

Abstract
Identification of the underlying genetic, cellular, and biochemical basis of lipid metabolic disorders provides an opportunity to deploy corrective, mechanism-targeted, topical therapy. We assessed this therapeutic approach in two patients with Congenital Hemidysplasia with Ichthyosiform Erythroderma and Limb Defects (CHILD) syndrome, an X-linked dominant disorder of distal cholesterol metabolism. Based upon the putative pathogenic role of both pathway-product deficiency of cholesterol and accumulation of toxic metabolic intermediates, we assessed the efficacy of combined therapy with lovastatin and cholesterol. We also evaluated the basis for the poorly understood, unique lateralization of the cutaneous and bone malformations of CHILD syndrome by analyzing gene activation in abnormal and unaffected skin. Ultrastructural analysis of affected skin showed evidence of both cholesterol depletion and toxic metabolic accumulation. Topical treatment with lovastatin/cholesterol (but not cholesterol alone) virtually cleared skin lesions by 3 months, accompanied by histologic and ultrastructural normalization of epidermal structure and lipid secretion. The unusual lateralization of abnormalities in CHILD syndrome reflects selective clearance of keratinocytes and fibroblasts that express the mutant allele from the unaffected side. These findings validate pathogenesis-based therapy that provides the deficient end-product and prevents accumulation of toxic metabolites, an approach of potential utility for other syndromic lipid metabolic disorders.
Keywords: ichthyosis, lipids, lateralization, X-inactivation, CHILD syndrome, statin, lovastatin
INTRODUCTION
The ichthyoses are a group of inherited scaling skin disorders of diverse etiologies (Oji et al., 2010), which share a permeability barrier abnormality that parallels the severity of the clinical phenotype (rev. in (Elias et al., 2008; Moskowitz et al., 2004; Schmuth et al., 2007)). Current treatment of the ichthyoses remains symptomatic and largely directed towards reducing the scaling component. This approach is often ineffective, may be accompanied by toxicity (e.g., with systemic retinoids), and is intuitively counterproductive, since the hyperkeratosis and hyperplasia likely reflect an epidermal homeostatic response to re-establishing a competent barrier (Elias, 2010). Use of recombinant protein replacement or gene therapy for genodermatoses remains remote, due to the: i) widespread nature of the cutaneous phenotype; ii) difficulties in transcutaneous drug delivery; iii) discomfort of injections needed to bypass the largely-impenetrable epidermal barrier; iv) enormous costs of the required ‘designer approach’; and v) uncertain long-term risks of viral transduction.
Recent advances in our understanding of the underlying genetic, cellular, and biochemical basis of many forms of ichthyosis (Elias et al., 2010) present an alternative, intriguing opportunity to initiate pathway- or mechanism-based therapy for many of these disorders. In syndromic lipid metabolic disorders, the cutaneous and extracutaneous manifestations often reflect not only deficiency of pathway product (i.e., cholesterol, free fatty acids, or ceramides), but also accumulation of toxic pathway metabolites (Cooper et al., 2003; Elias et al., 2008; Holleran et al., 1994; Kelley and Herman, 2001; Rizzo et al., 2010; Zettersten et al., 1998). In inherited disorders of distal cholesterol metabolism, attempts are underway to treat extracutaneous components by oral co-administration of cholesterol and/or statins (Chan et al., 2009; Haas et al., 2001; Szabo et al., 2010). Yet, this approach will inevitably be hampered by first-pass hepatic metabolism of statins, as well as intrahepatic incorporation of oral cholesterol into lipoprotein particles, which are unable to traverse the blood-brain barrier or access peripheral tissues without LDL receptors, such as the epidermis (Ponec et al., 1992).
We hypothesized that topical application of these agents could bypass hepatic first-pass metabolism, allowing direct access not only to the skin, but perhaps to extracutaneous tissues as well. If validated, such topical pathogenesis (pathway)-based therapy would display substantial inherent advantages, including: i) disease-targeted and mechanism-based specificity; ii) inherent safety; iii) relatively low cost, in comparison to small molecule, recombinant protein or gene therapy; and iv) exploitation of the ready accessibility and visibility of the skin to assess therapeutic efficacy and safety.
We have now validated this therapeutic approach in two unrelated patients with a syndromic disorder of distal cholesterol metabolism (Congenital Hemidysplasia with Ichthyosiform erythroderma and Limb Defects, CHILD syndrome, OMIM #308050), which includes a notoriously unresponsive ichthyosis. CHILD syndrome is an X-linked dominant disorder caused by mutations in NAD(P)H steroid dehydrogenase-like (NSDHL), which encodes a member of the enzyme complex that removes the C-4 methyl group from lanosterol (Fig. 1) (Grange et al., 2000; Kelley and Herman, 2001). Most affected individuals are female, since the disorder is largely embryonic lethal in males. Clinical manifestations range from localized skin disease (‘CHILD nevus’) to extensive limb malformations, which entirely or largely localize to one side of the body, often with a sharp midline demarcation (lateralization) (Happle, 2006). More limited skin lesions may follow one or more lines of Blaschko, a recognized pattern of ectodermal development (Sun and Tsao, 2008). The basis for the unique lateralization in CHILD syndrome is unknown.
Figure 1. The Cholesterol Biosynthetic Pathway.

HMG-CoA reductase is a rate-limiting step early in the cholesterol biosynthetic pathway, which is inhibited by statins. CHILD syndrome results from mutations in NAD(P)H steroid dehydrogenase-like (NSDHL), which prevent the post-lanosterol generation of cholesterol, potentially leading to pathway-product deficiency in addition to metabolite accumulation. Conradi-Hünermann-Happle syndrome is due to mutations in a gene encoding emopamil binding protein (EBP), which acts distal to NSDHL in cholesterol biosynthesis.
We report here that the skin lesions in both patients responded dramatically to topical co-application of cholesterol and lovastatin (but not to cholesterol alone). The striking improvement through topical delivery supports the use of such topical, pathogenesis-based therapy for cutaneous, and possibly extracutaneous, manifestations of syndromic disorders of lipid metabolism.
RESULTS
Description of Cases
Patient 1
A 13-year-old girl was born with marked soft tissue and bone deformities, including digit-like protrusions on the distal aspects of the markedly-reduced left upper and lower extremities. Her skin displayed ichthyosiform, erythrodermic plaques on the left side of her body. At 3 years of age, her rudimentary left lower leg was amputated to allow fitting of a prosthesis. However, her ichthyosis involved the entire left thigh and stump (Fig. 2a), preventing her wearing the prosthesis due to persistent skin breakdown. She showed no cutaneous or bone defects of the right side. The patient was treated with petrolatum-based emollients without benefit. She neither tolerated nor improved with topical applications of keratolytics, retinoids, corticosteroids, or calcineurin inhibitors. At 9 years of age she developed an enlarging, pendulous tumor of the vulva and left inner thigh. Tumor sections histologically revealed verruciform xanthomatosis, which was debulked at 12 years of age. DNA sequencing from blood leukocytes showed a c.248G>A transition in exon 3 of one allele of NSHDL at Xq28, predicting the substitution p.Gly83Asp (G83D) and verifying the diagnosis of CHILD syndrome.
Figure 2. Clinical and Histologic Improvement with Topical Applications of Lovastatin/Cholesterol.

a: Treated skin of Patient #1. b: Treated skin of Patient #2. c. Histopathologic evaluation of lesional skin of Patient #2 at baseline (left) showed marked epidermal acanthosis with psoriasiform hyperplasia. The granular layer was absent, and the stratum corneum showed parakeratosis and retention hyperkeratosis. Bar = 25 μm. By approximately 3 months after treatment initiation (right), the epidermis was of normal thickness with regular orthokeratinization and a normal granular layer. Bar = 7 μm.
Patient 2
A 22-year-old woman was born with a severely malformed right lower extremity, necessitating amputation during early childhood, and malformations of the right hand with nail dystrophy. During the first few years of life, she developed inflamed, thickened skin on the dorsal aspect of the right hand that gradually extended to the right axilla (Fig. 2b). She had no abnormalities on the left side of her body, except for a small, scaling plaque between the left 3rd and 4th fingers. The patient also had neurosensory deafness of the right ear and severe lumbar scoliosis. She was treated for 17 years with topical emollients, steroid ointments or cryotherapy without significant improvement. DNA sequencing from blood leukocytes showed a nonsense mutation in exon 4 on one allele of the NSDHL gene (c.317C>A; p.S106X), verifying the diagnosis of CHILD syndrome. Hphl digestion of the PCR fragment confirmed the predicted abnormal restriction pattern, which was not observed in controls (courtesy of Prof. K. Grzeschik, Marburg, Germany).
Efficacy and Mechanism of Pathogenesis (Pathway)-Based Therapy
Clinical observations
We first addressed the possibility that skin lesions in CHILD syndrome primarily reflect cholesterol (pathway end-product) deficiency alone, but found no clinical improvement after 3 months of treatment with twice daily topical 10% cholesterol in hydrophilic ointment. We next considered the possibility (supported by ultrastructural studies described below) that the cutaneous phenotype reflects both cholesterol deficiency and accumulation of toxic sterol metabolites. Lesional skin of both patients was treated twice daily with 2% lovastatin/2% cholesterol lotion. Decreased inflammation, skin thickening, and scaling was noted as early as 4–6 weeks in both patients, and the severity scores decreased by 72% (patient 1) and 79% (patient 2) by 3 months. By 6 months, treated skin largely normalized, but did not grow hair (Fig. 2a). Patient #1, now more than 1 year on therapy, uses the medication twice weekly with good control and is able to wear her prosthesis. Patient #2 has been treated for more than 17 months, now once to twice weekly, and shows residual hypopigmentation without inflammation or scaling (Fig. 2b). She has improved digital mobility, but little improvement in onychodystrophy, likely reflecting the inability of the compound to penetrate the nail. Patient #2 discontinued therapy for 15 days while on vacation without relapse. In both patients, the compounded lotion has been well-tolerated. Laboratory testing 6 months after initiation of dual therapy revealed no changes in blood counts, hepatic function, creatinine or serum lipids.
Light and electron microscopic alterations
Baseline biopsy sections of skin lesions from both patients prior to treatment showed marked hyperkeratosis and acanthosis with absence of the granular layer and dermal foam cells (Fig. 2c, left). Ultrastructural assessment of these biopsies showed severe alterations in the epidermal lamellar body (lipid) secretory system. Individual organelles displayed few internal lamellae, which often fused prematurely into intracellular, multivesicular bodies that were incompletely secreted (Figs. 3a-c). Partial failure of organelle secretion correlated with the presence of entombed organelle contents within corneocytes (Fig. 3c), and retention of acid lipase, an organelle content marker, within corneocytes (Fig. 4a). Finally, and as a result of impaired secretion, the stratum corneum extracellular matrix displayed a paucity of secreted lamellar material, interspersed with abundant non-lamellar vesicles (Fig. 3c; open arrows). In contrast, biopsy sections after 3 months of treatment showed a largely normal epidermis, with a prominent granular layer and orthokeratotic scale by light microscopy (Fig. 2c, right). Moreover, ultrastructural analysis showed nearly complete resolution of the structural abnormalities in the lamellar body secretory system, and normalization of secretion in both patients (Figs. 3d, 3e and 4b).
Figure 3. Ultrastructural Normalization from Pathogenesis-Based Therapy in CHILD Syndrome.

a and b: Pre-treatment biopsies show prominent abnormalities in epidermal lamellar body morphology (asterisks) and secretion in the stratum granulosum (SG). c: Impaired secretion results in entombment of unsecreted organelle contents within corneocytes (SC, solid arrows), reduced lamellar material in the SC interstices (open arrows), with massive distended foci of non-lamellar/amorphous material between corneocytes (asterisks). Post-treatment biopsies (d and e) show substantial improvement in lamellar body contents (e) and secretion, but some foci of non-lamellar material persist (d, open arrows). a, b, and e: osmium tetroxide post-fixation; c and d: ruthenium tetroxide post-fixation. Bars: a = 0.2 μm; b = 0.5 μm; c = 0.2 μm; d and e = 0.1 μm.
Figure 4. Ultrastructural Cytochemical Evidence of Improved Lamellar Body Secretion in Treated Skin.
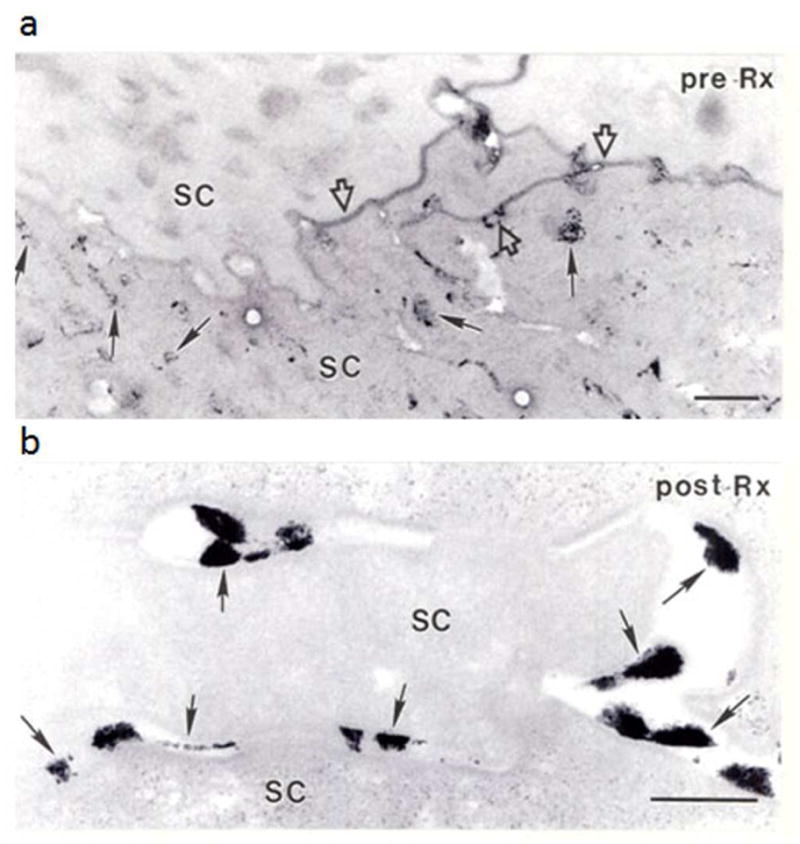
a: Pre-treatment biopsy, assessed with an ultrastructural marker of lamellar body contents, shows much retained (unsecreted) lamellar body contents within cornified cells (SC, arrows). Open arrows indicate focal sites where organelle contents have been secreted. b: Post-treatment biopsy reveals that most enzyme activity has been secreted (exteriorized) (arrows). Osmium tetroxide post-fixation. Bars: a: 0.5 μm; b = 0.25 μm.
A Subclinical Phenotype Persists in Clinically-Unaffected Sites
Pre-treatment biopsies also were obtained from a contralateral, ‘unaffected’ skin site in Patient #1. Although no visible ichthyosiform features could be appreciated either clinically or by light microscopy, ultrastructural images displayed mild abnormalities in lamellar body internal contents and lamellar bilayer organization, with the presence of lamellar/non-lamellar phase separation but not disordered secretion (Figs. 5a-c). These findings, though far less prominent than images from clinically-affected sites and without evidence of cytotoxicity, suggested that residual abnormalities persist in ‘uninvolved’ skin, prompting examination of gene expression in these affected vs. ‘unaffected’ contralateral sites.
Figure 5. Clinically ‘Unaffected’ Epidermis of CHILD Syndrome Shows Abnormalities of the Lamellar Body Secretory System.

Most of the lamellar bodies in the stratum granulosum (SG) display abnormal internal contents (c, asterisks). Lamellar body secretion is partially impaired, with entombment of some organelles (a, open arrows) and retained acidic lipase activity in some corneocytes, as shown by ultrastructural cytochemistry (b, open arrows). Note also the inhomogeneous distribution of secreted enzyme activity (b, arrows). Bars: a = 0.1 μm, b = 0.5 μm, c = 0.2 μm.
Genetic Basis for Clinically Normal Skin in CHILD Syndrome
X-inactivation studies on peripheral blood leukocytes showed preferential Inactivation of one allele (89+2% of normal allele), consistent with skewing of X-inactivation. Genomic DNA sequencing from both sets of keratinocytes showed equal representation of both alleles (Fig. 6). However, RT-PCR on cDNA from keratinocytes and fibroblasts isolated from the affected versus clinically unaffected side in Patient #1 demonstrated exclusive expression of the mutant allele by affected cultured keratinocytes, while unaffected keratinocytes solely expressed the wild type allele (Fig. 6). Only the normal allele was expressed by unaffected fibroblasts, but fibroblasts from the clinically affected area expressed an approximately even distribution of normal and mutant alleles.
Figure 6. NSDHL Gene Expression.

a. Sequences of cDNA obtained from cultured keratinocytes and fibroblasts in unaffected (upper panel) and affected (lower panel) areas of the skin. There is clear exclusive expression of the mutant c.248G>A allele in keratinocytes taken from affected skin, whereas fibroblasts from this area express both the mutant and the normal allele. In the unaffected areas only the wild type is found in keratinocytes as well as fibroblasts. b. Genomic DNA sequences obtained from peripheral blood leukocytes, unaffected and affected keratinocytes respectively. All traces show the presence of both wild type and mutant alleles. Arrows point to the mutant residue.
DISCUSSION
Discovery of genetic changes underlying thousands of disorders has increased our understanding of normal cell function, as well as the cellular and molecular abnormalities that lead to disease phenotypes. In addition, we can now consider the possibility of pathogenesis-based therapy. As our disease prototype to consider the feasibility of pathogenesis-based therapy, we chose CHILD syndrome, an X-linked dominant disorder of distal cholesterol synthesis. We show here that topical application of the deficient pathway end-product (cholesterol) is ineffective, but preventing the accumulation of toxic metabolites (with a statin) and replenishing end-product corrects the cutaneous phenotype.
The lack of efficacy of cholesterol alone is not due to the absence of LDL receptors in epidermis, given that the keratinocyte is able to incorporate NBD-labeled cholesterol (Man et al., 1993; Mao-Qiang et al., 1995). Rather, the response to dual- rather than mono-therapy probably reflects both the sterol depletion and metabolite accumulation that result from pathogenic mutations in enzymes of cholesterol synthesis, including CHILD syndrome (Porter and Herman, 2011). Blockade of metabolite production alone, however, is also insufficient. Indeed, normal mouse skin treated with topical statins develops ichthyosiform changes (Feingold et al., 1991; Menon et al., 1992a), reflecting the key role of cholesterol in the epidermal barrier (Elias, 2006; Feingold et al., 1990). Statin-induced cholesterol deficiency leads to decreased number and internal contents of epidermal lamellar bodies, as well as altered lamellar bilayer architecture, but no evidence of cytotoxicity (Feingold et al., 1990; Menon et al., 1992a). In contrast, lesional skin from both CHILD patients also display much more severe alterations in the lamellar body secretory system, indicative of metabolite-induced toxicity. In fact, the mild ultrastructural changes in clinically “unaffected” skin suggest cholesterol deficiency, not of accumulation of sterol precursors, and implicate toxic metabolites in causing the ichthyosiform changes. Accumulation of 4-methyl and 4,4-dimethyl sterol metabolites has been shown in tissue samples from heterozygous mutant female Bpa/Str mouse models of CHILD syndrome (Liu et al., 1999), although near-normal levels of cholesterol and only minimal elevations in sterol metabolites are found in serum in these mice and in individuals with CHILD syndrome (Hummel et al., 2003; Porter and Herman, 2011). This discrepancy likely reflects the functional mosaicism and skewed X-inactivation with widespread expression of the normal allele outside of affected sites. Insertion of sterol metabolites into cell membranes is known to induce membrane functional abnormalities, as well as toxic effects on oxysterol generation, altered hedgehog pathway signalling, and accelerated degradation of HMG CoA reductase (Porter and Herman, 2011). The lack of hair growth in affected areas, despite skin normalization with lovastain/cholesterol treatment, provides evidence of the important role of cholesterol and hedgehog pathway signaling in hair follicle development (Chiang et al., 1999; Cunningham et al., 2009; St-Jacques et al., 1998).
Our pathway-based therapeutic approach represents an entirely new concept for the topical therapy of disorders of distal cholesterol metabolism. Because it is pathway- rather than mutation-specific, it has broad applicability within this pathway. Cholesterol and lovastatin are generic (i.e., relatively inexpensive); have a known safety profile; and are lipid-soluble, facilitating penetration across the stratum corneum. The cholesterol/statin combination would likely be useful for another, closely-related disorder in this pathway (Fig. 1), Conradi-Hünermann-Happle syndrome (CDPX2), which similarly presents with bone malformations and patterned ichthyotic skin lesions, but without lateralization (Akiyama et al., 2009; Steijlen et al., 2007). In fact, topical application of an end-product plus an upstream pathway inhibitor could well be useful for the many syndromic and non-syndromic disorders of fatty acid and ceramide metabolism. Metabolite-induced toxicity (with variable levels of pathway product depletion) has been described in several other syndromic, lipid metabolic disorders leading to ichthyotic manifestations, including the relatively common recessive X-linked ichthyosis, SjÖgren-Larsson syndrome, neutral lipid storage disease (Chanarin-Dorfman syndrome), type II (acute infantile neuronopathic) Gaucher disease, and Refsum disease (rev in (Elias et al., 2010; Elias et al., 2008). In theory, a comparable transdermal approach might also benefit the extracutaneous manifestations of these syndromic disorders, since the topical route would allow both the statin and cholesterol to bypass hepatic first-pass metabolism.
We also addressed the distribution of CHILD syndrome lesions through ultrastructural analysis and gene inactivation studies. Given that the NSHDL gene resides on Xq28, its expression is subject to X-inactivation. Sequencing of genomic DNA from cultured keratinocytes of the “unaffected” and “affected” sides both showed approximately equal amounts of mutant and normal gene. However, sequencing of expressed DNA from keratinocytes from lesional skin showed mutant NSDHL, whereas from keratinocytes of clinically normal contralateral skin revealed only the wild type NSDHL. We speculate that the mild abnormalities seen in ultrastructural analysis of ‘uninvolved skin’, in which cultured keratinocytes only express the normal allele, could reflect a residual population of keratinocytes expressing the mutant allele that is below our limit of detection by sequencing (i.e., <5%). Interestingly, fibroblasts from the clinically affected skin demonstrated both mutant and wild type NSDHL sequences, and fibroblasts from the clinically-normal side expressed only the normal genotype, accounting for the concordance of the clinical and genotypic expression. The exclusive expression of the normal allele in keratinocytes and fibroblasts from the unaffected side explains the favorable outcome of full-thickness grafts from the unaffected to the affected side (König, et al., 2010). Indeed, our patient #1 had a full-thickness flap from the unaffected to the affected side at the midline of the abdomen performed during her procedure to debulk the verruciform xanthomatosis; this flapped skin was clinically normal at 6 months after surgery when the topical lovastatin/cholesterol therapy was initiated and remains clinically normal.
While culture of keratinocytes can offer a potential selective growth advantage of lesional or wild type cells, the strictly mutant or wild type sequence, respectively, in keratinocytes of lesional vs. clinically normal skin provides evidence that neither predominates in culture. Although one might consider segregation of affected keratinocytes as a mechanism for the observed expression patterns, exclusive expression of the wild type allele in both keratinocytes and fibroblasts on the unaffected side instead suggests preferential clearance of affected cells. Similarly, the skewing of X-inactivation in peripheral blood leukocytes is consistent with overall depletion of affected cells (Sun and Tsao, 2008). Preferential clearance of mutant cells postnatally has recently been described in the liver and brain of the Bpa mouse model of CHILD syndrome (Cunningham et al., 2009), although it should be noted that the mouse model does not show the lateralization of human CHILD syndrome. Although the mechanism of this preferential clearance and resultant lateralization remains unknown, its elucidation could increase our understanding of spatial organization during embryogenesis.
MATERIALS & METHODS
Patient Recruitment, Clinical Assessment and Genetic Analysis
Following institutional approval of experiments, written informed patient consent (and in the case of Patient 1 parental consent) and adherence to the Helsinki Guidelines, blood was drawn from both patients for genetic analysis to identify the NSHDL mutation. In addition, baseline photographs and biopsies of lesional and contralateral nonlesional skin were obtained for routine histology and electron microscopy. At both sites 3 physicians (3 dermatologists in the Canary Islands and a dermatologist and two pediatricians in the U.S.) each saw the patients at each visit. All independently judged the progress on a global basis, rating erythema, scaling and thickening as 0 to 4+. For mutation analysis, DNA was extracted from peripheral blood leukocytes by salt precipitation. All coding exons (from 3 to 9), including splice sites, were amplified by PCR (M13-tailed primer sequences and PCR protocol on request). PCR products were sequenced with universal M13-F and M13-R primers using the BigDye terminator v1.1 cycle sequencing kit (Applied Biosystems, Foster City, CA, USA). The mutation nucleotide position is based on NCBI-reference sequence BC007816.
Treatment Protocol
Prior to application, only bland emollients were applied; these were stopped for the trial and have not been restarted. A lovastatin 2%/cholesterol 2% suspension in paraben-preserved water was applied to the lesions of both patients twice daily for the first 6 months (total applications limited to 30 cc monthly), and then tapered to maintain control. The compounded lotion was made with an identical formulation at both sites by grinding up lovastatin tablets (5 g), combining with cholesterol NF powder 5g, and mixing for at least 3 mins with an electronic mortar and pestle to bring to full 240 cc in preserved water (www.markdrugs.com). Patients were observed at 6–8 weeks, 3 months, and then at 3-month intervals for clinical changes as well as potential adverse effects.
Assessment of Ultrastructural Pathology
After aldehyde pre-fixation, skin biopsies were processed for H&E staining, or post-fixed in either osmium tetroxide or ruthenium tetroxide (to visualize lamellar bilayer architecture), and embedded in Epoxy resin (Hou et al., 1991). An additional piece was processed for ultrastructural cytochemistry utilizing the lipid secretory marker, acidic lipase (Menon et al., 1992b). Ultrathin sections were examined in a Zeiss electron microscope, operated at 60 KV.
Investigation of Gene Inactivation
Skin samples for keratinocyte and fibroblast cultures were obtained from lesional (left inguinal) and nonlesional (right inguinal) sites of Patient #1. Epidermis and dermis were separated after overnight incubation in Dispase® at 4°C. Keratinocyte and fibroblast suspensions, obtained by trypsinization, were cultured (Rheinwald and Green, 1975). Genomic DNA extracted from affected and unaffected keratinocytes was analyzed for the c.248G>A mutation using the NSDHL exon 4 genomic PCR primers as described above. Total RNA was isolated and purified from the cultured cells with an RNeasy Kit (Qiagen, Valencia, CA, USA), and used to synthesize cDNA using a SuperScript® III First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA). In brief, 1.5-2 μg of total RNA was mixed with random primers and dNTPs, with incubation for 5 min at 65°C, followed by cooling. The cDNA was synthesized by adding SuperScript and incubation at 50°C for 50 min, followed by 5 min incubation at 85°C. After removal of the remaining RNA with RNase H, 10–200 ng of the cDNA was used for PCR reactions. RT-PCR was performed on the cDNA using primers in exon 4 and 5 flanking the mutation (NSDHL4cF 5’-TGGATTCCTGGGGCAGCAC-3’ and NSDHL5cR 5’-TTACTGGATGGTGGGGGTGAC-3’, PCR-protocol on request). PCR products were sequenced directly using the PCR primers with the BigDye terminator v1.1 cycle sequencing kit (Applied Biosystem). For analysis of X-inactivation, the polymorphic CAG-repeat in exon 1 of the AR gene was PCR-amplified after digestion with the methylation-sensitive enzymes, HhaI and HpaII (Fearon et al., 1987).
Acknowledgments
This work was supported by the National Institutes of Health Grants R01 AR44619 (AP) and R01 AR19098 (PE) and the Dutch Cancer Society (KWF) Grant KWF-UM2009-4352 (MvS). This research was supported in part by resources provided by the Northwestern University Skin Disease Research Center (5P30AR057216-02), Chicago, IL with support from the NIH/NIAMS (cultured patient keratinocytes and fibroblasts). We thank Drs. Bruce Bauer and Xiaoqi Wang for their assistance in procuring tissue/performing the flap and processing keratinocyte cDNA, respectively. We acknowledge Joan Wakefield and Nilda Reyes for providing superb editorial assistance.
Abbreviations
- CHILD
Congenital hemidysplasia, Ichthyosis and Limb Defects
- EBP
emopamil binding protein
- NSDHL
NAD(P)H steroid dehydrogenase-like
- SC
stratum corneum
- SG
stratum granulosum
Footnotes
Conflict of Interest
The authors state no conflict of interest.
References
- Akiyama M, Sakai K, Hayasaka K, et al. Conradi-Hunermann-Happle syndrome with abnormal lamellar granule contents. Br J Dermatol. 2009;160:1335–7. doi: 10.1111/j.1365-2133.2009.09110.x. [DOI] [PubMed] [Google Scholar]
- Chan YM, Merkens LS, Connor WE, et al. Effects of dietary cholesterol and simvastatin on cholesterol synthesis in Smith-Lemli-Opitz syndrome. Pediatr Res. 2009;65:681–5. doi: 10.1203/PDR.0b013e31819ea4eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Swan RZ, Grachtchouk M, et al. Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev Biol. 1999;205:1–9. doi: 10.1006/dbio.1998.9103. [DOI] [PubMed] [Google Scholar]
- Cooper MK, Wassif CA, Krakowiak PA, et al. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet. 2003;33:508–13. doi: 10.1038/ng1134. [DOI] [PubMed] [Google Scholar]
- Cunningham D, Spychala K, McLarren KW, et al. Developmental expression pattern of the cholesterogenic enzyme NSDHL and negative selection of NSDHL-deficient cells in the heterozygous Bpa(1H)/+ mouse. Mol Genet Metab. 2009;98:356–66. doi: 10.1016/j.ymgme.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias P, Williams M, Crumrine D, et al. Ichthyoses - clinical, biochemical, pathogenic, and diagnostic assessment. Vol. 39. S. Kargar AG; Basel: 2010. p. 144. [Google Scholar]
- Elias P, Williams M, Crumrine D, Schmuth M. Ichthyoses - Clinical, Biochemical, Pathogenic, and Diagnostic Assessment. Vol. 39. Karger; 2010. [Google Scholar]
- Elias PM. Defensive functions of the stratum corneum: integrative aspects. In: Elias PM, Feingold KR, editors. Skin Barrier. New York: Taylor & Francis; 2006. pp. 5–14. [Google Scholar]
- Elias PM, Williams ML, Holleran WM, et al. Pathogenesis of permeability barrier abnormalities in the ichthyoses: inherited disorders of lipid metabolism. J Lipid Res. 2008;49:697–714. doi: 10.1194/jlr.R800002-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon ER, Winkelstein JA, Civin CI, et al. Carrier detection in X-linked agammaglobulinemia by analysis of X-chromosome inactivation. N Engl J Med. 1987;316:427–31. doi: 10.1056/NEJM198702193160802. [DOI] [PubMed] [Google Scholar]
- Feingold KR, Man MQ, Menon GK, et al. Cholesterol synthesis is required for cutaneous barrier function in mice. J Clin Invest. 1990;86:1738–45. doi: 10.1172/JCI114899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold KR, Man MQ, Proksch E, et al. The lovastatin-treated rodent: a new model of barrier disruption and epidermal hyperplasia. J Invest Dermatol. 1991;96:201–9. doi: 10.1111/1523-1747.ep12461153. [DOI] [PubMed] [Google Scholar]
- Grange DK, Kratz LE, Braverman NE, et al. CHILD syndrome caused by deficiency of 3beta-hydroxysteroid-delta8, delta7-isomerase. Am J Med Genet. 2000;90:328–35. doi: 10.1002/(sici)1096-8628(20000214)90:4<328::aid-ajmg13>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Haas D, Kelley RI, Hoffmann GF. Inherited disorders of cholesterol biosynthesis. Neuropediatrics. 2001;32:113–22. doi: 10.1055/s-2001-16618. [DOI] [PubMed] [Google Scholar]
- Happle R. X-chromosome inactivation: role in skin disease expression. Acta Paediatr Suppl. 2006;95:16–23. doi: 10.1111/j.1651-2227.2006.tb02384.x. [DOI] [PubMed] [Google Scholar]
- Holleran WM, Ginns EI, Menon GK, et al. Consequences of beta-glucocerebrosidase deficiency in epidermis. Ultrastructure and permeability barrier alterations in Gaucher disease. J Clin Invest. 1994;93:1756–64. doi: 10.1172/JCI117160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou SY, Mitra AK, White SH, et al. Membrane structures in normal and essential fatty acid-deficient stratum corneum: characterization by ruthenium tetroxide staining and x-ray diffraction. J Invest Dermatol. 1991;96:215–23. doi: 10.1111/1523-1747.ep12461361. [DOI] [PubMed] [Google Scholar]
- Hummel M, Cunningham D, Mullett CJ, et al. Left-sided CHILD syndrome caused by a nonsense mutation in the NSDHL gene. Am J Med Genet A. 2003;122A:246–51. doi: 10.1002/ajmg.a.20248. [DOI] [PubMed] [Google Scholar]
- Kelley RI, Herman GE. Inborn errors of sterol biosynthesis. Annu Rev Genomics Hum Genet. 2001;2:299–341. doi: 10.1146/annurev.genom.2.1.299. [DOI] [PubMed] [Google Scholar]
- König A, Skrzypek J, Löffler H, et al. Donor dominance cures CHILD nevus. Dermatology. 2010;220:340–5. doi: 10.1159/000287253. [DOI] [PubMed] [Google Scholar]
- Liu XY, Dangel AW, Kelley RI, et al. The gene mutated in bare patches and striated mice encodes a novel 3beta-hydroxysteroid dehydrogenase. Nat Genet. 1999;22:182–7. doi: 10.1038/9700. [DOI] [PubMed] [Google Scholar]
- Man MQ, Feingold KR, Elias PM. Exogenous lipids influence permeability barrier recovery in acetone-treated murine skin. Arch Dermatol. 1993;129:728–38. [PubMed] [Google Scholar]
- Mao-Qiang M, Brown BE, Wu-Pong S, et al. Exogenous nonphysiologic vs physiologic lipids. Divergent mechanisms for correction of permeability barrier dysfunction. Arch Dermatol. 1995;131:809–16. doi: 10.1001/archderm.131.7.809. [DOI] [PubMed] [Google Scholar]
- Menon GK, Feingold KR, Mao-Qiang M, et al. Structural basis for the barrier abnormality following inhibition of HMG CoA reductase in murine epidermis. J Invest Dermatol. 1992a;98:209–19. doi: 10.1111/1523-1747.ep12555880. [DOI] [PubMed] [Google Scholar]
- Menon GK, Ghadially R, Williams ML, et al. Lamellar bodies as delivery systems of hydrolytic enzymes: implications for normal and abnormal desquamation. Br J Dermatol. 1992b;126:337–45. doi: 10.1111/j.1365-2133.1992.tb00675.x. [DOI] [PubMed] [Google Scholar]
- Moskowitz DG, Fowler AJ, Heyman MB, et al. Pathophysiologic basis for growth failure in children with ichthyosis: an evaluation of cutaneous ultrastructure, epidermal permeability barrier function, and energy expenditure. J Pediatr. 2004;145:82–92. doi: 10.1016/j.jpeds.2004.03.052. [DOI] [PubMed] [Google Scholar]
- Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Soreze 2009. J Am Acad Dermatol. 2010;63:607–41. doi: 10.1016/j.jaad.2009.11.020. [DOI] [PubMed] [Google Scholar]
- Ponec M, te Pas MF, Havekes L, et al. LDL receptors in keratinocytes. J Invest Dermatol. 1992;98:50S–6S. doi: 10.1111/1523-1747.ep12462204. [DOI] [PubMed] [Google Scholar]
- Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res. 2011;52:6–34. doi: 10.1194/jlr.R009548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975;6:331–43. doi: 10.1016/s0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- Rizzo WB, S'Aulis D, Jennings MA, et al. Ichthyosis in Sjogren-Larsson syndrome reflects defective barrier function due to abnormal lamellar body structure and secretion. Arch Dermatol Res. 2010;302:443–51. doi: 10.1007/s00403-009-1022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmuth M, Gruber R, PM E, et al. Ichthyosis update: towards a function-driven model of pathogenesis of the disorders of cornification and the role of corneocyte proteins in these disorders. Adv Dermatol. 2007;23:231–56. doi: 10.1016/j.yadr.2007.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Jacques B, Dassule HR, Karavanova I, et al. Sonic hedgehog signaling is essential for hair development. Curr Biol. 1998;8:1058–68. doi: 10.1016/s0960-9822(98)70443-9. [DOI] [PubMed] [Google Scholar]
- Steijlen PM, van Geel M, Vreeburg M, et al. Novel EBP gene mutations in Conradi-Hunermann-Happle syndrome. Br J Dermatol. 2007;157:1225–9. doi: 10.1111/j.1365-2133.2007.08254.x. [DOI] [PubMed] [Google Scholar]
- Sun BK, Tsao H. X-chromosome inactivation and skin disease. J Invest Dermatol. 2008;128:2753–9. doi: 10.1038/jid.2008.145. [DOI] [PubMed] [Google Scholar]
- Szabo GP, Olah AV, Kozak L, et al. A patient with Smith-Lemli-Opitz syndrome: novel mutation of the DHCR7 gene and effects of therapy with simvastatin and cholesterol supplement. Eur J Pediatr. 2010;169:121–3. doi: 10.1007/s00431-009-0987-z. [DOI] [PubMed] [Google Scholar]
- Zettersten E, Man MQ, Sato J, et al. Recessive x-linked ichthyosis: role of cholesterol-sulfate accumulation in the barrier abnormality. J Invest Dermatol. 1998;111:784–90. doi: 10.1046/j.1523-1747.1998.00386.x. [DOI] [PubMed] [Google Scholar]