

Abstract
Squamous cell cancers account for more than half of all human cancers, and esophageal cancer is the sixth leading cause of cancer death worldwide. The majority of esophageal squamous cell carcinomas have identifiable p53 mutations, yet the same p53 mutations are found at comparable frequencies in pre-cancerous dysplasia, indicating that transformation requires additional somatic changes yet to be defined. Here we show that the zinc finger transcription factor KLF5 transactivates NOTCH1 in the context of p53 mutation or loss. KLF5 loss limited NOTCH1 activity and was sufficient on its own to transform primary human keratinocytes harboring mutant p53, leading to formation of invasive tumors. Restoration of NOTCH1 blocked transformation of KLF5-deficient and p53 mutant keratinocytes. While human dysplastic epithelia accumulated KLF5, KLF5 expression was lost concurrently with NOTCH1 in squamous cell cancers. Taken together, these results define KLF5 loss as a critical event in squamous cell transformation and invasion. Our findings suggest that KLF5 may be a useful diagnostic and therapeutic target in esophageal squamous carcinomas and possibly more generally in other cancers associated with p53 loss-of-function.
Keywords: KLF5, p53, NOTCH1, squamous cell cancer, esophageal cancer
Introduction
More than half of all human cancers arise from squamous epithelia (1, 2). In particular, esophageal squamous cell cancer accounts for more than 400,000 new cases annually and is almost universally fatal. While alterations in a number of genes have been linked to the development of human esophageal squamous cell cancer, most genetic changes were identified by examining existing cancers (3). Thus, little is known about the molecular changes underlying transformation and carcinogenesis in humans. p53 mutations are common in esophageal malignancy yet are early events and do not induce invasive squamous cell cancer by themselves, suggesting that part of the oncogenic pathway is missing (4-6). In addition, the development of esophageal squamous cell cancer models that of other squamous cell cancers, which together account for nearly 1 million deaths annually (2, 7, 8) and in which p53 mutation or loss-of-function is also frequent but not sufficient for transformation (9-12).
p53 status may influence the function of Krüppel-like factor 5 (KLF5) (13), a zinc finger transcription factor which is frequently pro-proliferative, including in primary esophageal keratinocytes (14), but is growth inhibitory in esophageal cancer cells (15). KLF5 has been implicated in transformation in several contexts (16-18), but no evidence of cellular transformation is seen with constitutive Klf5 expression in esophageal epithelial cells (14), and overexpression of Klf5 in esophageal epithelia in vivo leads to increased proliferation but no dysplasia or cancer (19). A context dependent role of KLF5 is seen in other tissues but is not well understood (20, 21). We hypothesized that KLF5 loss might synergize with p53 mutation or loss in squamous cells to promote transformation and tumorigenesis.
Materials and Methods
Cell Culture
Isolation of primary human esophageal keratinocytes (EPC2) and retroviral transduction of hTERT, SV40 T-antigen, H-RasG12V, and p53R175H to generate EPC2-hTERT, T-Te, T-TeRas, and EPC2-hTERT-p53R175H cells, respectively, have been previously described (22, 23). EPC1 and EPC2 cells were a gift of Dr. Anil Rustgi (University of Pennsylvania). EPC1 cells were transduced with hTERT (EPC1-hTERT) and p53R175H (EPC1-hTERT-p53R175H) as for EPC2 cells (24). Human primary epidermal keratinocytes were purchased from the American Type Culture Collection, Manassas, VA (ATCC product #PCS-200-011). The following cell lines were used: TE1, TE2, TE5, TE7, TE8, TE9, TE10, TE11, TE12. TE15, and HCE4 for esophageal squamous cell cancer (25, 26); SCC9, SCC15, SCC25, Detroit 562, A253, and OCTT2 (gift of Dr. Devraj Basu, University of Pennsylvania) for oropharyngeal squamous cell cancer (27-30); HaCat for skin squamous cell cancers (31); H1263 for lung squamous cell cancer (gift of Dr. Sunil Singhal, University of Pennsylvania); and SiHa and HeLa for cervical squamous cancer (32, 33). Primary keratinocytes were grown at 37°C and 5% CO2 with keratinocyte serum-free medium (K-SFM; Invitrogen, Carlsbad, CA), with 40 μg/ml bovine pituitary extract (Invitrogen), 1.0 ng/ml epidermal growth factor (Invitrogen), 100 u/ml penicillin, 100 μg/ml streptomycin (Invitrogen). Squamous cancer cell lines were grown in 1:1 DMEM/Ham's F12 medium (Invitrogen) with 10% fetal bovine serum (Sigma-Aldrich, Saint Louis, MO). For demethylation treatment, cells were grown to 60% confluence in 100 mm dishes, and 5-azacytidine, dissolved in sterile water, was added at 1 or 5 μM into growth media. Media was changed every 24 hours for 5 days.
Viral constructs and infection
To express human KLF5 and NOTCH1 intracellular domain (NICD), cDNAs were subcloned into the pRevTre retroviral vector (Clontech, Mountain View, CA). Lentiviral vector pTripz (Thermo Scientific Open Biosystems, Huntville, AL, USA) was used to express three distinct short RNA hairpins against KLF5 (Thermo Scientific Open Biosystems; Catalog #RHS4743). shRNA against p53 were cloned into the pLKO.1-TRC lentiviral cloning vector (Addgene, Cambridge, MA; Addgene plasmid #10878) (34). Inserts were verified by DNA sequencing. For retroviral packaging, pRevTre and pRevTet-on retroviral vectors (Clontech) were transfected into Phoenix-Ampho cells (ATCC product #SD 3443) with Lipofectamine™2000 (Invitrogen) according to the manufacturer's instructions. Virus-containing medium was harvested 48 and 72 hours after transfection, and filtered with 0.45 μm MicroFunnel Filters (Pall Life Sciences, Ann Arbor, MI). Keratinocytes were infected with culture supernatants from individual Phoenix-Ampho cells at a 1:6 dilution in K-SFM. Cells were passaged after 24 hours and selected with 300 μg/ml G418 and hygromycin 1 μg/ml for 14 days. Lentivirus was packaged by co-transfecting constructs into HEK 293T cells with the lentiviral packaging plasmids pCMV-dR8.74 (35) and pMD2.VSVG (Addgene plasmid #12259). Non-silencing TRIPZ lentiviral inducible shRNAmir (Thermo Scientific Open Biosystems; Catalog #RHS4743) was used as a control for shRNA experiments. The core sequences of the shRNA are shown in Table S1. After confirming successful knockdown with all constructs, KLF5 shRNA#2 and p53 shRNA#1 were used for subsequent experiments, except as noted.
Quantitative real-time PCR
Total RNA was isolated and quantitative real time PCR was performed Total RNA was isolated with the RNeasy micro kit (Qiagen, Valencia, CA), and cDNA was synthesized with Superscript II reverse transcriptase (Invitrogen), as described previously (36). Quantitative real time PCR was performed in triplicate on three samples for each experimental condition using an ABI Prism 7000 sequence detection system (Applied Biosystems, Carlsbad, CA) and SyBr Green PCR master mix (Applied Biosystems). TATA-box-binding protein was used as an internal control. Primer sequences are listed in Table S2.
Western-blotting
For each sample, 30 μg of total protein were separated on a NuPAGE 4–12% bis-tris acrylamide gel (Invitrogen) and transferred onto polyvinylidene difluoride membrane (Millipore, Billerica, MA), as described previously (14). After blocking with 5% non-fat milk in PBS with 0.2% Tween-20, membranes were incubated overnight at 4 °C with 1:5000 rabbit anti-KLF5 (generated against amino acids 106-122 of human KLF5 by Biosource International/QCB) (15) or 1:1000 goat anti-human NOTCH1 (Santa Cruz Biotechnology, Santa Cruz, CA); Membranes were then incubated with a 1:3000 dilution of anti-rabbit/horseradish peroxidase or anti-mouse/horseradish peroxidase (GE Healthcare Life Sciences, Piscataway, NJ) and developed with the enhanced chemiluminescence plus Western blot analysis kit (GE Healthcare Life Sciences).
Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays were performed in triplicate with the ChIP assay kit (Millipore) (14). Cells were treated with 1% formaldehyde for 10 min to cross-link associated protein to DNA, then lysed, and sonicated. After a 10-fold dilution, the samples were pre-cleared with protein A-agarose/salmon sperm DNA for 30 minutes at 4 °C and incubated overnight at 4 °C with 1:5000 rabbit anti-Klf5 (15), 1:1000 mouse anti-p53 (Vector Laboratories, Burlingame, CA), or 1:500 anti-mouse IgG (Sigma-Aldrich), as a negative control. Protein-DNA complexes were then precipitated with protein A-agarose for 1 hour at 4 °C, heated at 65 °C for 4 hours, and treated with proteinase K. Final DNA was extracted with phenol/chloroform. Quantitative PCR was performed with Power SyBrGreen MasterMix (Applied Biosystems) for 25 cycles. A DNA fragment was amplified with primers corresponding to the region from -989 to -780 bp upstream from the translation start site of the NOTCH1 gene. Primer sequences are listed in Table S3.
Luciferase reporter assays
N1PR-Luc, containing the 5′ regulatory region of the human NOTCH1 gene from -961 to -1 upstream of the translation start site, and N1PRmt-Luc, created by mutating multiple p53-binding repeats located between nucleotides −880 and −783, were gifts of Dr. Tohru Kiyono (National Cancer Research Institute, Tokyo, Japan) (37). Using the using the computational program TESS (38), we identified four putative KLF5 binding sites in the region from -894 to -806 (Figure S1). N1PRmt2-Luc was created by introducing mutations into two putative, proximal KLF5 binding sites in N1PRmt-Luc using the Stratagene QuikChange Multi Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). The CBF1 luciferase reporter plasmid (8′CBF1) was as described (39). Cells infected with shRNA expressing lentivirus were induced with doxycycline 2 μg/ml for 48 hours and then transfected at 50% confluence with the different reporter plasmids in triplicate on 24-well plates using FuGENE 6 transfection reagent (Roche Applied Science, Indianapolis, IN). Cells were lysed at 48 hours with Cell Lysis Buffer (BD Biosciences, San Jose, CA), and luciferase reporter activity was analyzed using Dual-Luciferase Reporter Assay System (Promega, Madison, WI) with a microtiter plate luminometer (Dynex Technologies, Chantilly, VA). Luciferase activity was normalized to Renilla activity and expressed as relative luciferase activity. For studies of KLF5 overexpression, cells were co-transfected with pCMV6-KLF5 (OriGene, Rockville, MD) and the different reporter plasmids.
In vitro transformation assays
For studies of transformation in soft agar, 6-well plates were coated with 1% low-melting agarose (Lonza Rockland, Rockland, ME) in a 1:1 ratio of DMEM (Invitrogen) and KBM (Lonza Rockland) supplemented with BPE 30 μg/ml (Invitrogen), EGF 0.5 ng/ml (Invitrogen), 5% FBS (Sigma-Aldrich) and Penicillin-Streptomycin (Invitrogen). 2.5 × 104 cells were suspended per well and overlaid with 1% low-melting agarose (Lonza Rockland) in DMEM/KBM at 1:1 ratio supplemented with BPE 30 μg/ml, EGF 0.5 ng/ml, 5% FBS and Penicillin-Streptomycin, and plates were incubated in 5% CO2 at 37°C for two weeks. Colonies was visualized by staining with 0.001% crystal violet and counted in at least 10 low-power fields from three individual experiments.
Bisulfite conversion PCR sequencing
Genomic DNA was extracted from cell lines using the QIAamp DNA Mini Kit (Qiagen), and 2 μg of DNA was used for the bisulfite conversion reaction with EpiTec Bisulfite Kit (Qiagen) following the manufacture's instructions. Primers were generated by MethPrimer (40), and the bisulfite treated DNA was amplified in a 25 μl reaction volume with primer pairs from -686 to -457 or from -476 to +17 in the upstream regulatory region of the human KLF5 gene. After cleanup with the QiAquick PCR Purification Kit (Qiagen), PCR products were directly sequenced, and data were analyzed using BiQ-Analyzer software (41). Primer sequences are listed in Table S4.
Animal studies
All animal studies were approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC). Mice were maintained under pathogen-free conditions, and 1 × 107 cells in 100 μl of PBS were injected subcutaneously into each lower flank of 5-8 week-old female SCID/NCr mice (NCRNU-M; Taconic, Hudson, NY). All animals received doxycycline at a concentration of 1 mg/ml with 1% sucrose in their daily drinking water. Tumor diameters were measured with digital calipers, and the tumor volume was calculated in mm3. Animals were followed for 35 days, and all animals were sacrificed at the conclusion of the experiment. A total of five mice were used for each cell line.
Organotypic culture
Matrix was premade 7 days prior to organotypic culture experiments by mixing Matrigel (BD Biosciences) and bovine tendon acid-extracted collagen (Organogenesis, Canton, MA) at a 1:1 ratio with 7.5 × 104 human skin fibroblast cells in 1 × minimal essential medium with Earle's salts (Lonza Rockland), 1.68 mm l-glutamine (Cellgro, Herndon, VA), 10% fetal bovine serum (Hyclone, Logan, UT), and 0.15% sodium bicarbonate (Lonza Rockland). 5 × 105 keratincoytes were seeded onto matrix using 6-well plates and maintained for a total of 7 days (42). Cells were then fed with Epidermalization I medium for 2 days, which is a 3:1 mixture of Dulbecco's modified Eagle's medium (JRH Biosciences, Lenexa, KS) and Ham's F-12 (Invitrogen), supplemented with 4 mm l-glutamine, 0.5 μg/ml hydrocortisone, 0.1 mm O-phosphorylethanolamine, 20 pmtriiodothyronine, 0.18 mm adenine, 1.88 mmCaCl2, 4 pm progesterone (Sigma); 10 μg/ml insulin, 10 μg/ml transferrin, 10 mm ethanolamine, 10 ng/ml selenium (ITES) (Lonza Rockland); and 0.1% chelated newborn calf serum (Hyclone). For the following 2 days, cells were fed with Epidermalization II medium, which is identical to Epidermalization I except that it contains 0.1% unchelated newborn calf serum. Cells were then raised to the air-liquid interface and cultured in Epidermalization III medium for 3 days containing the same growth supplements as Epidermalization II except 2% newborn calf serum. At the conclusion of the experiment, cells were fixed with 10% formaldehyde, embedded in paraffin, sectioned at 5 μm, applied to Probe-on Plus slides (Thermo Fisher Scientific, Rockford, IL), and stained with hematoxylin and eosin. Images were captured on a Nikon Eclipse E600 microscope (Nikon Instruments, Melville, NY) with a Photometrics CoolSNAP charge-coupled device camera (Roper Scientific, Tucson, AZ).
Human tissues
All studies involving human tissues were evaluated by the University of Pennsylvania Institutional Review Board (IRB). Tissues (n=7) containing adjacent normal, dysplastic, and esophageal squamous cell cancer were obtained as paraffin blocks, sectioned at 5 μm, and applied to Probe-on Plus slides. A representative sample was stained with hematoxylin and eosin and evaluated for normal epithelia, high-grade dysplasia, and invasive squamous cell cancer by a trained gastrointestinal pathologist (A.R.S.). For immunochemistry, after wax removal and rehydration, slides were immersed in 10 mM citric acid buffer (pH 6.0) and microwaved for 15 minutes for antigen retrieval. Nonspecific binding of avidin-biotin was blocked with the Avidin/Biotin Blocking Kit (Vector Laboratories), and non-specific antibody binding was blocked with StartingBlock T20 Blocking Buffer (Thermo Fisher Scientific). Sections were incubated over night at 4 °C with 1:2000 rabbit anti-KLF5 (15) or 1:500 goat anti-NOTCH1 (Santa Cruz Biotechnology), 5% BSA, and 0.2% Triton X-100 in PBS. Biotinylated species-specific secondary antibodies (Vector Laboratories, Burlingame, CA) were added, and antibody binding was detected with the Vectastain Elite ABC Kit (Vector Laboratories) and the DAB Peroxidase Substrate Kit (Vector Laboratories), followed by counterstaining with hematoxylin. KLF5 expression was scored by a trained gastrointestinal pathologist (A.G) using the following scale: 0, no staining; 1, very weak staining; 2, weak staining; 3, moderate staining; 4, strong staining; and 5, very strong staining. For each normal tissue, high-grade dysplasia, and invasive squamous cell cancer, 100 cells were scored.
Statistical analyses
Results were expressed as mean ± SEM, with statistical significance of differences between experimental conditions established at 95%. Student's t-test, two sample F-test, and R-squared linear regression were performed using the Analysis ToolPak for Excel (Microsoft, Redmond, WA). Fisher's exact test p-value was used for comparison of tumor numbers in mice. For tumor xenograft studies in mice, we estimated 5 mice per group (N=10 per group because of 2 tumor sites per mouse) in order to detect differences in the proportion of mice having tumor formation, using 90% power and an alpha-level of 0.05, based upon assumptions detailed in Table S5.
Results
Initially, we examined KLF5 expression in an established, step-wise esophageal transformation model in which primary human esophageal keratinocytes (EPC2 cells) were immortalized by hTERT without (EPC2-hTERT cells) or with SV40 T-antigen (T-Te cells), then transformed by H-rasV12G resulting in the creation of a tumorigenic, transformed cell line (T-TeRas cells) (22, 23). Parental EPC2 and EPC2-hTERT cells, which retain normal growth control and senescence and show no malignant changes (23, 24), expressed KLF5 at similar levels to T-Te cells (Figure S2), which lack functional p53 due to the presence of SV40 T-antigen (43) but are not transformed (22). In contrast, KLF5 expression was lost in transformed (22) T-TeRas cells; the mechanism of this loss appeared to be increased methylation of KLF5. The proximal promoter of KLF5 contains a large number of CpG islands (Figure S3A), which were hypermethylated in T-TeRas compared to parental EPC2-hTERT cells (Figure S3B-C), and demethylation treatment of T-TeRas cells reestablished KLF5 expression (Figure S3D), consistent with a role for promoter hypermethylation in silencing KLF5.
To determine the functional consequences of KLF5 and p53 loss in keratinocytes, we used shRNA to silence KLF5 in EPC2-hTERT cells, as well as EPC2-hTERT cells containing the hotspot mutation p53R175H (Figure S4A-B) (24) or knockdown of p53 with shRNA (Figure S4C). Of note, codon 175 is the most frequent mutation site in esophageal squamous cell cancers (11), and p53R175H inhibits p53 DNA binding, while also exerting some gain-of-function properties (44). The keratinocyte tumor suppressor NOTCH1 is a direct transcriptional target of p53 (37, 45), and we postulated that KLF5, an important transcriptional regulator (20), might be critical for maintenance of NOTCH1 expression in the context of p53 mutation or loss. In esophageal keratinocytes with wild-type p53, expression of NOTCH1 protein (Figure 1A) and mRNA (Figure 1B) was unchanged with KLF5 suppression alone, while cells with p53R175H or p53 knockdown had reduced full-length and intracellular NOTCH1 when KLF5 was suppressed. KLF5 suppression also inhibited Notch signaling in cells with p53R175H or p53 knockdown, as indicated by decreased expression of the Notch target HES1 (Figure 1C) and reduced CBF1 reporter activity (Figure 1D).
Figure 1.

KLF5 knockdown with p53 mutation or loss decreases NOTCH1 expression and signaling. (A) By Western blot, both full-length (FL) and intracellular (IC) NOTCH1 were decreased when KLF5 shRNA was expressed along with p53R175H or p53 shRNA. (B) NOTCH1 mRNA was also reduced by KLF5 knockdown in the context of p53R175H mutation or p53 suppression. Non-silencing shRNA was used as a control for p53 shRNA and for KLF5 shRNA. (C) Expression of the Notch target gene HES1 was markedly reduced by KLF5 knockdown in keratinocytes with mutant p53R175H or p53 shRNA. (D) KLF5 knockdown also decreased activity of a CBF1 luciferase reporter, a marker of Notch pathway activation, in cells containing p53R175H or p53 shRNA.
The NOTCH1 promoter contains a number of overlapping p53 (37, 45) and putative KLF5 binding sites (Figure S1). In EPC2-hTERT cells, p53 preferentially bound NOTCH1 (Figure 2A), but in cells with p53R175H or p53 suppression, we observed a reciprocal rise in KLF5 binding to NOTCH1 (Figure 2B). KLF5 induction in EPC2-hTERT-p53R175H cells (Figure 2C), but not parental EPC2-hTERT cells (Figure S5), activated both wild-type NOTCH1 reporter (N1PR) and a NOTCH1 reporter with mutations in the p53 binding sites (37) (N1PRmt), while KLF5 suppression downregulated N1PR and N1PRmt (Figure 2D). The effects of KLF5 on NOTCH1 were blocked by additional mutations to two KLF5 binding sites of the NOTCH1 reporter (N1PRmt2).
Figure 2.

KLF5 and p53 bind and coordinately regulate NOTCH1. (A-B) As expected, quantitative chromatin immunoprecipitation (ChIP) revealed binding of p53 (A) to NOTCH1. However, when p53 binding was lost, with p53R175H or p53 shRNA, there was a reciprocal rise in KLF5 binding (B) to the same region. (C-D) NOTCH1 luciferase reporter (N1PR) was activated by KLF5 (C) and inhibited by KLF5 suppression (D) in EPC2-hTERT-p53R175H cells. Mutations within the p53 binding sites of N1PR (N1PRmt) failed to abolish these effects, but NOTCH1 responsiveness to KLF5 was abolished by additional mutations in two putative KLF5 binding sites.
To determine whether KLF5 loss had a functional role in keratinocyte transformation, we examined keratinocytes for 14 days after KLF5 suppression (Figure S6A). Keratinocytes typically maintained a rounded, epithelioid appearance in culture, but EPC2-hTERT-p53R175H keratinocytes with KLF5 knockdown developed a spindle-shaped appearance (Figure 3A-B, Figure S6B-C) consistent with epithelial-mesenchymal transition (EMT) (46). EMT in keratinocytes with KLF5 suppression and p53R175H was confirmed by induction of Snail, Twist, FOXC2, ZEB1, and ZEB2, the mesenchymal markers MMP2 and MMP3, downregulation of Desmoplakin, and the cadherin “switch” (Table 1, Figure S6D) (46). In addition, EPC2-hTERT-p53R175H cells with KLF5 knockdown formed colonies in soft agar, in contrast to keratinocytes with either KLF5 suppression or p53R175H alone, and restoration of NOTCH1 by induction of the NOTCH1 intracellular domain (NICD) effectively blocked colony formation in EPC2-hTERT-p53R175H cells with KLF5 knockdown, as well as T-TeRas, HCE4, and TE8 cells (Figure 3C, Figure S7A-C). shRNA knockdown of KLF5 and p53 also transformed keratinocytes as indicated by induction of EMT markers and by colony formation (Figure S8A). These findings were confirmed in a second independent human esophageal keratinocyte line, EPC1 (24) (Figure S8B-D).
Figure 3.
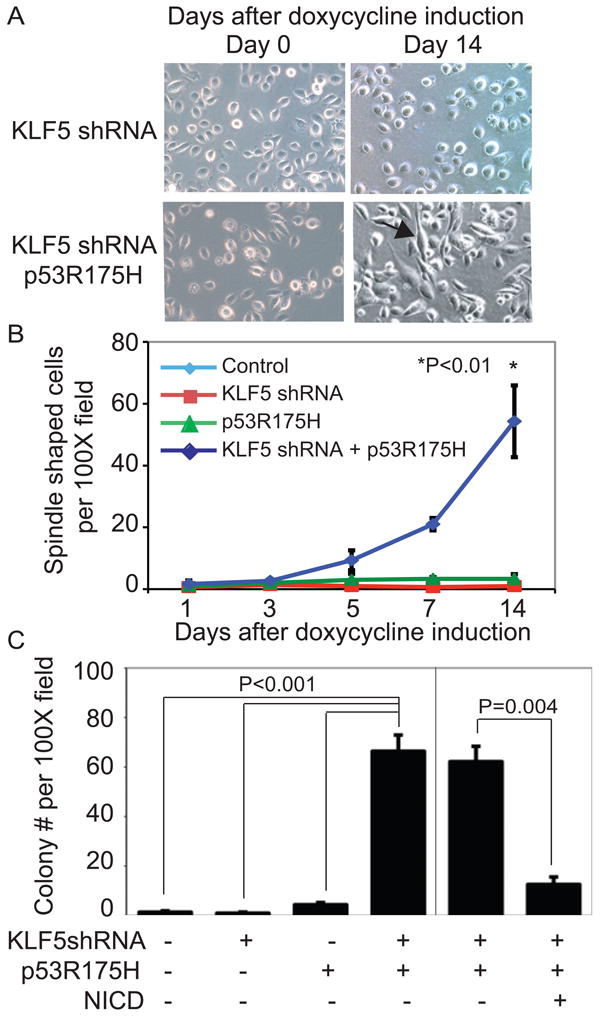
KLF5 knockdown with p53R175H promotes keratinocyte transformation, tumor growth, and invasion. (A). By 14 days, numerous cells with both KLF5 knockdown and p53R175H became spindle-shaped (arrows), compared to the rounded, epithelioid appearance with KLF5 knockdown alone, consistent with a mesenchymal phenotype and EMT. (B) Quantification revealed a statistically significant difference in the number of spindle shaped cells with both KLF5 knockdown and p53R175H, compared to KLF5 knockdown or p53R175H alone. (C) Growth in soft agar was seen with both KLF5 knockdown and p53R175H but not with either KLF5 knockdown or p53R175H alone. Restoration of Notch signaling with NICD inhibited colony formation in soft agar of EPC2-hTERT cells with KLF5 knockdown and p53R175H.
Table 1. KLF5 shRNA and p53R175H induced EMT in human primary keratinocytes.
| Gene name | Fold change relative to control | p-value |
|---|---|---|
| Snail | 103.2 | 0.004 |
| Twist | 8.8 | 0.02 |
| FOXC2 | 191.0 | 0.01 |
| ZEB1 | 214.6 | 0.01 |
| ZEB2 | 5574.4 | 0.01 |
| MMP2 | 1685.9 | 0.01 |
| MMP3 | 46.6 | 0.02 |
| N-cadherin | 4.8 | 0.01 |
| E-cadherin | 0.000006 | 0.003 |
| Desmoplakin | 0.0004 | 0.0009 |
To investigate the effects of KLF5 loss on tumor formation and invasion, we conducted xenograft studies in mice and grew keratinocytes in three-dimensional organotypic culture. When EPC2-hTERT-p53R175H-KLF5shRNA cells were injected into SCID/NCr mice, tumors developed in 4/10 sites, compared to 0/10 sites for mice with EPC2-hTERT cells or cells with KLF5 suppression or p53R175H alone (p=0.009), with a significant difference in average tumor volume (Figure 4A). Tumors from EPC2-hTERT-p53R175H-KLF5shRNA xenografts were characteristic of invasive squamous cell carcinoma (Figure 4B). In organotypic culture, keratinocytes with both KLF5 suppression and p53 mutation but not either alone showed neoplastic changes and invasion (Figure 4C), while induction of NICD in EPC2-hTERT-p53R175H-KLF5shRNA cells blocked these effects. Interestingly, EPC2-hTERT cells with KLF5 knockdown formed thin epithelia which lacked normal stratification, consistent with loss of the established functions of KLF5 in proliferation and migration of non-transformed esophageal keratinocytes (14, 36).
Figure 4.
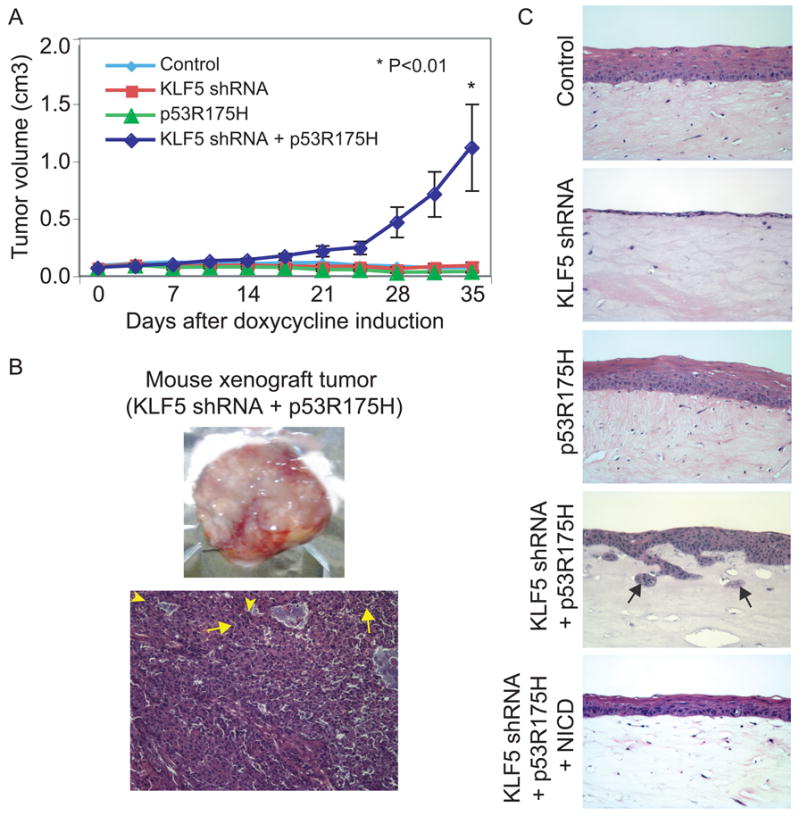
KLF5 loss in the presence of mutant p53 leads to tumor growth and invasion in vivo and in vitro. (A) Primary human esophageal keratinocytes with both KLF5 knockdown and p53R175H but not either alone formed tumors in SCID/NCr mice. (B) Xenograft tumors from keratinocytes with both KLF5 knockdown and p53R175H had islands of invasive tumor cells containing abundant eosinophilic cytoplasm and large hyperchromatic and pleomorphic nuclei with numerous mitoses (arrowheads), in a background of desmoplastic stroma. Tumor cells also showed intercellular bridges (arrows), consistent with squamous cell differentiation. Magnification=100×. (C) In organotypic culture, primary human esophageal keratinocytes with p53R175H underwent normal stratification; cells with KLF5 knockdown failed to stratify normally, consistent with the roles of KLF5 in proliferation and migration of non-transformed keratinocytes. When KLF5 was silenced and p53R175H was expressed, keratinocytes were less mature with increased nuclear-cytoplasmic ratios and microinvasion, characterized by invasive nests and small, detached islands of neoplastic squamous epithelia (arrows). Expression of NICD in cells with KLF5 suppression and p53R175H blocked transformation and invasion. Magnification=200×.
These findings suggested that KLF5 loss might be a critical event in human esophageal squamous cell carcinogenesis. Thus, we examined KLF5 expression in esophageal squamous cell cancers and adjacent dysplastic and normal epithelia. In normal epithelia, KLF5 was expressed in a gradient from the basal layer towards the surface, with highest levels in the proliferative zones (Figure 5A). Unlike p53, which is mutated or lost early in esophageal tumorigenesis (4, 6), KLF5 was highly expressed in dysplastic epithelia. However, KLF5 expression was decreased markedly in invasive squamous cell carcinoma. NOTCH1 was expressed in a similar pattern in normal epithelia and was also absent from invasive cancer (Figure S9A). Quantitatively, KLF5 expression increased significantly from normal to high-grade dysplasia but decreased significantly in invasive cancer (Figure 5B). KLF5 expression was also decreased or absent in 13/13 human esophageal squamous cell cancer lines (Figure 5C), and loss of KLF5 correlated with decreased NOTCH1 expression and loss of p53 function, assessed by expression of the p53 target p21Waf1/Cip1 (Figure 5D). As in T-TeRas cells, KLF5 expression in squamous cancer cells was restored by demethylation treatment.
Figure 5.
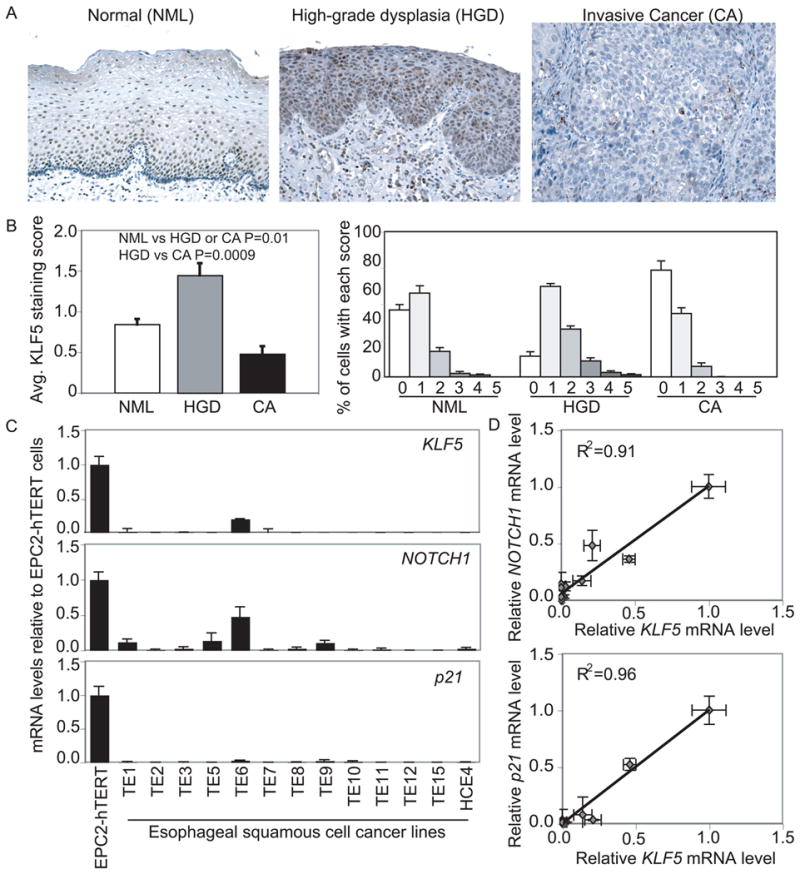
KLF5 is lost in human esophageal squamous cell cancer. (A) KLF5 was normally expressed in a gradient with highest levels in basal and suprabasal cells. In high-grade dysplasia, strong nuclear KLF5 expression was seen throughout esophageal epithelia; expression was lost in invasive esophageal squamous cell cancers. Magnification=100×. (B) Quantitatively, the average score for KLF5 staining per cell increased by 70% from normal (NML, n=6) epithelia to high-grade dysplasia (HGD, n=5), but decreased by 45% and 70%, respectively from NML or HGD to invasive squamous cell cancer (CA, n=6). Furthermore, 37% of NML, 11% of HGD, and 59% of CA cells had no staining, while the percentage with scores of 2 or greater was 17% for NML, 39% for HGD, and 6% for CA. (C) Esophageal squamous cell cancer lines lost KLF5, NOTCH1, and p21Waf1/Cip1 expression by quantitative real-time PCR. (D) KLF5 expression in esophageal squamous cell cancer lines was highly correlated with NOTCH1 and p21Waf1/Cip1 expression.
The pathobiology of esophageal squamous cell cancer is typical of cancers of stratified squamous epithelia (3, 4), and p53 inactivation is common but not causative in these cancers (9). We thus examined whether KLF5 loss in the context of p53 loss or mutation was broadly applicable to squamous cell carcinogenesis. In fact, KLF5 was decreased in 8 of 9 squamous cell cancer lines from oropharynx, lung, skin, and cervix (Figure S9B). Most of these established cell lines contain known p53 mutations; in other cases, such as SiHa and HeLa, p53 is wild-type but is functionally inactivated by HPV (11). Interestingly, only a fraction of women infected with high-risk HPV types develop cervical cancer, indicating that additional factors contribute to malignant progression (12). Similar to our findings in esophageal keratinocytes, KLF5 suppression in combination with p53R175H transformed primary epidermal keratinocytes (Figure S9C-D).
Discussion
p53 mutations are relatively early events in squamous cell carcinogenesis but by themselves are not sufficient for the development of human squamous cell cancer (4-6). Here, we identify the zinc finger protein KLF5 as a key determinant of invasive squamous cell cancer. KLF5 loss together with a naturally occurring p53 mutation or p53 loss leads to keratinocyte transformation and invasion by loss of NOTCH1 transactivation. While human dysplastic epithelia accumulate KLF5, esophageal squamous cell cancers have concurrent loss of KLF5, functional p53, and NOTCH1.
The effects of p53 mutations are variable, and almost 80% of p53 mutations in cancer are missense mutations, leading to the synthesis of a stable protein which lacks typical DNA-binding activity (10). The p53R175H mutation, in particular, results from a single amino acid substitution and confers both gain- and loss-of-function properties (44). These gain-of-function properties may explain subtle differences in the effects of p53R175H versus p53 suppression in keratinocytes with KLF5 suppression. Nonetheless, since NOTCH1 is decreased and KLF5 binding to the NOTCH1 promoter increases in cells with knockdown of p53 by shRNA, these effects are not specific to p53R175H gain-of-function properties. Moreover, while NOTCH1 may not be the only common target for KLF5 and p53, it appears that regulation of NOTCH1 is especially critical for squamous cell transformation, as NICD induction can reverse the effects of KLF5 and p53 loss. The KLF family member KLF4 is also expressed in esophageal epithelia and could theoretically compensate for loss of KLF5 function. However, KLF4 and KLF5 generally have opposing effects (47) and, within esophageal squamous epithelia, are expressed in different cell populations (19).
While we have focused here on squamous cell biology, p53 status might similarly determine KLF5 function during epithelial transformation in the intestine or other tissues. In the intestine, KLF5 promotes proliferation in non-transformed epithelial cells but inhibits proliferation in cancer cell lines (21). In mice with ApcMin/+ and KRASV12, Klf5 haploinsufficiency decreases the formation of premalignant intestinal adenomas (48, 49); an effect of KLF5 on adenocarcinoma development in vivo has not been reported. Importantly, although NOTCH1 is a tumor suppressor in keratinocytes, Notch signaling may be tumor-promoting in the intestine and other tissues (50-52). Thus, in these tissues, other KLF5 and p53 targets may be of greater importance. Notably, KLF5 is deleted or down-regulated in other cancers, including breast and prostate (53, 54).
In sum, we propose a model (Figure 6) in which p53 normally binds to NOTCH1 but can be replaced by KLF5 when p53 binding is lost in dysplasia, an early event in squamous cell carcinogenesis. Subsequent KLF5 loss leads to failure to transactivate NOTCH1 and, potentially, other target genes and drives squamous cell transformation and invasive cancer. Thus, our findings explain why p53 loss or mutation may be necessary but not sufficient for development of human squamous cell cancers and suggest that KLF5 loss is a valuable diagnostic and therapeutic target for esophageal cancer, a significant cause of morbidity and mortality, for cancers of other stratified squamous epithelia, and, potentially, for other cancers associated with p53 mutation or loss.
Figure 6.
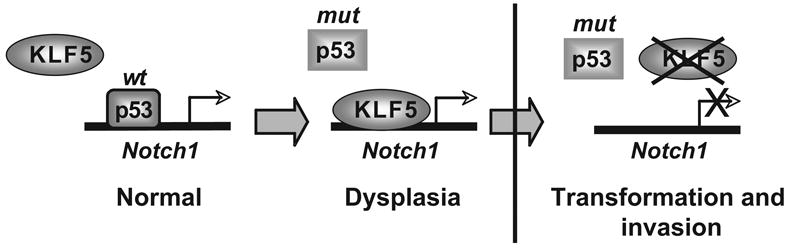
A model for KLF5 and p53 in the regulation of NOTCH1 and esophageal squamous cell carcinogenesis.
Supplementary Material
Acknowledgments
We thank the following: Dr. Anil Rustgi (University of Pennsylvania) for the EPC-1 and EPC-2 cell lines; Dr. Tohru Kiyono (National Cancer Research Institute, Tokyo, Japan) for the N1PR-Luc and N1PRmt-Luc reporters; Dr. Sunil Singhal (University of Pennsylvania) for reagents and advice with the xenograft experiments; Drs. Devraj Basu, Constantinos Koumenis, and John Seykora (University of Pennsylvania), and Dr. Andres Klein-Szanto (Fox Chase Cancer Center) for help with cell lines; Colleen Brensinger (University of Pennsylvania) for statistical support; and Dr. Mitchell Lazar (University of Pennsylvania) for discussions and critical review of the manuscript.
Grant Support: This work was supported by NIH NIDDK R01 DK080031 and DK080031-02S1 to JPK and by the University of Pennsylvania Center for Molecular Studies in Digestive and Liver Diseases (NIH NIDDK P30 DK050306) through the Morphology Core, the Cell Culture Core, and the Molecular Biology Core, and by NIH NCI P01 CA098101 (“Mechanisms of Esophageal Carcinogenesis”).
Footnotes
Conflicts of interest: The authors have no conflicts of interest to disclose
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer Statistics, 2010. CA Cancer J Clin. 2010 doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Stoner GD, Gupta A. Etiology and chemoprevention of esophageal squamous cell carcinoma. Carcinogenesis. 2001;22:1737–46. doi: 10.1093/carcin/22.11.1737. [DOI] [PubMed] [Google Scholar]
- 4.Mandard AM, Hainaut P, Hollstein M. Genetic steps in the development of squamous cell carcinoma of the esophagus. Mutat Res. 2000;462:335–42. doi: 10.1016/s1383-5742(00)00019-3. [DOI] [PubMed] [Google Scholar]
- 5.Hollstein MC, Metcalf RA, Welsh JA, Montesano R, Harris CC. Frequent mutation of the p53 gene in human esophageal cancer. Proc Natl Acad Sci U S A. 1990;87:9958–61. doi: 10.1073/pnas.87.24.9958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao H, Wang LD, Zhou Q, Hong JY, Huang TY, Yang CS. p53 tumor suppressor gene mutation in early esophageal precancerous lesions and carcinoma among high-risk populations in Henan, China. Cancer Res. 1994;54:4342–6. [PubMed] [Google Scholar]
- 7.Goessel G, Quante M, Hahn WC, Harada H, Heeg S, Suliman Y, et al. Creating oral squamous cancer cells: a cellular model of oral-esophageal carcinogenesis. Proc Natl Acad Sci U S A. 2005;102:15599–604. doi: 10.1073/pnas.0409730102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rustgi AK. Models of esophageal carcinogenesis. Semin Oncol. 2006;33:S57–8. doi: 10.1053/j.seminoncol.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 9.Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26:2157–65. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- 10.Soussi T. p53 alterations in human cancer: more questions than answers. Oncogene. 2007;26:2145–56. doi: 10.1038/sj.onc.1210280. [DOI] [PubMed] [Google Scholar]
- 11.Soussi T, Asselain B, Hamroun D, Kato S, Ishioka C, Claustres M, et al. Meta-analysis of the p53 mutation database for mutant p53 biological activity reveals a methodologic bias in mutation detection. Clin Cancer Res. 2006;12:62–9. doi: 10.1158/1078-0432.CCR-05-0413. [DOI] [PubMed] [Google Scholar]
- 12.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–60. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 13.Zhu N, Gu L, Findley HW, Chen C, Dong JT, Yang L, et al. KLF5 Interacts with p53 in regulating survivin expression in acute lymphoblastic leukemia. J Biol Chem. 2006;281:14711–8. doi: 10.1074/jbc.M513810200. [DOI] [PubMed] [Google Scholar]
- 14.Yang Y, Goldstein BG, Nakagawa H, Katz JP. Krüppel-like factor 5 activates MEK/ERK signaling via EGFR in primary squamous epithelial cells. Faseb J. 2007;21:543–50. doi: 10.1096/fj.06-6694com. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y, Goldstein BG, Chao HH, Katz JP. KLF4 and KLF5 regulate proliferation, apoptosis and invasion in esophageal cancer cells. Cancer Biol Ther. 2005;4:1216–21. doi: 10.4161/cbt.4.11.2090. [DOI] [PubMed] [Google Scholar]
- 16.Sun R, Chen X, Yang VW. Intestinal-enriched Kruppel-like factor (Kruppel-like factor 5) is a positive regulator of cellular proliferation. J Biol Chem. 2001;276:6897–900. doi: 10.1074/jbc.C000870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nandan MO, Chanchevalap S, Dalton WB, Yang VW. Kruppel-like factor 5 promotes mitosis by activating the cyclin B1/Cdc2 complex during oncogenic Ras-mediated transformation. FEBS Lett. 2005;579:4757–62. doi: 10.1016/j.febslet.2005.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nandan MO, Yoon HS, Zhao W, Ouko LA, Chanchevalap S, Yang VW. Kruppel-like factor 5 mediates the transforming activity of oncogenic H-Ras. Oncogene. 2004;23:3404–13. doi: 10.1038/sj.onc.1207397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldstein BG, Chao HH, Yang Y, Yermolina YA, Tobias JW, Katz JP. Overexpression of Kruppel-like factor 5 in esophageal epithelia in vivo leads to increased proliferation in basal but not suprabasal cells. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1784–92. doi: 10.1152/ajpgi.00541.2006. [DOI] [PubMed] [Google Scholar]
- 20.McConnell BB, Ghaleb AM, Nandan MO, Yang VW. The diverse functions of Kruppel-like factors 4 and 5 in epithelial biology and pathobiology. Bioessays. 2007;29:549–57. doi: 10.1002/bies.20581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bateman NW, Tan D, Pestell RG, Black JD, Black AR. Intestinal tumor progression is associated with altered function of KLF5. J Biol Chem. 2004;279:12093–101. doi: 10.1074/jbc.M311532200. [DOI] [PubMed] [Google Scholar]
- 22.Kim SH, Nakagawa H, Navaraj A, Naomoto Y, Klein-Szanto AJ, Rustgi AK, et al. Tumorigenic conversion of primary human esophageal epithelial cells using oncogene combinations in the absence of exogenous Ras. Cancer Res. 2006;66:10415–24. doi: 10.1158/0008-5472.CAN-06-2104. [DOI] [PubMed] [Google Scholar]
- 23.Harada H, Nakagawa H, Oyama K, Takaoka M, Andl CD, Jacobmeier B, et al. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol Cancer Res. 2003;1:729–38. [PubMed] [Google Scholar]
- 24.Takaoka M, Harada H, Deramaudt TB, Oyama K, Andl CD, Johnstone CN, et al. Ha-Ras(G12V) induces senescence in primary and immortalized human esophageal keratinocytes with p53 dysfunction. Oncogene. 2004;23:6760–8. doi: 10.1038/sj.onc.1207923. [DOI] [PubMed] [Google Scholar]
- 25.Nishihira T, Hashimoto Y, Katayama M, Mori S, Kuroki T. Molecular and cellular features of esophageal cancer cells. J Cancer Res Clin Oncol. 1993;119:441–9. doi: 10.1007/BF01215923. [DOI] [PubMed] [Google Scholar]
- 26.Banks-Schlegel SP, Quintero J. Growth and differentiation of human esophageal carcinoma cell lines. Cancer Res. 1986;46:250–8. [PubMed] [Google Scholar]
- 27.Peterson WD, Jr, Stulberg CS, Simpson WF. A permanent heteroploid human cell line with type B glucose-6-phosphate dehydrogenase. Proc Soc Exp Biol Med. 1971;136:1187–91. doi: 10.3181/00379727-136-35455. [DOI] [PubMed] [Google Scholar]
- 28.Rheinwald JG, Beckett MA. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultures from human squamous cell carcinomas. Cancer Res. 1981;41:1657–63. [PubMed] [Google Scholar]
- 29.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, et al. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. 1973;51:1417–23. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- 30.Basu D, Nguyen TT, Montone KT, Zhang G, Wang LP, Diehl JA, et al. Evidence for mesenchymal-like sub-populations within squamous cell carcinomas possessing chemoresistance and phenotypic plasticity. Oncogene. 2010;29:4170–82. doi: 10.1038/onc.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–71. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puck TT, Marcus PI, Cieciura SJ. Clonal growth of mammalian cells in vitro; growth characteristics of colonies from single HeLa cells with and without a feeder layer. J Exp Med. 1956;103:273–83. doi: 10.1084/jem.103.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedl F, Kimura I, Osato T, Ito Y. Studies on a new human cell line (SiHa) derived from carcinoma of uterus. I. Its establishment and morphology. Proc Soc Exp Biol Med. 1970;135:543–5. doi: 10.3181/00379727-135-35091a. [DOI] [PubMed] [Google Scholar]
- 34.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–98. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 35.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–71. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y, Tetreault MP, Yermolina YA, Goldstein BG, Katz JP. Krüppel-like factor 5 controls keratinocyte migration via the integrin-linked kinase. J Biol Chem. 2008;283:18812–20. doi: 10.1074/jbc.M801384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yugawa T, Handa K, Narisawa-Saito M, Ohno S, Fujita M, Kiyono T. Regulation of Notch1 gene expression by p53 in epithelial cells. Mol Cell Biol. 2007;27:3732–42. doi: 10.1128/MCB.02119-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schug J. Using TESS to Predict Transcription Factor Binding Sites in DNA Sequence. In: Baxevanis AD, editor. Current Protocols in Bioinformatics. J. Wiley and Sons; 2003. [DOI] [PubMed] [Google Scholar]
- 39.Ronchini C, Capobianco AJ. Notch(ic)-ER chimeras display hormone-dependent transformation, nuclear accumulation, phosphorylation and CBF1 activation. Oncogene. 2000;19:3914–24. doi: 10.1038/sj.onc.1203719. [DOI] [PubMed] [Google Scholar]
- 40.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–31. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 41.Bock C, Reither S, Mikeska T, Paulsen M, Walter, Lengauer T. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics. 2005;21:4067–8. doi: 10.1093/bioinformatics/bti652. [DOI] [PubMed] [Google Scholar]
- 42.Andl CD, Mizushima T, Nakagawa H, Oyama K, Harada H, Chruma K, et al. Epidermal growth factor receptor mediates increased cell proliferation, migration, and aggregation in esophageal keratinocytes in vitro and in vivo. J Biol Chem. 2003;278:1824–30. doi: 10.1074/jbc.M209148200. [DOI] [PubMed] [Google Scholar]
- 43.Levine AJ. The common mechanisms of transformation by the small DNA tumor viruses: The inactivation of tumor suppressor gene products: p53. Virology. 2009;384:285–93. doi: 10.1016/j.virol.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 44.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010;2:a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lefort K, Mandinova A, Ostano P, Kolev V, Calpini V, Kolfschoten I, et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 2007;21:562–77. doi: 10.1101/gad.1484707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghaleb AM, Nandan MO, Chanchevalap S, Dalton WB, Hisamuddin IM, Yang VW. Kruppel-like factors 4 and 5: the yin and yang regulators of cellular proliferation. Cell Res. 2005;15:92–6. doi: 10.1038/sj.cr.7290271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McConnell BB, Bialkowska AB, Nandan MO, Ghaleb AM, Gordon FJ, Yang VW. Haploinsufficiency of Kruppel-Like Factor 5 Rescues the Tumor-Initiating Effect of the ApcMin Mutation in the Intestine. Cancer Research. 2009;69:4125–33. doi: 10.1158/0008-5472.CAN-08-4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nandan MO, Ghaleb AM, McConnell BB, Patel NV, Robine S, Yang VW. Kruppel-like factor 5 is a crucial mediator of intestinal tumorigenesis in mice harboring combined ApcMin and KRASV12 mutations. Mol Cancer. 2010;9:63. doi: 10.1186/1476-4598-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dotto GP. Notch tumor suppressor function. Oncogene. 2008;27:5115–23. doi: 10.1038/onc.2008.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L. Rational targeting of Notch signaling in cancer. Oncogene. 2008;27:5124–31. doi: 10.1038/onc.2008.226. [DOI] [PubMed] [Google Scholar]
- 52.Wilson A, Radtke F. Multiple functions of Notch signaling in self-renewing organs and cancer. FEBS Letters. 2006;580:2860–8. doi: 10.1016/j.febslet.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 53.Chen C, Bhalala HV, Qiao H, Dong JT. A possible tumor suppressor role of the KLF5 transcription factor in human breast cancer. Oncogene. 2002;21:6567–72. doi: 10.1038/sj.onc.1205817. [DOI] [PubMed] [Google Scholar]
- 54.Chen C, Bhalala HV, Vessella RL, Dong JT. KLF5 is frequently deleted and down-regulated but rarely mutated in prostate cancer. Prostate. 2003;55:81–8. doi: 10.1002/pros.10205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.