Biochim Biophys Acta. 2006;1759:141–151. doi: 10.1016/j.bbaexp.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006;13:941–950. doi: 10.1038/sj.cdd.4401925. [DOI] [PubMed] [Google Scholar]
- Lee KC, Crowe AJ, Barton MC. p53-mediated repression of alpha-fetoprotein gene expression by specific DNA binding. Mol Cell Biol. 1999;19:1279–1288. doi: 10.1128/mcb.19.2.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol. 2004;11:1068–1075. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- Maltzman W, Czyzyk L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol Cell Biol. 1984;4:1689–1694. doi: 10.1128/mcb.4.9.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori A, Zauberman A, Doitsh G, Paran N, Oren M, Shaul Y. p53 binds and represses the HBV enhancer: an adjacent enhancer element can reverse the transcription effect of p53. EMBO J. 1998;17:544–553. doi: 10.1093/emboj/17.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, DeBerardinis RJ, Goodier JL, Zhang Y, Yang N, Gerton GL, et al. A mouse model of human L1 retrotransposition. Nat Genet. 2002;32:655–660. doi: 10.1038/ng1022. [DOI] [PubMed] [Google Scholar]
- Ostertag EM, Prak ET, DeBerardinis RJ, Moran JV, Kazazian HH., Jr Determination of L1 retrotransposition kinetics in cultured cells. Nucleic Acids Res. 2000;28:1418–1423. doi: 10.1093/nar/28.6.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochampally R, Fodera B, Chen L, Lu W, Chen J. Activation of an MDM2-specific caspase by p53 in the absence of apoptosis. J Biol Chem. 1999;274:15271–15277. doi: 10.1074/jbc.274.21.15271. [DOI] [PubMed] [Google Scholar]
- Resnick-Silverman L, St Clair S, Maurer M, Zhao K, Manfredi JJ. Identification of a novel class of genomic DNA-binding sites suggests a mechanism for selectivity in target gene activation by the tumor suppressor protein p53. Genes Dev. 1998;12:2102–2107. doi: 10.1101/gad.12.14.2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchenio T, Casella JF, Heidmann T. Members of the SRY family regulate the human LINE retrotransposons. Nucleic Acids Res. 2000;28:411–415. doi: 10.1093/nar/28.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Zeng J, Lowe CB, Sellers RG, Salama SR, Yang M, et al. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc Natl Acad Sci USA. 2007;104:18613–18618. doi: 10.1073/pnas.0703637104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock DM, Lawler CB, Linsenmeyer ME, Doherty JP, Warren WD. Asymmetric methylation in the hypermethylated CpG promoter region of the human L1 retrotransposon. J Biol Chem. 1997;272:7810–7816. doi: 10.1074/jbc.272.12.7810. [DOI] [PubMed] [Google Scholar]
- Zhao R, Gish K, Murphy M, Yin Y, Notterman D, Hoffman WH, et al. Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes Dev. 2000;14:981–993. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.