

Abstract
Background:
Macroalgae can be viewed as a potential antioxidant and anti-inflammatory sources owing to their capability of producing compounds for its protection from environmental factors such as heat, pollution, stress, oxygen concentration, and UV radiations.
Objective:
To isolate major compounds which are mainly responsible for the pharmacological activity of brown alga under investigation, Sargassum sp.
Materials and Methods:
Algal material was air dried, extracted with a mixture of organic solvents, and fractionated with different adsorbents. The structures of obtained pure compounds were elucidated with different spectroscopic techniques, and two pure materials were tested for protection of DNA from damage, antioxidant, antitumor, and cytotoxicity.
Results:
Four pure compounds were obtained, of which fucosterol (1) and fucoxanthin (4) were tested; it was found that fucoxanthin has strong antioxidant and cytotoxicity against breast cancer (MCF-7) with IC50 = 11.5 μg/ml.
Conclusion:
The naturally highly conjugated safe compound fucoxanthin could be used as antioxidant and as an antitumor compound.
Keywords: Antioxidant, cytotoxicity, fucoxanthin, Sargassum sp
INTRODUCTION
The genus Sargassum belongs to brown algae of class Phaeophyceae in the order Fucales. Numerous species are distributed throughout the tropical and subtropical oceans of the world, where they generally inhabit shallow water and coral reefs. However, the genus may be well known for its planktonic (free-floating) species.[1] It was clear from the literatures that more than 62 species of the genus Sargassum has been investigated,[2] and indicated its productivity with impressive diversity of natural compounds. For instances, the secondary metabolites contain different structural classes such as plastoquinones,[3–5] chromanols,[6] chromenes,[7,8] steroids,[9] and glycerides.[10] The publications showed that, Sargassum metabolites have wide range of biological activities, which include antibiotic, anti-HIV, anticoagulant, anticonvulsant, anti-inflammatory, antineoplastic, and antitumor.[11–14]
In continuation of our search program, which interested in the isolation of secondary metabolites from marine sources, especially macroalgae collected from Red Sea,[15–17] a marine macroalgae, identified as Sargassum sp., collected from El- Shuaiba lagoon 80 km south of Jeddah, was investigated. The total extract (Pet. ether: Chloroform: Methanol [1 : 1 : 1]) had been fractionated using different chromatographic techniques and afforded four metabolites; fucosterol (1), saringosterone (2), saringosterol (3), and fucoxanthin (4). 1 and 4 had been tested toward the Bleomycin-dependent DNA damage, cytotoxicity against HepG2 (human hepatocellular liver carcinoma cell line), WI 38 (Skin carcinoma cell line), Vero (cell line was initiated from the kidney of a normal adult African green monkey), MCF-7 (breast cancer cell lines), antitumor, and antioxidant using 2,2‘-azino-bis-3-ethylbenzthiazoline-6-sulfonic acid (ABTS).
MATERIALS AND METHODS
Apparatus and material
Chromatographic material: Aluminum oxide type 60-120 mesh was used for column chromatography. Thin-layer chromatography (TLC) silica gel GF 254 was used for TLC. Preparative thin-layer chromatography (PTLC) was performed on aluminum oxide plates (20 × 20 cm) of 250-mm thickness. Electron impact mass spectra were determined at 70 eV on a Kratos MS-25 instrument. 1D and 2D NMR (Nuclear Magnetic Resonance) spectra were recorded on Bruker AVANCE III WM 600 MHz spectrometers and 13C NMR at 150 MHz Chemical shifts are given in δ (ppm) relative to TMS (Tetramethyl silane) as internal standard. The Infra Red (IR) spectra were recorded on a Perkin Elmer spectrometer model 100. The spray reagent used is 50%-sulfuric acid in methanol as spraying reagent. The chromatoplate was heated after spraying at 100 to 105°C until the spots attained maximum color intensity. The alga was described as Sargassum (Family Sargassaceae) and was collected by hand from El- Shuaiba lagoon 80 km south of Jeddah, Saudi Arabia, in the Red Sea, in June 2010.
Extraction of Sargassum sp
The air-dried algal material (350 g) was extracted by equal volume of mixture of Pet. ether, chloroform, and methanol (2 × 6 l, 24 hours for each batch) at room temperature. The extract was concentrated under reduced pressure to obtain 10 g residue. This material was chromatographed on a column of Silica gel.
Column chromatography
Silica gel column (500 g, 80 × 2.5 cm) was used for resolution. The residue (10 g) was homogenized with small amount of silica gel (50 g) and poured on to the top of the column which was packed in Pet. ether (40: 60). The eluent were used successively (Pet. ether- Ether, Pet. ether- Ethyl acetate). Fractions were collected (50 ml); Pet. ether- Ether mixture of increasing polarity by diethyl ether and then ethyl acetate and followed by TLC using silica-gel chromatoplates, appropriate solvent system and 50% sulfuric acid in methanol as spraying reagent. If the material was not pure, preparative TLC was applied using the appropriate solvent system and the adsorbent aluminum oxide for purification.
Isolated compounds
The fraction A eluted by Pet. ether-Ether (6 : 4) was collected and rechromatographed over PTLC of silica gel using Pet. Ether-ethyl acetate (8 : 2) to give three compounds: 1 , 2, and 3. The fraction B eluted by Pet. ether-Ethyl acetate (6 : 4) was collected and rechromatographed on Sephadex LH-20 using a mixture of MeOH-CHCl3 (9 : 1) and then finally purified by preparative TLC of silica gel using chloroform methanol (9.5 : 0.5) to afford a pure compound 4.
Fucosterol (1), (24E)-Stigmasta-5, 24(28)-diene-3β-ol: white solid (30 mg, 0.008% dry wt) m.p. 133-135°C. IR (cm-1): 3480 (OH), 1945 (C = C-H), 1640 (C = C), 1370 (gem. Di-Me); EI-MS m/z: 412 [M, C29H48O] + , 397 [M-CH3]+, 379 [M-CH3-H2O]+, 314 (100) [M-C6H10O]+. 1H NMR (CDCl3) δ (ppm) 0.86- 0.88 (d, J = 6.6 Hz, 6H, Me-26 and Me-27), 0.91 (d, J = 6.6 Hz, 3H, Me-21), 0.68 (s, 3H, Me-18), 1.00 (s, 3H, Me-19), 1.59 (d, J = 6.6 Hz, 3H, Me-29), 3.53 (m, H, H-3), 5.35 (m, H, H-6), 5.11 (q, H, H-28), 2.22 (sep, J = 6.6 Hz, H, H-25). 13C NMR (CDCl3) δ (ppm) (39.75, C-1), (35.20, C-2), (71.80, C-3), (42.33, C-4), (140.74, C-5), (121.70, C-6), (36.14, C-7), (35.78, C-8), (50.10, C-9), (39.72, C-10), (28.23, C-11), (42.28, C-12), (42.30, C-13), (56.74, C-14), (31.64, C-15), (34.78, C-16), (56.72, C-17), (11.85, C-18), (19.40, C-19), (39.50, C-20), (24.32, C-21), (37.23, C-22), (31.90, C-23), (146.00, C-24), (33.90, C-25), (22.50, C-26), (22.23, C-27), (115.94, C-28), (18.90, C-29).
Saringosterone (2), 24-vinyl cholest -4-ene -3-one: a colorless oil (10 mg, 0.003% dry wt). IR (cm-1): 3420 (OH), 1675 (C = O), 1630 (C = C); EIMS m/z (rel. int.): 426 (12) [M, C29H46O2]+, 383 (21) [M+-C3H7], 313 (30), 271(35), 269 (100). HREIMS: m/z 426.3413 (calcd. 426.3349) C29H46O2; 1H-NMR (CDCl3) δ 5.76 (dd, J = 17, 12 Hz, H, H-28), 5.73 (br s, H, H-4), 5.27 (d, J = 12 Hz, H, H-29), 5.15 (d, J = 17 Hz, H, H-29), 1.16 (s, 3H, Me-19), 0.95 (d, J = 6.5 Hz, 3H, Me-21), 0.86 (d, J = 7.0 Hz, 3H, Me-26), 0.84 (d, J = 7.0 Hz, 3H, Me-27), 0.72 (s, 3H, Me-18). 13C-NMR δ (36.54, C-1), (34.65, C-2), (200.35, C-3), (124.43, C-4), (172.35, C-5), (33.64, C-6), (32.65, C-7), (36.32, C-8), (54.41, C-9), (39.25, C-10), (21.65, C-11), (40.22, C-12), (43.08, C-13), (56.26, C-14), (24.85, C-15), (28.86, C-16), (56.55, C-17), (12.63, C-18), (17.34, C-19), (36.84, C-20), (18.35, C-21), (32.54, C-22), (28.95, C-23), (89.76, C-24), (29.05, C-25), (19.46, C-26), (18.07, C-27), (137.78, C-28), (117.12, C-29).
Saringosterol (3), 24-vinyl cholest-5-ene-3β, 24-diol, saringosterol: colorless oil (5 mg, 0.001% dry wt.). IR(cm-1): 3445 (OH), 1644 (C=C); EIMS m/z (rel. int.): 428 (12) [M, C29H48O2]+, 410 (6) [M+-H 2O], 314 (40), 273(20), 271 (100), 255 (28), 228 (22), 213 (40), 145 (64). 1H NMR (CDCl3) d 5.73 (dd, J=17, 12 Hz, H, H-28), 5.34 (br s, H, H-6), 5.28 (d, J = 12 Hz, H, H-29), 5.17 (d, J = 17 Hz, H, H-29), 3.53 (m, H, H-3), 1.02 (s, 3H, Me-19), 0.97 (d, J = 6.6 Hz, 3H, Me-21), 0.88 (d, J = 6.6 Hz, 3H, Me-26], 0.86 (d, J = 6.6 Hz, 3H, Me-27), 0.67 (s, 3H, Me-18], 13C-NMR d (37.17,C-1), (32.54, C-2), (72.43, C-3), (37.94, C-4), (141.43, C-5), (122.31, C-6), (32.35, C-7), (36.84, C-8), (50.74, C-9), (37.93, C-10), (21.75, C-11), (40.41, C-12), (42.97, C-13), (57.43, C-14), (24.95, C-15), (29.06, C-16), (56.55, C-17), (12.54, C-18), (17.32, C-19), (36.54, C-20), (18.35, C-21), (32.33, C-22), (28.95, C-23), (89.85, C-24), (29.19, C-25), (20.06, C-26), (19.53, C-27), (137.82, C-28), (117.04, C-29).
Fucoxanthin (4): red residue (60 mg, 0.017% dry wt.). IR (cm-1): 3439 (OH), 2361 and 2332 (sp-hybrid carbon [allenic]), 1721 and 1253 (ester), and 1654 and 1605 (polyene); HRFAB: 658.4224 (calcd. 658.4233) [C42H58O6], 460(5), 391(5), 307(50), 154(100), 1H-NMR (CDCl3) d 7.15(d, J = 11.0 Hz, H, H-10), 6.75 (m, H, H-15), 6.66 (d, J = 15.0 Hz, H, H-12), 6.63 (m, H, H-15’), 6.60 (m, H, H-11’), 6.57 (m, H, H-11), 6.41 (d, J = 11.5 Hz, H, H-14), 6.35 (d, J = 15.0 Hz, H, H-12’), 6.27 (d, J = 11.5 Hz, H, H-14’), 6.13 (d, J = 11.0 Hz, H, H-10’), 6.05(br s, H, H-8’), 5.38 (m, H, H-3’), 3.81(m, H, H-3), 3.65 and 2.60 (qAB, J = 18.5 Hz, 2H, H-7), 2.33 and 1.78 (m, 2H, H-4), 2.27 and 1.51 (m, 2H, H-4’), 2.04 (s, 3H, Ac), 2.01 and 1.41(m, 2H, H-2’), 1.99 (s, 6H, H-20, H-20’), 1.95 (s, 3H, H-19), 1.81 (s, 3H, H-19’), 1.47 and 1.37 (m, 2H, H-2), 1.38 (3H, s, H-18’), 1.35 (s, 3H, H-17’), 1.22(s, 3H, H-18), 1.07 (s, 3H, H-16’), 1.03 (s, 3H, H-17), 0.96 (s, 3H, H-16). 13C-NMR (CDCl 3) δ (35.18, C-1), (47.11, C-2), (64.34, C-3), (41.68, C-4), (66.19, C-5), (67.11, C-6), (40.83, C-7), (197.87, C-8), (134.54, C-9), (139.14, C-10), (123.39, C-11), (145.05, C-12), (136.67, C-13), (135.44, C-14), (132.19, C-15), (28.16, C-16), (25.06, C-17), (21.46, C-18), (11.85, C-19), (12.80, C-20), (35.79, C-1’), (45.45, C-2’), (68.03, C-3’), (45.25, C-4’), (72.71, C-5’), (117.52, C-6’), 202.36, C-7’), (103.39, C-8’), (132.19, C-9’), (128.54, C-10’), (125.71, C-11’), (138.09, C-12’), (137.11, C-13’), (132.52, C-14’), (129.44, C-15’), (31.31, C-16’), (32.14, C-17’), (29.22, C-18’), (14.04, C-19’), (12.94, C-20’), (170.45, Ac-1), (21.18, Ac-2).
Biological evaluation of the isolated compounds
DNA (Calf Thymus type1), bleomycin sulfate, butylated hydroxyanisole, thiobarbituric acid (TBA), ethylenediaminetetraacetic acid (EDTA), and ascorbic acid were obtained from sigma. 2,2’-azo-bis-(2-amidinopropane) dihydrochloride and ABTS were purchased from Wako Co., USA.
Cell viability and Bleomycin-dependent DNA damage assay in negative control (Fucoxanthin on MCF-7)
The reaction mixture contained DNA (0.5 mg/ml), bleomycin sulfate (0.05 mg/ml), MgCl2 (5 mM), FeCl3 (50 mM), and samples to be tested in a conc. of 0.1 mg/ml. L-ascorbic acid was used as positive control. The mixture was incubated at 37°C for 1 hour. The reaction was terminated by addition of 0.05 ml EDTA (0.1 M). The color was developed by adding 0.5 ml TBA (1% w/v) and 0.5 ml HCl (25% v/v), followed by heating at 80°C for 10 minutes. After centrifugation, the extent of DNA damage was measured by increase in absorbance at 532 nm.[18,19] The viability of the cells was determined by the microscopical examination[20] using a hemocytometer and using trypan blue stain (stains only the dead cells).
Antioxidant activity screening assay 2,2’-azino-bis-3-ethylbenzthiazoline-6-sulfonic acid method
For each of the investigated compounds, 2 ml of ABTS solution (60 μM) was added to 3 ml MnO2 solution (25 mg/ml), all prepared in 5 ml aqueous phosphate buffer solution (pH, 7; 0.1 M). The mixture was shaken, centrifuged, filtered, and the absorbance of the resulting green-blue solution (ABTS radical solution) at λ 734 nm was adjusted to approximately ca. 0.5. Then, 50μl of (2 mM) solution of the tested compound in spectroscopic grade MeOH/phosphate buffer (1: 1) was added. The absorbance was measured and the reduction in color intensity was expressed as inhibition percentage. L-ascorbic acid was used as standard antioxidant (positive control). Blank sample was run without ABTS and using MeOH/phosphate buffer (1: 1) instead of tested compounds. Negative control was run with ABTS and MeOH/phosphate buffer (1: 1) only.[21]
Cytotoxicity and antitumor assay
Samples were prepared for assay by dissolving in 50 μl of DMSO (Dimethyl Sulfoxide), and diluting aliquots into sterile culture medium at 0.4 mg/ml. These solutions were sub-diluted to 0.02 mg/ml in sterile medium and the two solutions were used as stocks to test samples at 100, 50, 20, 10, 5, 2, and 1 mg/ml in triplicate in the wells of microtiter plates. The compounds were assayed in triplicate on monolayers grown in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) calf serum (HyClone Laboratories, Ogden, UT), 60 mg/ml Penicillin G, and 100 mg/ml streptomycin sulfate maintained at 37°C in a humidified atmosphere containing about 15% (v/v) CO 2 in air. All medium components were obtained from Sigma Chemical Co., St. Louis, MO, unless otherwise indicated. Cell stocks were maintained at 34°C in culture flasks filled with medium supplemented with 1% (v/v) calf serum. Subcultures of cells for screening were grown in the wells of microtiter trays (Falcon Microtest III 96-wells trays, Becton Dickinson Labware, Lincolin Park, NJ) by suspending cells in medium following trypsin-EDTA treatment, counting the suspension with a hemocytometer, diluting in medium containing 10% calf serum to 2 × 104 cells per 200 ml culture, aliquoting into each well of a tray, and culturing until confluent. Microtiter trays with confluent monolayer cultures of cells were inverted, the medium shaken out, and replaced with serial dilutions of sterile compounds in triplicate in 100 l medium followed by titered virus in 100 ml medium containing 10% (v/v) calf serum in each well. In each tray, the last row of wells was reserved for controls that were not treated with compounds. Trays were cultured for 96 hours. Trays were inverted onto a paper towel pad, the remaining cells rinsed carefully with medium, and fixed with 3.7% (v/v) formaldehyde in saline for at least 20 minutes. The fixed cells were rinsed with water, and examined visually. The cytotoxic activity is identified as confluent, relatively unaltered monolayers of stained cells treated with the investigated compounds. Cytotoxicity was estimated as the concentration that caused approximately 50% loss of the monolayer. 5-fluorouracil was used as a positive control.[22]
RESULTS AND DISCUSSION
Compound 1 showed in its EI MS a molecular ion at m/z 412, (Liebermann-burchard reaction gave the typical slow reacting green color of a Δ5 sterol) which[23] together with 13C NMR data suggested a molecular formula of C29 H48O. The loss of part of the side chain (C7H14) is characteristic of sterols[23] with a Δ24(28). 1H NMR spectroscopic data were typical of sterols, in which a signal at δ 0.88 (6H, d, J= 6.6 Hz) attributable to the two methyls of an isopropyl group, a signal at δ 0.91 (3H, d, J= 6.6 Hz) and two signals at δ 1.00 (3H, s) and at δ 0.68 (3H, s) assigned to two methyl groups attached to two quaternary carbons are assigned to C-26, C-27, C-21, C-19 and C-18, respectively. In addition, a doublet at δ 1.59 ppm (J= 6.6 Hz) for the proton at C-29 (δc 18.70), a quartet δ 5.18 ppm (J = 6.6 Hz) at C-28 proton (δc 115.54), a multiplet at δ 5.35 for C-6 proton (δc 121.70), and a multiplet at 3.53 for C-3 proton (δc 71.80) were observed. 13C NMR of compound 1 indicated 29 carbons, of which two double bonds and a secondary alcohol (cf exp.) were present. The DEPT spectra showed that compound 1 contains six methyl, ten methylene, nine methane, and four quaternary carbons. The stereochemistry at the side chain double bond can be explained by observing the 1HNMR spectra at the C-25 proton which resonates at δ 2.32 ppm (δc 36.50) in case of (E) isomer (Frost, ward, 1968). The position of the ethylidene group was proved from HMQC, HMBC correlations, and COSY experiment. These data are in agreement with literature[24] which can allow us to assign compound 1 as fucosterol [Figure 1].
Figure 1.
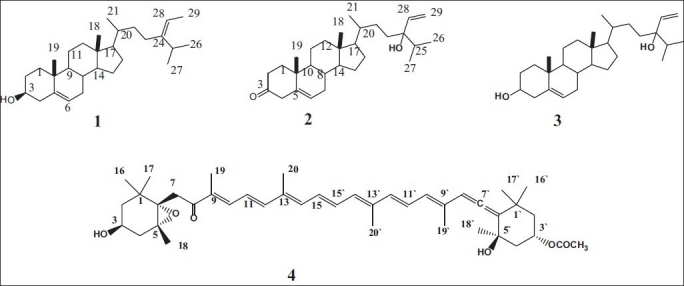
Structure of compounds 1-4
Compound 2 showed in its EI MS a molecular ion at m/z 426, which together with 13C-NMR and HREI MS suggested a molecular formula of C29H46O2 (m/z 426.3413; calcd. 426.3349). The double doublet at δ 5.75 (J = 18, 13 Hz), doublet at 5.29 (J = 13 Hz), and doublet at 5.17 (J = 18 Hz) in the 1H-NMR and signals at δc 137.79 and 117.10 in the 13C NMR were attributable to terminal vinyl protons, three methyl doublets at 0.96, 0.87, 0.85 in the 1H-NMR spectrum. These data, together with the presence of a fragment at m/z 271 in the mass spectrum due to the loss of the side-chain C10H19O, suggest a stigmastane skeleton with unsaturation at C-28. The IR spectrum showed strong bands at 3420 and 1675 cm-1. The above data, together with the presence of signals at δc 200.38, 172.38, 124.42, and 89.75 in the 13C NMR spectrum and a signal at δ 5.74 in the 1H NMR spectrum, suggested the presence of α,β -unsaturated carbonyl group and a tertiary hydroxyl function in the molecule. As the compound contains two double bonds and two oxygen atoms, one as carbonyl and the second as tertiary hydroxyl, also has seven degrees of unsaturation revealed by mass spectrometry, it must be a tetracyclic product. The structure of known compound 2 [Figure 1] was established by comparison of its data (cf exp.) with literature.[25]
Compound 3, its structure was established by comparing their physical and spectral data (cf exp.) with those in the literature[25,26] as 24-vinyl cholest -5-ene -3β, 24-diol.
Compound 4 was isolated as a bright orange solid and the IR showed the presence of the hydroxyl (3439 cm-1), sp-hybrid carbon (allenic) (2361, 2332 cm-1), ester (1721, 1253 cm-1), and polyene (1654, 1605 cm-1) groups. Its molecular formula was deduced as C42H58O6 based on HRFAB-MS analysis [M+], m/z: 658.4224 (Calcd. for C42H58O6, 658. 4233), and NMR spectroscopic data (cf exp.). The 1H and 13C NMR spectra of active compound revealed signals assignable to polyene having acetyl, conjugated ketone, two quaternary geminal dimethyls, two quaternary geminal methyls of oxygen, four olefinic methyls, and allene functionalities. The physicochemical features outlined above suggested that the active compound was a carotenoid in which one of the hydroxyl groups was acetylated. This suggestion was further supported by the UV spectrum [448 (ε560,000), 248 (24,000)]. From detailed comparison of the data for the active compound with those of fucoxanthin [Figure 1], the active compound was in agreement with an authentic fucoxanthin in all aspects.[27–29]
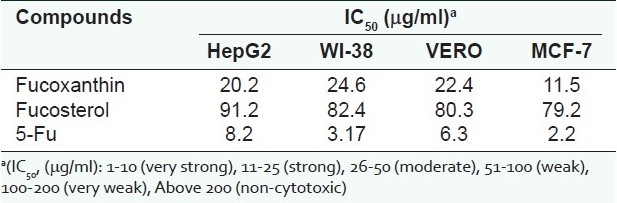
The data obtained from Table 1 indicate that fucoxanthin (4) have some protective activity to DNA by certain mechanism . The isolated compounds were screened for their antitumor activity against MCF-7. The viability of the cells used in control experiments exceeded 95%; compound 4 proved to have cytotoxic activity sustained for 48 hours after adding the compound to MCF-7, as shown in Table 1. Compounds 1 and 4 were tested for antioxidant activity, where fucoxanthin (4) exhibited the highest antioxidant activity by 72% inhibition [Table 2]. On the other hand, compound 1 exhibited a weak to moderate activity.
Table 1.
Cell viability and bleomycin-dependent DNA damage assay of fucoxanthin (4)

Table 2.
Antioxidant activity of tested compounds by ABTS method

Compounds 1 and 4 displayed significant antioxidant and anticancer activities [Table 3] against HepG2 (human hepatocellular liver carcinoma cell line), WI 38 (Skin carcinoma cell line), Vero (cell line was initiated from the kidney of a normal adult African green monkey), and MCF-7 (breast cancer cell lines). Fucoxanthin reduced the viability of MCF-7 cells with IC50 (μg/ml) = 11.5.
Table 3.
Cytotoxicity of tested compounds on different cancer cell lines
ACKNOWLEDGMENT
The authors would like to acknowledge SABIC, the Saudi Arabian Company for Basic Industries, for the financial support of this work (SP-11-2) through the collaboration with the Deanship of Scientific Research (DSR) at King Abdul-Aziz University.
Footnotes
Source of Support: SABIC, the Saudi Arabian Company for Basic Industries, through the collaboration with the Deanship of Scientifi c Research (DSR) at King Abdul-Aziz University
Conflict of Interest: None declared.
REFERENCES
- 1.Blunt JW, Munro MH. Christchurch, New Zealand: Department of Chemistry, University of Canterbury; 2007. Marin Literature Database. [Google Scholar]
- 2.Blunt JW, Copp BR, Hu WP, Munro MH, Northcote PT, Prinsep MR. Marine natural products. Nat Prod Rep. 2008;25:35–94. doi: 10.1039/b701534h. [DOI] [PubMed] [Google Scholar]
- 3.Segawa M, Shirahama H. New Plastoquinones from the Brown Alga Sargassum sagamianum var. yezoense. Chemical Letters. 1987;1:1365–66. [Google Scholar]
- 4.Mori J, Iwashima M, Wakasugi H, Saito H, Matsunaga T, Ogasawara M, et al. New plastoquinones isolated from the brown alga, Sargassum micracanthum. Chem Pharm Bull (Tokyo) 2005;53:1159–63. doi: 10.1248/cpb.53.1159. [DOI] [PubMed] [Google Scholar]
- 5.Ishitsuka M, Kusumi T, Nomura Y, Konno T, Kakisawa H. New geranylgeranylbenzoquinone derivatives from Sargassum tortile. Chemical Letters. 1979;8:1269–72. [Google Scholar]
- 6.Kato T, Kumanireng AS, Ichinose I, Kitahara Y, Kakinuma Y, Kato Y. Structure and synthesis of the active component from a marine alga, Sargassum tortile, which induces the settling of swimming larvae of Coryne uchidai. Chemical Letters. 1975;4:335–8. doi: 10.1007/BF02026361. [DOI] [PubMed] [Google Scholar]
- 7.Jang KH, Lee BH, Choi BW, Lee HS, Shin J. Chromenes from the brown alga Sargassum siliquastrum. J Nat Prod. 2005;68:716–23. doi: 10.1021/np058003i. [DOI] [PubMed] [Google Scholar]
- 8.Kikuchi T, Mori Y, Yokoi T, Nakazawa S, Kuroda H, Masada Y, et al. Structure of sargatriol, a new isoprenoid chromenol from a marine alga, Sargassum tortile. Chem Pharm Bull (Tokyo) 1975;23:690–2. [Google Scholar]
- 9.Tang H, Yi Y, Yao X, Zhou D, Lu T, Jiang Y. Studies on bioactive steroid constituents from Sargassum carpophyllum. Chin Pham J. 2002;37:262–5. [Google Scholar]
- 10.Tang H, Yi Y, Yao X, Zhang S, Zou Z, Li L. Glycerides from marine brown algae Sargassum carpophyllum. Chinese Journal of Marine Drugs. 2002;21:5–9. [Google Scholar]
- 11.Ayyad S-EN, Abdel-Halim OB, Shier WT, Hoye TR. Cytotoxic hydroazulene diterpenes from brown alga Cystoseira myrica. Z Naturforsch C. 2003;58:33–8. doi: 10.1515/znc-2003-1-205. [DOI] [PubMed] [Google Scholar]
- 12.Lincolon RA, Strupinski K, Walker JM. Bioactive compounds from algae. Life Chem Rep. 1991;8:97–183. [Google Scholar]
- 13.Ayyad S-EN, Slama MO, Mokhtar AH, Anter AF. Cytotoxic bicyclic diterpene from the brown alga Sargassum crispum. Boll Chim Farm. 2001;140:155–9. [PubMed] [Google Scholar]
- 14.Banais B, Francisco C, Gonzalez E, Fenical W. Diterpenoid metabolites from the marine alga Cystoseira elegans. Tetrahedron. 1983;39:629–38. [Google Scholar]
- 15.Alarif WM, Ayyad S-EN, Al-lihaibi SS. Acyclic Diterpenoid from the Red Alga Gracilaria Foliifera. Rev Latinoamer Quím. 2010;38:52–8. [Google Scholar]
- 16.Alarif WM, Abou-Elnaga Z Sh, Ayyad S-EN, Al-lihaibi SS. Insecticidal Metabolites from the Green Alga Caulerpa racemosa. CLEAN – Soil, Air and Water. 2010;38:548–57. [Google Scholar]
- 17.Ayyad S-EN, Makki MS, Al-kayal NS, Basaif SA, El-Foty KO, Asiri AM, et al. Cytotoxic and Protective DNA Damage of Three New Diterpenoids from the Brown Alga Dictoyota dichotoma. Eur J Med Chem. 2011;46:175–82. doi: 10.1016/j.ejmech.2010.10.033. [DOI] [PubMed] [Google Scholar]
- 18.Gutterdge J, Rowley D, Halliwell B. Superoxide dependent formation of hydroxyl radicals in the presence of iron salts.Detection of free ion in biological systems using bleomycin-dependent degradation of DNA. Biochem J. 1981;199:263–5. doi: 10.1042/bj1990263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdel-Wahab BF, El-Ahl AA, Badria FA. Synthesis of new 2-naphthyl ethers and their protective activities against DNA damage induced by bleomycin-iron. Chem Pharm Bull (Tokyo) 2009;57:1348–51. doi: 10.1248/cpb.57.1348. [DOI] [PubMed] [Google Scholar]
- 20.Badria F, Ameen M, Akl MR. Evaluation of Cytotoxic Compounds from Calligonum comosum L. growing in Egypt. Z. 2007;62:656–60. doi: 10.1515/znc-2007-9-1005. [DOI] [PubMed] [Google Scholar]
- 21.El-Gazzar AB, Youssef MM, Abu-Hashem AA, Badria FA. Design and synthesis of azolopyrimidoquinolines, pyrimidoquinazolines as anti-oxidant, anti-inflammatory and analgesic activities. Eur J Med Chem. 2009;44:609–24. doi: 10.1016/j.ejmech.2008.03.022. [DOI] [PubMed] [Google Scholar]
- 22.El-Subbagh HI, Abu-Zaid SM, Mahran MA, Badria FA, Al-obaid AM. Synthesis and biological evaluation of certain alpha, beta-unsaturated ketones and their corresponding fused pyridines as antiviral and cytotoxic agents. J Med Chem. 2000;43:2915–21. doi: 10.1021/jm000038m. [DOI] [PubMed] [Google Scholar]
- 23.Gibbons GF, Goad LJ, Goodwin TW. The identification of 28- isofucosterol in the marine green algae Enteromorpha intestinalis and Ulva lactuca. Phytochemistry. 1968;7:983–8. [Google Scholar]
- 24.Goad JL, Akihisa T. 1st ed. London: Blackie Academic and Professional; 1977. Analysis of Sterols; pp. 382–3. [Google Scholar]
- 25.Ayyad S-EN, Sowellim SZ, el-Hosini MS, Abo-Atia A. The structural determination of a new steroidal metabolite from the brown alga Sargassum asperifolium. Z Naturforsch C. 2003;58:333–6. doi: 10.1515/znc-2003-5-607. [DOI] [PubMed] [Google Scholar]
- 26.Tsuda K, Hayatsu R, Kashida Y, Akagi S. Steroid studies IV. Studies on the constitution of sargasterol. J Am Chem Soc. 1958;80:921–5. [Google Scholar]
- 27.Haugan JA, Englert G, Glinz E, Liaaen-Jensen S. Algal carotenoids. 48. Structural assignments of geometrical isomers of fucoxanthin. Acta Chem Scand. 1992;46:389–95. [Google Scholar]
- 28.Yan X, Chuda Y, Suzuki M, Nagata T. Fucoxanthin as the major antioxidant in Hizikia fusiformis, a common edible seaweed. Biosci Biotechnol Biochem. 1999;63:605–7. doi: 10.1271/bbb.63.605. [DOI] [PubMed] [Google Scholar]
- 29.Ayyad S-EN, Diab M. NMR Application of Mosher's Method: The absolute configurations of fucoxanthin from Turbinaria triquatra. Alex J Pharm Sci. 2002;16:27–30. [Google Scholar]