

Abstract
To understand the process of cardiac aging, it is of crucial importance to gain insight into the age-related changes in gene expression in the senescent failing heart. Age-related cardiac remodeling is known to be accompanied by changes in extracellular matrix (ECM) gene and protein levels. Small noncoding microRNAs regulate gene expression in cardiac development and disease and have been implicated in the aging process and in the regulation of ECM proteins. However, their role in age-related cardiac remodeling and heart failure is unknown. In this study, we investigated the aging-associated microRNA cluster 17–92, which targets the ECM proteins connective tissue growth factor (CTGF) and thrombospondin-1 (TSP-1). We employed aged mice with a failure-resistant (C57Bl6) and failure-prone (C57Bl6 × 129Sv) genetic background and extrapolated our findings to human age-associated heart failure. In aging-associated heart failure, we linked an aging-induced increase in the ECM proteins CTGF and TSP-1 to a decreased expression of their targeting microRNAs 18a, 19a, and 19b, all members of the miR-17–92 cluster. Failure-resistant mice showed an opposite expression pattern for both the ECM proteins and the microRNAs. We showed that these expression changes are specific for cardiomyocytes and are absent in cardiac fibroblasts. In cardiomyocytes, modulation of miR-18/19 changes the levels of ECM proteins CTGF and TSP-1 and collagens type 1 and 3. Together, our data support a role for cardiomyocyte-derived miR-18/19 during cardiac aging, in the fine-tuning of cardiac ECM protein levels. During aging, decreased miR-18/19 and increased CTGF and TSP-1 levels identify the failure-prone heart.
Keywords: cardiac aging, connective tissue growth factor, matricellular proteins, microRNA, miR-18, miR-19, thrombospondin-1
Introduction
Aging is considered a multifactorial process that is controlled by genetic and environmental factors and ultimately leads to deterioration of body and organ functions. Heart failure (HF) is a typical age-associated disease and is characterized by unique physiological and morphological changes in aged myocardium [reviewed in (Lakatta & Sollott, 2002)]. Although the rate and mechanism through which an animal or tissue ages differ among species, the constant remodeling and accumulation of the extracellular matrix (ECM) are recognized as a key feature of cardiac aging in humans, mice, and rats and contributes to the structural changes that lead to HF with advancing age (Capasso et al., 1990; Barasch et al., 2009; Boyle et al., 2011). In particular, the expression of the ECM proteins SPARC (Bradshaw et al., 2010), fibronectin (Burgess et al., 2001), thrombospondin-2 (TSP-2) (Swinnen et al., 2009), and connective tissue growth factor (CTGF) (Wang et al., 2010) increases with aging. Both TSP-2 knockout mice and cardiomyocyte-specific CTGF transgenic animals develop spontaneous age-related cardiomyopathy (Panek et al., 2009; Swinnen et al., 2009), confirming a role of ECM proteins in age-related cardiac remodeling. However, the underlying mechanisms that drive the age-related gene expression of ECM molecules remain elusive.
The identification of small noncoding microRNAs (miRNAs) opened new doors for investigating the regulation of gene expression by adding another layer of control at the post-transcriptional level. MiRNAs are approximately 22 nucleotides long RNA molecules that can target mRNAs for translational repression or degradation by complementary binding to specific sequences in the protein-coding gene transcript. The first implication of miRNAs in aging was provided by a study that showed that lin-4 and its target protein lin-14 determine longevity in C. elegans by affecting the insulin-like signaling pathway (Boehm & Slack, 2005). Now, miRNAs have emerged as important mediators of diverse age-associated pathologies, ranging from cancer and diabetes to neurodegenerative disorders (Grillari & Grillari-Voglauer, 2010), and increasing evidence demonstrates altered miRNA expression profiles in aging muscle, brain, lung, and liver [reviewed in (Chen et al., 2010)]. In addition, miRNAs are regulated during cellular senescence, and complete loss of miRNA function caused premature senescence in embryonic fibroblasts (Mudhasani et al., 2008), putting miRNAs in the forefront of cellular senescence in different organs and cell types (Chen et al., 2010; Grillari and Grillari-Voglauer, 2010). Their role in the heart was first suggested by expression profiling studies showing changes in the expression of specific miRNAs in failing human hearts (van Rooij et al., 2006; Thum et al., 2007). Further animal models proved the involvement of specific miRNAs in cardiac development, function, and under pathological conditions (van Rooij et al., 2007; Zhao et al., 2007; da Costa Martins et al., 2010). Nevertheless, despite their proven importance in aging, cardiac disease, and development, a role for miRNAs during cardiac aging and age-related HF remains to be elucidated.
This study investigated the role of the miR-17–92 cluster in the aged heart, in view of its central role in regulation of matrix genes and in cellular aging (Dews et al., 2006; Suarez et al., 2008; Shan et al., 2009). This cluster encodes six miRNAs (miR-17, miR-18a, miR-19a, miR-19b, miR-20a, and miR-92a-1) that are located within an 800-base pair region of human chromosome 13. Originally, the miR-17–92 cluster was linked to tumor genesis, and transcription of the cluster was found to be directly activated by the proto-oncogene c-Myc (He et al., 2005) [reviewed in(van Haaften & Agami, 2010)]. The pro-oncogenic activity of miR-17–92 partially involves the regulation of the ECM proteins CTGF and thrombospondin-1 (TSP-1) by the cluster members miR-18 and miR-19, through sequence-specific targeting within the 3′-untranslated region (3′-UTR) of these gene transcripts (Supporting information Fig. S1) (Dews et al., 2006). Interestingly, cardiogenesis was severely hampered in mice deficient for miR-17–92, suggesting an important role for this cluster in cardiac development (Ventura et al., 2008). This, together with miR-18 and miR-19 targeting CTGF and TSP-1 and the fact that ECM proteins are crucial for healthy cardiac aging, has led us to hypothesize that these miRNAs play a role in age-related cardiac remodeling. Therefore, we investigated whether age-related changes in miR-18a, miR-19a, and miR-19b expression regulate CTGF, TSP-1, and collagen levels in rodent models of aging-associated heart failure and in the human failing heart.
Results
HF-prone mice develop cardiac dilation and dysfunction at old age
As a model for age-related HF in rodents, we examined age-associated changes in cardiac function in two genetically different mouse strains, i.e. C57Bl6 (HF resistant) and C57Bl6 × 129Sv (HF prone). Both strains developed normally, and echocardiographic analysis showed no differences in cardiac function at 12 and 52 weeks of age. However, fractional shortening was significantly compromised in 104-week-old HF-prone mice compared to age-matched HF-resistant hearts (Table 1). Indices of left ventricular wall thickness, LVPW and IVS, indicated progressive thinning of the LV wall with aging in HF-prone mice, followed by ventricular dilation at old age (Table 1). HF-resistant mice on the other hand had no ventricular dilation and showed a tendency to increased wall thickness with aging, resulting in a preserved cardiac function at old age (Table 1). Age-associated cardiac dysfunction in HF-prone mice was further shown by significantly higher heart, kidney, and lung weights at old age (Table 1). Thus, poor cardiac aging in HF-prone mice was characterized by a dilated cardiomyopathy-like phenotype as is seen in human HF.
Table 1.
Echocardiographic and morphometric analysis of male mice at different ages
| HF resistant | HF prone | |||||
|---|---|---|---|---|---|---|
| 12 weeks n = 8 | 52 weeks n = 8 | 104 weeks n = 9 | 12 weeks n = 9 | 52 weeks n = 7 | 104 weeks n = 8 | |
| FS (%) | 28 ± 1.5 | 29 ± 1.6 | 29 ± 1.7 | 28 ± 2.0 | 29 ± 3.5 | 20 ± 2.2*†‡ |
| LVIDd (mm) | 4.0 ± 0.1 | 4.2 ± 0.1 | 4.2 ± 0.1 | 3.8 ± 0.2 | 3.7 ± 0.2 | 4.6 ± 0.2*† |
| LVIDs (mm) | 2.9 ± 0.1 | 3.0 ± 0.1 | 3.0 ± 0.1 | 2.7 ± 0.1 | 2.7 ± 0.2 | 3.7 ± 0.3*†‡ |
| LVPWd (mm) | 0.75 ± 0.03 | 0.84 ± 0.03* | 0.90 ± 0.03* | 0.99 ± 0.11 | 0.91 ± 0.05 | 0.88 ± 0.06 |
| LVPWs (mm) | 1.16 ± 0.06 | 1.18 ± 0.06 | 1.27 ± 0.04 | 1.40 ± 0.08‡ | 1.22 ± 0.04 | 1.02 ± 0.07*†‡ |
| IVSd (mm) | 0.72 ± 0.01 | 0.81 ± 0.03* | 0.81 ± 0.04* | 0.96 ± 0.15 | 0.91 ± 0.05 | 0.83 ± 0.03 |
| IVSs (mm) | 1.03 ± 0.01 | 1.22 ± 0.04* | 1.18 ± 0.07 | 1.41 ± 0.18 | 1.37 ± 0.09 | 1.06 ± 0.06† |
| Heart rate (bpm) | 482 ± 14 | 473 ± 11 | 493 ± 8 | 506 ± 15 | 521 ± 9‡ | 502 ± 25 |
| Body weight (g) | 25.0 ± 0.3 | 30.8 ± 0.5* | 33.2 ± 0.8*† | 23.5 ± 1.1 | 29.8 ± 0.7* | 29.8 ± 0.7*‡ |
| HW/BW ratio (mg g−1) | 4.8 ± 0.1 | 4.8 ± 0.2 | 4.4 ± 0.1* | 5.4 ± 0.2‡ | 5.5 ± 0.1‡ | 6.0 ± 0.3‡ |
| LuW/BW ratio (mg g−1) | 5.7 ± 0.3 | 5.7 ± 0.2 | 6.2 ± 0.2 | 5.9 ± 0.3 | 5.9 ± 0.3 | 7.5 ± 1.0 |
| LiW/BW ratio (mg g−1) | 48.0 ± 0.7 | 47.7 ± 1.1 | 48.1 ± 0.9 | 47.0 ± 2.8 | 46.6 ± 1.6 | 53.7 ± 3.8 |
| KiW/BW ratio(mg g−1) | 8.0 ± 0.2 | 8.7 ± 0.3 | 8.7 ± 0.2 | 8.8 ± 0.4 | 11.9 ± 0.3*‡ | 11.3 ± 0.4*‡ |
FS, fractional shortening; HF, Heart failure; LVIDd and LVIDs, left ventricular internal diameter in diastole and systole; LVPWd and LVPWs, left ventricular posterior wall thickness in diastole and systole; IVSd and IVSs, intraventricular septum thickness in diastole and systole; HW, heart weight; BW, body weight; LuW, lung weight; LiW, liver weight; KiW, kidney weight.
P≤0.05 vs 12 weeks of age with the same genotype.
P≤0.05 vs 52 weeks of age with the same genotype.
P≤0.05 vs HF-resistant mice with the same age.
HF-prone hearts have more interstitial fibrosis and increased levels of CTGF and TSP-1
Dilated cardiomyopathy is characterized by increased deposition of interstitial collagen, stiffening the heart, and compromising its contractility (Kemi et al., 2000; Burgess et al., 2001). Histological examination of cardiac sections showed significant interstitial fibrosis in HF-prone mice as compared to 52-week littermates and to 104-week-old HF-resistant hearts (Fig. 1A, B). On the other hand, the mild increase in collagen in aged HF-resistant mice was not significant (Fig. 1A, B), while accumulation of interstitial collagen was similar in 12- and 52-week-old HF-resistant and HF-prone mice. As expected, perivascular fibrosis increased with aging, but was not different between HF-prone and HF-resistant hearts (Fig. 1A, B).
Fig. 1.
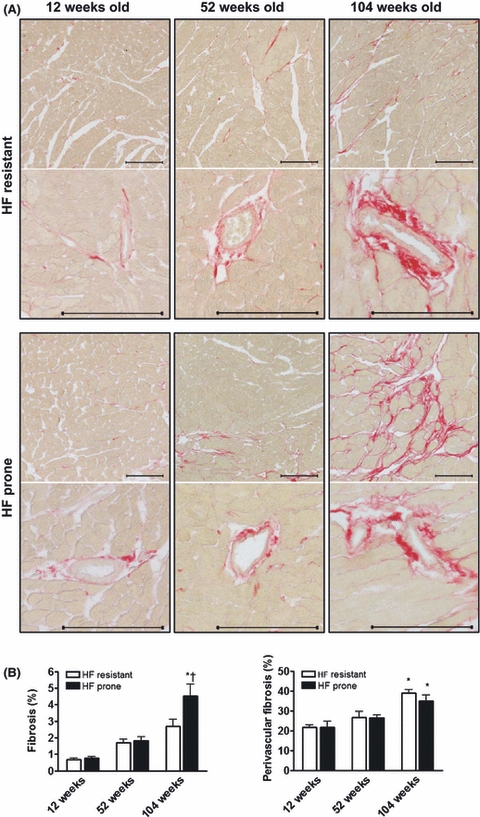
Enhanced left ventricular interstitial fibrosis in old heart failure (HF) prone mice. (A) Histological analysis of the hearts of HF-resistant and HF-prone mice by Sirius Red staining. Photographs show representative areas of interstitial fibrosis (upper panel) and collagen deposition in the perivascular area (lower panel). Scale bars represent 100 μm. (B) Quantitative analysis of the interstitial and perivascular collagen content in HF-resistant (12 weeks, n = 8; 52 weeks, n = 8; and 104 weeks, n = 9) and HF-prone mice (12 weeks, n = 6; 52 weeks, n = 11; and 104 weeks, n = 9) revealed increased interstitial fibrosis in the left ventricles of 104-week-old HF-prone mice. Perivascular fibrosis was significantly increased in 104-week-old hearts, but was not different between HF-resistant and HF-prone mice. Data are presented as mean ± SEM. *P≤0.05 vs 52-week-old HF-prone mice; †P≤0.05 vs 104-week-old HF-resistant mice.
The ECM proteins CTGF and TSP-1 are recognized as powerful regulators of ECM remodeling and are mediators of tissue fibrosis in humans, mice, and rats (Belmadani et al., 2007; Shi-Wen et al., 2008). We found significantly increased cardiac transcript and protein levels of CTGF and TSP-1 in 104-week-old HF-prone mice (Fig. 2A, B; Supporting information Fig. S2). Vice versa, the hearts of aged HF-resistant mice had no induction of CTGF and TSP-1 transcript or protein levels (Fig. 2A, B). Therefore, increased interstitial fibrosis and CTGF and TSP-1 expression characterize the HF-prone aged heart.
Fig. 2.

Opposite expression profiles in heart failure (HF)-resistant versus HF-prone mice. CTGF, TSP-1, miR-18a, miR-19a, and miR-19b levels in aged HF-resistant (12 weeks, n = 8; 52 weeks, n = 8; and 104 weeks, n = 9) and HF-prone mice (12 weeks, n = 6; 52 weeks, n = 11; and 104 weeks, n = 9). (A and B) Immunoblotting was performed on four mice per age-group and revealed significant induction of CTGF and TSP-1 protein expression in failing hearts of 104-week-old HF-prone mice, whereas CTGF and TSP-1 levels were reduced in old HF-resistant mice. Immunoblots show representative protein bands of TSP-1, CTGF, and GAPDH. (C) RT-PCR analysis showed increased expression of miR-18a, miR-19a, and miR-19b in 104-week-old HF-resistant hearts, whereas age-matched HF-prone mice had decreased expressions. miRNA expression and CTGF and TSP-1 protein levels were normalized for GAPDH expression and presented as mean ± SEM. *P≤0.05 vs 12 weeks of age; †P≤0.05 vs 52 weeks of age; $P = 0.05 vs 12 weeks of age.
Opposite cardiac miR-17–92 cluster expression profiles in HF-resistant and HF-prone aging
CTGF and TSP-1 have been identified as target genes of the miR-17–92 cluster (Dews et al., 2006), more specifically of the cluster members miR-18a and miR-19a/b (Suarez et al., 2008; Ohgawara et al., 2009). We found opposite expression profiles of the miR-17–92 cluster in HF-prone aging compared to aging with preserved cardiac function. At 104 weeks of age, HF-prone mice had significantly reduced expression levels of miR-17, miR-18a, miR-19a, miR-19b, miR-20a, and miR-92a-1 as compared to 12-week littermates (Fig. 2C and Supporting information Table S1), coinciding with the observed increased presence of their targets TSP-1 and CTGF. Aging of HF-resistant mice, on the other hand, was accompanied by significantly enhanced expression of these miRNAs, except for miR-17 and miR-20a (Supporting information Table S1).
The miR-18/19 – CTGF/TSP-1 axis is regulated in human age-associated heart failure
The three miR-17–92 cluster members miR-18a, miR-19a, and miR-19b specifically target the ECM proteins CTGF and TSP-1. To investigate the role of these genes in human HF, we studied their expression profiles in cardiac biopsies of idiopathic cardiomyopathy (ICM) patients at old age with a moderately decreased or preserved systolic function (ejection fraction (EF) between 40 and 55%) (Paulus et al., 2007) and severely impaired cardiac function (EF < 30%) and compared them to young ICM subjects. In line with reduced expression levels in failing hearts of old mice, decreased miR-18a, miR-19a, and miR-19b expression was associated with severe heart failure at old age (Fig. 3A), while miRNA expression in old patients with a preserved function was not different from young ICM patients (Fig. 3A). Additionally, CTGF and TSP-1 transcript levels were significantly induced in old failing ICM hearts, which further corroborates our findings in old HF-prone mice (Fig. 3B). Together, these data suggest that regulation of CTGF and TSP-1 is the result of the shared expression of miR-18a, miR-19a, and miR19b, enabling modest changes in miRNA expression to control transcriptional repression.
Fig. 3.

CTGF and TSP-1 expression are elevated in human heart failure (HF). RT-PCR analysis of miR-18a, miR-19a, miR-19b, CTGF, and TSP-1 transcript levels in myocardial biopsies from idiopathic cardiomyopathy (ICM) patients at older age with normal (n = 5) and severely impaired (n = 9) cardiac function. Transcript levels were compared to the expression in young ICM subjects with a preserved cardiac function (n = 5). (A) MiR-18a, miR-19a, and miR-19b expression was significantly decreased in older ICM patients with HF. (B) CTGF and TSP-1 transcript levels were significantly induced in older patients with a compromised cardiac function, when compared to younger ICM subjects. All data were normalized for GAPDH expression and presented as mean ± SEM. *P≤0.05 vs young ICM patients. $Pa = 0.06 in failing vs nonfailing hearts of older ICM patients.
The miR-18/19 – CTGF/TSP-1 axis is regulated in aged cardiomyocytes in vitro
To gain further insight into the role of the miR-17–92 cluster in aging of cardiomyocytes, neonatal rat cardiomyocytes (NRCMs) were aged in vitro, and miRNA levels were determined.
Aging of cardiomyocytes in vitro was validated by comparing the lipofuscin content in 4- and 21-day-old NRCMs to the intracellular accumulation of lipofuscin in the hearts of 12- and 104-week-old C57Bl6 mice. Lipofuscin, being autofluorescent, nondegradable biological ‘garbage’, is a hallmark of aging in postmitotic cells, such as cardiomyocytes and neurons (Terman & Brunk, 2004). On high magnification examination using electron microscopy, accumulation of lipofuscin was rarely observed in the hearts of 12-week-old mice, whereas a large amount of intralysosomal lipofuscin inclusions was detected in 104-week-old mice (Fig. 4A). Prolonged culturing of NRCMs resembles the aging process in mice, as lipofuscin was hardly detected at 4 days but had accumulated at 21 days, while the characteristic sarcomere structures were maintained (Fig. 4A). Using optical imaging lipofuscin content per cardiomyocyte increased >150-fold after 21 days compared to 4-day-old NRCMs (Fig. 4B).
Fig. 4.

Aging-induced expression profiles in cardiomyocytes in vitro. (A) Electron microscopic images of the left ventricle of 12- and 104-week-old mice, and 4- and 21-day-old neonatal rat cardiomyocytes (NRCMs) showing perinuclear accumulation of lipofuscin in cardiomyocytes of 104-week-old mice and NRCMs after 21 days in culture. Scale bars represent 2 μm. (B) Two-photon/confocal images and quantification of autofluorescent lipofuscin granules (green) in cultured cardiomyocytes. Nuclei are stained with Hoechst (blue). Scale bars represent 100 μm. (C) RT-PCR analysis showed significant induction of collagen type 3A1 (COL3A1), but not collagen type 1A1 (COL1A1) in cultured NRCMs. (D) RT-PCR analysis revealed decreased miR-18a, miR-19a, and miR-19b expression in aged NRCMs after 21 days in culture. (E) CTGF and TSP-1 transcript levels increased with NRCM aging in vitro. (F) Immunoblotting confirmed the increase in CTGF and TSP-1 protein induction during cardiomyocyte aging. All in vitro experiments were performed with n = 3 per group, and protein and transcript levels were normalized for GAPDH expression. Data were presented as mean ± SEM. *P≤0.05 vs 4-day-old NRCMs.
Beside lipofuscin accumulation, the aged myocardium is hallmarked by increased matrix deposition. To study the production of collagen in aged cardiomyocytes, we determined collagen type 1A1 and 3A1 levels. RT-PCR analysis showed significant induction of the thin collagen type 3A1, but not the thicker type 1A1, in 21-day-old NRCMs (Fig. 4C), further strengthening the aged phenotype of these cells. Taken together, prolonged culturing of NRCMs resembles two common characteristics of cardiac aging in vivo and was therefore used to study age-related changes in cardiomyocytes in vitro.
RT-PCR analysis showed that the expression levels of all members of the miR-17–92 cluster were reduced in aged cardiomyocytes, except miR-92a-1 (Fig. 4D and Supporting information Table S2). Importantly, miR-18a, miR-19a, and miR-19b were among the most strongly repressed miRNAs. Along with reduced expression of these miRNAs, CTGF and TSP-1 transcript and protein levels were significantly induced in aged cardiomyocytes (Fig. 4E, F). These findings confirm the expression profiles in aged HF-prone mice and again suggest that miR-18a, miR-19a, and miR-19b could transcriptionally repress CTGF and TSP-1 levels in cardiomyocyte aging and HF at old age.
Cardiomyocyte CTGF and TSP-1 and collagen production are regulated by miR-18/19
To investigate the cardiac localization of miR-18 and -19, we performed in situ hybridization. MiR-18a and miR-19b are abundantly expressed in the adult mouse heart and are predominantly localized in the perinuclear area of cardiomyocytes (Fig. 5A–C). This was corroborated by the finding that miR-18a and miR-19b expression was higher in cardiomyocytes compared to cardiac fibroblasts (Fig. 5D). Importantly, the abundant expression of miR-18a and miR-19b in cardiomyocytes coincides with low levels of CTGF and TSP-1, whereas in cardiac fibroblasts, relatively low levels of miR-18a and miR-19b were associated with high CTGF and TSP-1 transcription (Fig. 5E).
Fig. 5.

MiR-18a and miR-19b regulate CTGF and TSP-1 expression in cardiomyocytes. (A–C) In situ hybridization showed the abundant expression of miR-18a and miR-19b in the myocardium of adult C57Bl6 mice, most of it expressed by cardiomyocytes. Black arrows indicate the cardiomyocyte nucleus and illustrate the perinuclear localization of these miRNAs. (D and E) Comparison of the expression profiles in cultured neonatal rat cardiomyocytes (NRCMs) and neonatal rat cardiac fibroblasts (NRCFs) shows that abundant expression of miR-18a and miR-19b in NRCMs is paralleled by low CTGF and TSP-1 transcript levels. (F and G) Immunoblotting revealed that manipulating miR-18a and miR-19b function by overexpression of these miRNAs using mimics in NRCMs was sufficient to decrease CTGF and TSP-1 protein expression, while inhibition with antagomirs enhanced CTGF and TSP-1 levels. (H and I) In contrast to NRCMs, immunoblotting in cultured NRCFs showed that CTGF and TSP-1 protein expression was not suppressed by overexpression of miR-18a and miR-19b, nor did inhibition of these miRNAs result in increased CTGF and TSP-1 levels. Mimic and antagomir experiments were performed with n = 4 per group, and data were normalized for GAPDH expression. Data were presented as mean ± SEM. *P≤0.05 vs scrambled control oligonucleotides.
Next, we performed a series of functional studies to determine the role of miR-18a and miR-19b in the regulation of CTGF and TSP-1 and collagen production in cardiomyocytes and cardiac fibroblasts. Overexpression of miR-18a and miR-19b, using miRNA mimics, resulted in significant repression of CTGF and TSP-1 mRNA and protein expression in cardiomyocytes (Fig. 5F and G; Supporting information Fig. S3A). Vice versa, blunting of miR-18a and miR-19b using antagomirs was sufficient to increase CTGF and TSP-1 transcript and protein levels in cardiomyocytes (Fig. 5F, G; Supporting information Fig. S3A). Cardiac fibroblasts demonstrated decreased CTGF and TSP-1 transcript levels upon introduction of miR-18a and miR-19b; however, this did not result in reduced protein levels (Fig. 5H, I). Along the same line, endogenous miRNA inhibition was not sufficient to enhance CTGF and TSP-1 transcript and protein expression in cardiac fibroblasts (Fig. 5H, I; Supporting information Fig. S3B). These results show that regulation of CTGF and TSP-1 by miR-18a and miR-19b is uniquely restricted to the cardiomyocyte.
CTGF and TSP-1 are pro-fibrotic (Belmadani et al., 2007; Shi-Wen et al., 2008), and therefore, we investigated whether their regulating miRs-18a and miR-19b were also capable of affecting collagen production. Indeed, overexpression of miR18a and miR-19b in cardiomyocytes repressed collagen 1A1 and 3A1 mRNA levels, while inhibition of these miRNAs using antagomirs significantly enhanced collagen transcription (Fig. 6A). In contrast, collagen 1A1 and 3A1 transcription was not affected by either miR-18a and miR-19b overexpression or inhibition in neonatal rat cardiomyocytes and cardiac fibroblasts (NRCFs), indicating that collagen expression in cardiac fibroblasts is unrelated to these miRNAs (Fig. 6B). Thus, in concordance with CTGF and TSP-1 regulation by miR-18a and miR-19b in cardiomyocytes, these data strongly imply that miR-18a and miR-19b contribute to the induction of collagen synthesis in aged cardiomyocytes via the regulation of the pro-fibrotic CTGF and TSP-1.
Fig. 6.

MiR-18a and miR-19b regulate collagen 1A1 and 3A1 expression in cardiomyocytes in vitro. RT-PCR analysis for the induction collagen 1A1 (COL1A1) and collagen 3A1 (COL3A1) in cultured neonatal rat cardiomyocytes and cardiac fibroblasts after manipulation with miR-18a and miR-19b mimics and antagomirs. (A) Overexpression of miR-18a and miR-19b in cardiomyocytes significantly reduces collagen 1A1 and collagen 3A1 transcript levels, while inhibition of these miRNAs using antagomirs significantly induced transcription of both collagen types. (B) Collagen 1A1 and 3A1 expression in cultured cardiac fibroblasts seemed unrelated to miR-18a and miR-19b, as no significant repression or induction was observed in NRCFs after treatment with miR-18a and miR-19b mimics or antagomirs, respectively. All experiments were performed with n = 4 per group, and data were normalized for GAPDH transcript levels. Data were presented as mean ± SEM. *P≤0.05 vs scrambled control oligonucleotides.
Discussion
With aging, cardiac function declines as the result of cardiomyocyte loss, left ventricular hypertrophy, dilation and accumulation and remodeling of the extracellular matrix (Lakatta & Sollott, 2002). It is well accepted that genetic changes significantly contribute to the aging process in the heart (Bodyak et al., 2002; Park & Prolla, 2005). Because of the highly accessible mRNA microarray technique, most studies primarily focused on the regulation of gene expression by transcriptional control. With the discovery of miRNAs, now the emphasis is being expanded to the post-transcriptional level of regulation that gives rise to these age-specific gene profiles.
A study showing that the miRNA lin-4 and its target protein lin-14 determine life span in C. elegans has paved the way for exploring the role of miRNAs in aging (Boehm & Slack, 2005). Now, increasing evidence on miRNAs as mediators of age-associated pathologies, together with reports of age-related changes in miRNA expression in aged organs and senescent cells, points toward miRNA regulation as a common biological mechanism that underlies aging and cellular senescence in different organs and cell types (Mudhasani et al., 2008; Chen et al., 2010; Grillari and Grillari-Voglauer, 2010). Nevertheless, so far no studies have addressed the role of miRNAs in the old heart and aging of cardiomyocytes.
In the present study, we showed extensive changes in the expression of the miR-17–92 cluster in a model of age-related heart failure in mice. We were able to extrapolate our findings to patients, which confirm the well-recognized highly conserved nature of microRNAs and their target sites. Initially, the miR-17–92 cluster was linked to cancer pathogenesis and was thought to be pro-tumorgenic because of its regulation by c-Myc (Dews et al., 2006). However, the cluster was later found to be tumor suppressive in multiple forms of cancer (Zhang et al., 2006). Tumor suppressor mechanisms can induce cellular senescence and contribute to the aging process (Campisi, 2003), turning the miR-17–92 cluster into a potential mediator of aging. Indeed, overexpression of miR-17 and miR-20a inhibited senescence in primary human fibroblasts by blunting the activation of p21WAF1, while inhibition of miR-17 caused senescence in anaplastic thyroid cancer cells (Takakura et al., 2008; Hong et al., 2010). Furthermore, miR-17–92 expression is consistently down-regulated in multiple models of aging, i.e. after irradiation (Maes et al., 2008), p53 induction (Brosh et al., 2008), or stress-induced senescence (Li et al., 2009), and in old human skin, bone-marrow-derived mesenchymal stem cells, T cells (Hackl et al., 2010), and peripheral blood mononuclear cells (Noren Hooten et al., 2010). These reports are in line with our data showing repression of the miR17–92 cluster in old failing hearts. In addition, we demonstrate that the miR-17–92 cluster is part of the senescence signature of the aged cardiomyocyte.
From the six members of the miR-17–92 cluster, miR-18a, miR-19a, and miR-19b were among the most strongly repressed miRNAs in aged cardiomyocytes and hearts of old failure-prone mice. Several studies have linked these specific cluster members, and not the other cluster members, to the matricellular proteins CTGF and TSP-1 (Dews et al., 2006; Suarez et al., 2008; Ohgawara et al., 2009). Interestingly, CTGF upregulation in age-induced cardiac disease correlates with TGF-β induction, cardiac fibrosis, and left ventricular stiffening (Koitabashi et al., 2007; Wang et al., 2010). The role of TSP-1 in cardiac aging was not reported so far, but another member of the family of thrombospondins, TSP-2, was recently shown to be up-regulated in aged hearts and to protect against age-related cardiac dysfunction (Swinnen et al., 2009). Here, we showed increased expression of CTGF and TSP-1 in age-related HF both in mice and human.
Traditionally, it has been assumed that CTGF and TSP-1 are predominantly expressed by fibroblasts in the heart. However, several studies have recognized that during cardiac remodeling, CTGF and TSP-1 are also secreted by cardiomyocytes to regulate their surrounding ECM environment (Ohnishi et al., 1998; Chen et al., 2000; Frangogiannis et al., 2005). Moreover, a role for miRNAs in regulating CTGF expression in cardiomyocytes has been established by Duisters et al., (2009), who showed that increased CTGF transcription during pathological LV hypertrophy in (young) hearts is controlled by miR-30 and miR-133. Our in vitro results support a role for miR-18a, miR-19a, and miR-19b in regulating CTGF and TSP-1 expression in the aged cardiomyocyte. Here, miRNA mimics of miR-18a and miR-19b blunted the expression of CTGF and TSP-1, and vice versa, inhibition of these miRNAs enhanced CTGF and TSP-1 levels. In cardiac fibroblasts, overexpression of miR-18a and miR-19b also decreased CTGF and TSP-1 transcription; however, inhibition of these miRNAs was not sufficient to increase CTGF and TSP-1. This may be attributed to the fact that a fibroblast produces large amounts of CTGF and TSP-1 while it contains relatively low amounts of miR-18a and miR-19b. These results imply that the age-related regulation of CTGF and TSP-1 expression by miR-18a and miR-19b in the heart is uniquely restricted to the cardiomyocyte to control its surrounding ECM.
In conclusion, our study is the first to show that miRNA expression of the miR-17–92 cluster changes with cardiac aging and associates decreased miR-18a, miR-19a, and miR-19b expression with age-related remodeling in the heart. Our results suggest that up-regulation of these miRNAs in the aged failure-protected heart blunts the expression of TSP-1 and CTGF to dampen the fibrotic remodeling process that contributes to the functional decline with age. Although we do not show direct regulation of CTGF and TSP-1 by these miRNAs, previous data have proven a direct mechanism (Suarez et al., 2008; Ohgawara et al., 2009). This study provides evidence for the involvement of miRNAs in regulating cardiac aging and identifies them as potential new therapeutic targets for the modulation of aging-induced cardiac remodeling.
Methods
An expanded Methods section is available in the Data S1.
Mice
Male C57Bl6 mice were obtained from Janvier (Le Genest Saint Isle, France), and mice on a C57Bl6 × 129Sv genetic background were generated within the animal facility of the University of Maastricht. All animals were housed under standard day–night rhythm and ad libitum conditions until 12, 52, and 104 weeks of age. Cardiac function was assessed under sedation (2% isoflurane), followed by removal of the hearts for further histological and molecular analyses. For histology, hearts were fixed in 1% paraformaldehyde, embedded in paraffin, and stained with Sirius red. For high magnification analysis, hearts were fixed in 2.5% glutaraldehyde. Electron microscopic images were made with a Philips CM100 (F.E.I., Eindhoven, The Netherlands).
This study was approved by the Institutional Animal Research Committee and conforms with the guidelines for the use of laboratory animals formulated in the Dutch law on care and use of experimental animals.
Patients
Nineteen subjects diagnosed with ICM were included based on age and cardiac function. Five old patients (60.5 years ± 0.8) with preserved cardiac function (ejection fraction between 40 and 55%) and no signs of coronary artery disease (EF: 45.6% ± 9.2) and nine ICM patients at older age (67.0 years ± 4.3) with a severely compromised cardiac function (EF: 18.9% ± 3.0) were compared to a group of five young patients (33.2 years ± 4.4) with preserved cardiac function (EF: 46.5% ± 4.4). This study occurred in line with the recommendations of the institutional ethics committee of the University Hospital Maastricht.
In vitro experiments
Neonatal rat cardiomyocytes and cardiac fibroblasts were isolated from 1- to 3-day-old Lewis rats as described previously (De Windt et al., 1997).
For aging of cardiomyocytes in vitro, NRCMs were cultured for 4 and 21 days, and autofluorescent lipofuscin was excited using a laser at 488 nm, with confocal detection at 510–560 nm (Nikon Eclipse E600FN, Tokyo, Japan). Hoechst-stained nuclei were excited with a two-photon Spectra-Physics Tsunami laser (Spectra-Physics, Irvine, CA, USA) centered at 800 nm and visualized at 400–480 nm. The lipofuscin content per cardiomyocyte was determined using Leica Qwin image processing and analysis software (Leica. Microsystems Cambridge Ltd, Cambridge, UK).
For the overexpression or inhibition of miR-18a and miR-19b, NRCMs and NRCFs were transfected with 80 nm miRIDIAN hairpin inhibitor (antagomiR) miR-18a (#IH-300487-06) or miR-19b (#IH-300489-05), or with miRIDIAN mimic miR-18a (#C-300487-05), or miR-19b (#C-300489-03) (Dharmacon, Colorado, CO, USA). Cells transfected with miRIDIAN microRNA hairpin inhibitor negative control #1 (#IN-001005-01) and miRIDIAN microRNA mimic negative control #2 (#CN-002000-01) served as control groups.
RNA isolation and Real-time PCR
Total RNA was extracted from homogenized cells and heart tissues using the mirVANA miRNA isolation kit (Ambion, Austin, TX, USA) according to the manufacturer's protocol. Gene transcript levels of CTGF, TSP-1, collagen 1A1, and collagen 3A1 (primer sequences are listed in Supporting information Table S3), or miRNA expression were detected with the MyIQ Single Color Real-Time PCR detection System (Bio-Rad, Hercules, CA, USA). MiR-17, miR18a, miR-19a, miR-19b, miR-20, and miR-92a-1 expression was measured using miRNA-specific miScript Primer Assays (Qiagen, Hilden, Germany). All expression levels were presented relative to glyceraldehydes-3-phosphate dehydrogenase (GAPDH).
Immunoblotting
Proteins were extracted from left ventricular heart tissue and in vitro from NRCM and NRCFs. Protein lysates were separated by SDS–PAGE and transferred to a polyvinylidene fluoride membrane (Immobilon-P, 0.45 μm pore size). The membranes were probed overnight at 4°C with a primary antibody to detect CTGF (#GTX26992; GeneTex Inc., Irvine, CA, USA), TSP-1 (in-house rabbit anti-human TSP-1 was kindly provided by MF Hoylaerts (Moura et al., 2008), University of Leuven, Belgium), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (#RDI-TRK5G4-6C5; Fitzgerald Inc., Concord, MA, USA). Protein levels were determined using Quantity One software (Bio-Rad, Hercules, CA, USA) and presented relative to GAPDH protein expression.
In Situ Hybridization
Mouse left ventricular heart tissue was used for miRNA in situ hybridization as described previously (Nuovo, 2010). Double DIG-labeled locked nucleic acid (LNA) hybridization probes complementary to mouse mature miR-18a (5DIGN/CTATCTGCACTAGATGCACCTTA/3DIG_N) (#38462-15), miR-19b (5DIGN/TCAGTTTTGCATGGATTTGCACA/3DIG_N) (#38092-15), and a scrambled probe (5DIGN/GTGTAACACGTCTATACGCCCA/3DIG_N) (#99004-15) were purchased from Exiqon (Vedbaek, Denmark).
Statistical analysis
All data are expressed as mean ± standard error of the mean (SEM). Differences between groups were evaluated by Student's t-test or one-way anova with Bonferroni post hoc test when appropriate. Differences in interstitial and perivascular fibrosis were analyzed by two-way anova and Bonferroni post hoc test. Probability values <0.05 were considered statistically significant.
Acknowledgments
The authors thank Hans Duimel for his excellent technical support in electron microscopy and Wim Engels for his assistance in designing a method to measure lipofuscin accumulation in cardiomyocytes in vitro using a combination of 2-photon and confocal microscopy. Robert Dennert collected and provided the human cardiac biopsy specimens. Marc F. Hoylaerts (Katholieke Universiteit Leuven, Belgium) kindly provided the rabbit anti-human TSP-1 antibody.
Funding sources
This work was supported by a Veni grant (016.096.126) from the Netherlands Organisation for Scientific Research (NWO) to BS, a Horizon grant (93519017) from the Netherlands Genomics Initiative (BS), and a research grant from the Netherlands Heart Foundation (BS) (NHS 2009B025). Additional support was from NWO Vidi grant (SH) and research grants from the Netherlands Heart Foundation (SH) (NHS 2007B036, 2008B011).
Disclosures
None declared.
Author contributions
G.C.A., M.W.M.S., and M.S. performed echocardiographic analysis in mice. W.V. and R.E.W.L. designed and performed in vitro experiments with NRCMs and NRCFs. J.P.M.C. and M.A.M.J.Z. designed and assisted with microscopic studies. C.E. provided the human cardiac biopsy specimens. G.C.A., S.H., and B.S. planned the study, interpreted the data, and wrote the manuscript.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1 CTGF and TSP-1 3′-UTRs have conserved miR-18 and miR-19 target sites.
Fig. S2 CTGF and TSP-1 transcript levels in HF resistant and HF prone mice.
Fig. S3 CTGF and TSP-1 transcripts are regulated by miR-18a and miR-19b in cardiomyocytes.
Table S1 Fold change expression profiles of miR-17–92 cluster members in HF resistant and HF prone mice at different ages.
Table S2 Fold change expression profiles of miR-17–92 cluster members in cardiomyocytes in vitro.
Table S3 Real-Time PCR primers.
Data S1 Methods.
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
References
- Barasch E, Gottdiener JS, Aurigemma G, Kitzman DW, Han J, Kop WJ, Tracy RP. Association between elevated fibrosis markers and heart failure in the elderly: the cardiovascular health study. Circ. Heart Fail. 2009;2:303–310. doi: 10.1161/CIRCHEARTFAILURE.108.828343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmadani S, Bernal J, Wei CC, Pallero MA, Dell'italia L, Murphy-Ullrich JE, Berecek KH. A thrombospondin-1 antagonist of transforming growth factor-beta activation blocks cardiomyopathy in rats with diabetes and elevated angiotensin II. Am. J. Pathol. 2007;171:777–789. doi: 10.2353/ajpath.2007.070056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodyak N, Kang PM, Hiromura M, Sulijoadikusumo I, Horikoshi N, Khrapko K, Usheva A. Gene expression profiling of the aging mouse cardiac myocytes. Nucleic Acids Res. 2002;30:3788–3794. doi: 10.1093/nar/gkf497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm M, Slack F. A developmental timing microRNA and its target regulate life span in C. elegans. Science. 2005;310:1954–1957. doi: 10.1126/science.1115596. [DOI] [PubMed] [Google Scholar]
- Boyle AJ, Shih H, Hwang J, Ye J, Lee B, Zhang Y, Kwon D, Jun K, Zheng D, Sievers R, Angeli F, Yeghiazarians Y, Lee R. Cardiomyopathy of aging in the mammalian heart is characterized by myocardial hypertrophy, fibrosis and a predisposition towards cardiomyocyte apoptosis and autophagy. Exp. Gerontol. 2011 doi: 10.1016/j.exger.2011.02.010. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw AD, Baicu CF, Rentz TJ, Van LaerAO, Bonnema DD, Zile MR. Age-dependent alterations in fibrillar collagen content and myocardial diastolic function: role of SPARC in post-synthetic procollagen processing. Am. J. Physiol. Heart Circ. Physiol. 2010;298:H614–H622. doi: 10.1152/ajpheart.00474.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh R, Shalgi R, Liran A, Landan G, Korotayev K, Nguyen GH, Enerly E, Johnsen H, Buganim Y, Solomon H, Goldstein I, Madar S, Goldfinger N, Borresen-Dale AL, Ginsberg D, Harris CC, Pilpel Y, Oren M, Rotter V. p53-Repressed miRNAs are involved with E2F in a feed-forward loop promoting proliferation. Mol. Syst. Biol. 2008;4:229. doi: 10.1038/msb.2008.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess ML, McCrea JC, Hedrick HL. Age-associated changes in cardiac matrix and integrins. Mech. Ageing Dev. 2001;122:1739–1756. doi: 10.1016/s0047-6374(01)00296-2. [DOI] [PubMed] [Google Scholar]
- Campisi J. Cancer and ageing: rival demons? Nat. Rev. Cancer. 2003;3:339–349. doi: 10.1038/nrc1073. [DOI] [PubMed] [Google Scholar]
- Capasso JM, Palackal T, Olivetti G, Anversa P. Severe myocardial dysfunction induced by ventricular remodeling in aging rat hearts. Am. J. Physiol. 1990;259:H1086–H1096. doi: 10.1152/ajpheart.1990.259.4.H1086. [DOI] [PubMed] [Google Scholar]
- Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J. Mol. Cell. Cardiol. 2000;32:1805–1819. doi: 10.1006/jmcc.2000.1215. [DOI] [PubMed] [Google Scholar]
- Chen LH, Chiou GY, Chen YW, Li HY, Chiou SH. microRNA and aging: a novel modulator in regulating the aging network. Ageing Res. Rev. 2010;9(Suppl 1):S59–S66. doi: 10.1016/j.arr.2010.08.002. [DOI] [PubMed] [Google Scholar]
- da Costa Martins PA, Salic K, Gladka MM, Armand AS, Leptidis S, el Azzouzi H, Hansen A, Coenen-de Roo CJ, Bierhuizen MF, van der Nagel R, van Kuik J, de Weger R, de Bruin A, Condorelli G, Arbones ML, Eschenhagen T, De Windt LJ. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat. Cell Biol. 2010;12:1220–1227. doi: 10.1038/ncb2126. [DOI] [PubMed] [Google Scholar]
- De Windt LJ, Willemsen PH, Popping S, Van der Vusse GJ, Reneman RS, Van Bilsen M. Cloning and cellular distribution of a group II phospholipase A2 expressed in the heart. J. Mol. Cell. Cardiol. 1997;29:2095–2106. doi: 10.1006/jmcc.1997.0444. [DOI] [PubMed] [Google Scholar]
- Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat. Genet. 2006;38:1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, Herias V, van Leeuwen RE, Schellings MW, Barenbrug P, Maessen JG, Heymans S, Pinto YM, Creemers EE. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ. Res. 2009;104:170–178. doi: 10.1161/CIRCRESAHA.108.182535. 176p following 178. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, Winkelmann K, Michael LH, Lawler J, Entman ML. Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111:2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- Grillari J, Grillari-Voglauer R. Novel modulators of senescence, aging, and longevity: small non-coding RNAs enter the stage. Exp. Gerontol. 2010;45:302–311. doi: 10.1016/j.exger.2010.01.007. [DOI] [PubMed] [Google Scholar]
- van Haaften G, Agami R. Tumorigenicity of the miR-17-92 cluster distilled. Genes Dev. 2010;24:1–4. doi: 10.1101/gad.1887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackl M, Brunner S, Fortschegger K, Schreiner C, Micutkova L, Muck C, Laschober GT, Lepperdinger G, Sampson N, Berger P, Herndler-Brandstetter D, Wieser M, Kuhnel H, Strasser A, Rinnerthaler M, Breitenbach M, Mildner M, Eckhart L, Tschachler E, Trost A, Bauer JW, Papak C, Trajanoski Z, Scheideler M, Grillari-Voglauer R, Grubeck-Loebenstein B, Jansen-Durr P, Grillari J. miR-17, miR-19b, miR-20a, and miR-106a are down-regulated in human aging. Aging Cell. 2010;9:291–296. doi: 10.1111/j.1474-9726.2010.00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong L, Lai M, Chen M, Xie C, Liao R, Kang YJ, Xiao C, Hu WY, Han J, Sun P. The miR-17-92 cluster of microRNAs confers tumorigenicity by inhibiting oncogene-induced senescence. Cancer Res. 2010;70:8547–8557. doi: 10.1158/0008-5472.CAN-10-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemi M, Keenan KP, McCoy C, Hoe CM, Soper KA, Ballam GC, van Zwieten MJ. The relative protective effects of moderate dietary restriction versus dietary modification on spontaneous cardiomyopathy in male Sprague-Dawley rats. Toxicol. Pathol. 2000;28:285–296. doi: 10.1177/019262330002800208. [DOI] [PubMed] [Google Scholar]
- Koitabashi N, Arai M, Kogure S, Niwano K, Watanabe A, Aoki Y, Maeno T, Nishida T, Kubota S, Takigawa M, Kurabayashi M. Increased connective tissue growth factor relative to brain natriuretic peptide as a determinant of myocardial fibrosis. Hypertension. 2007;49:1120–1127. doi: 10.1161/HYPERTENSIONAHA.106.077537. [DOI] [PubMed] [Google Scholar]
- Lakatta EG, Sollott SJ. Perspectives on mammalian cardiovascular aging: humans to molecules. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2002;132:699–721. doi: 10.1016/s1095-6433(02)00124-1. [DOI] [PubMed] [Google Scholar]
- Li G, Luna C, Qiu J, Epstein DL, Gonzalez P. Alterations in microRNA expression in stress-induced cellular senescence. Mech. Ageing Dev. 2009;130:731–741. doi: 10.1016/j.mad.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes OC, An J, Sarojini H, Wu H, Wang E. Changes in MicroRNA expression patterns in human fibroblasts after low-LET radiation. J. Cell. Biochem. 2008;105:824–834. doi: 10.1002/jcb.21878. [DOI] [PubMed] [Google Scholar]
- Moura R, Tjwa M, Vandervoort P, Van Kerckhoven S, Holvoet P, Hoylaerts MF. Thrombospondin-1 deficiency accelerates atherosclerotic plaque maturation in ApoE-/- mice. Circ. Res. 2008;103:1181–1189. doi: 10.1161/CIRCRESAHA.108.185645. [DOI] [PubMed] [Google Scholar]
- Mudhasani R, Zhu Z, Hutvagner G, Eischen CM, Lyle S, Hall LL, Lawrence JB, Imbalzano AN, Jones SN. Loss of miRNA biogenesis induces p19Arf-p53 signaling and senescence in primary cells. J. Cell Biol. 2008;181:1055–1063. doi: 10.1083/jcb.200802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren Hooten N, Abdelmohsen K, Gorospe M, Ejiogu N, Zonderman AB, Evans MK. microRNA expression patterns reveal differential expression of target genes with age. PLoS ONE. 2010;5:e10724. doi: 10.1371/journal.pone.0010724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuovo GJ. In situ detection of microRNAs in paraffin embedded, formalin fixed tissues and the co-localization of their putative targets. Methods. 2010;52:307–315. doi: 10.1016/j.ymeth.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Ohgawara T, Kubota S, Kawaki H, Kondo S, Eguchi T, Kurio N, Aoyama E, Sasaki A, Takigawa M. Regulation of chondrocytic phenotype by micro RNA 18a: involvement of Ccn2/Ctgf as a major target gene. FEBS Lett. 2009;583:1006–1010. doi: 10.1016/j.febslet.2009.02.025. [DOI] [PubMed] [Google Scholar]
- Ohnishi H, Oka T, Kusachi S, Nakanishi T, Takeda K, Nakahama M, Doi M, Murakami T, Ninomiya Y, Takigawa M, Tsuji T. Increased expression of connective tissue growth factor in the infarct zone of experimentally induced myocardial infarction in rats. J. Mol. Cell. Cardiol. 1998;30:2411–2422. doi: 10.1006/jmcc.1998.0799. [DOI] [PubMed] [Google Scholar]
- Panek AN, Posch MG, Alenina N, Ghadge SK, Erdmann B, Popova E, Perrot A, Geier C, Dietz R, Morano I, Bader M, Ozcelik C. Connective tissue growth factor overexpression in cardiomyocytes promotes cardiac hypertrophy and protection against pressure overload. PLoS ONE. 2009;4:e6743. doi: 10.1371/journal.pone.0006743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Prolla TA. Gene expression profiling studies of aging in cardiac and skeletal muscles. Cardiovasc. Res. 2005;66:205–212. doi: 10.1016/j.cardiores.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Paulus WJ, Tschope C, Sanderson JE, Rusconi C, Flachskampf FA, Rademakers FE, Marino P, Smiseth OA, De Keulenaer G, Leite-Moreira AF, Borbely A, Edes I, Handoko ML, Heymans S, Pezzali N, Pieske B, Dickstein K, Fraser AG, Brutsaert DL. How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur. Heart J. 2007;28:2539–2550. doi: 10.1093/eurheartj/ehm037. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl Acad. Sci. USA. 2006;103:18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- Shan SW, Lee DY, Deng Z, Shatseva T, Jeyapalan Z, Du WW, Zhang Y, Xuan JW, Yee SP, Siragam V, Yang BB. MicroRNA MiR-17 retards tissue growth and represses fibronectin expression. Nat. Cell Biol. 2009;11:1031–1038. doi: 10.1038/ncb1917. [DOI] [PubMed] [Google Scholar]
- Shi-Wen X, Leask A, Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008;19:133–144. doi: 10.1016/j.cytogfr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Suarez Y, Fernandez-Hernando C, Yu J, Gerber SA, Harrison KD, Pober JS, Iruela-Arispe ML, Merkenschlager M, Sessa WC. Dicer-dependent endothelial microRNAs are necessary for postnatal angiogenesis. Proc. Natl Acad. Sci. USA. 2008;105:14082–14087. doi: 10.1073/pnas.0804597105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen M, Vanhoutte D, Van Almen GC, Hamdani N, Schellings MW, D'Hooge J, Van der Velden J, Weaver MS, Sage EH, Bornstein P, Verheyen FK, VandenDriessche T, Chuah MK, Westermann D, Paulus WJ, Van de Werf F, Schroen B, Carmeliet P, Pinto YM, Heymans S. Absence of thrombospondin-2 causes age-related dilated cardiomyopathy. Circulation. 2009;120:1585–1597. doi: 10.1161/CIRCULATIONAHA.109.863266. [DOI] [PubMed] [Google Scholar]
- Takakura S, Mitsutake N, Nakashima M, Namba H, Saenko VA, Rogounovitch TI, Nakazawa Y, Hayashi T, Ohtsuru A, Yamashita S. Oncogenic role of miR-17-92 cluster in anaplastic thyroid cancer cells. Cancer Sci. 2008;99:1147–1154. doi: 10.1111/j.1349-7006.2008.00800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman A, Brunk UT. Aging as a catabolic malfunction. Int. J. Biochem. Cell Biol. 2004;36:2365–2375. doi: 10.1016/j.biocel.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–267. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Zhang J, Walker SJ, Dworakowski R, Lakatta EG, Shah AM. Involvement of NADPH oxidase in age-associated cardiac remodeling. J. Mol. Cell. Cardiol. 2010;48:765–772. doi: 10.1016/j.yjmcc.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Huang J, Yang N, Greshock J, Megraw MS, Giannakakis A, Liang S, Naylor TL, Barchetti A, Ward MR, Yao G, Medina A, O'Brien-Jenkins A, Katsaros D, Hatzigeorgiou A, Gimotty PA, Weber BL, Coukos G. microRNAs exhibit high frequency genomic alterations in human cancer. Proc. Natl Acad. Sci. USA. 2006;103:9136–9141. doi: 10.1073/pnas.0508889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.