

Abstract
This paper addresses the question of whether one can economically improve the robustness of a molecular phylogeny estimate by increasing gene sampling in only a subset of taxa, without having the analysis invalidated by artifacts arising from large blocks of missing data. Our case study stems from an ongoing effort to resolve poorly understood deeper relationships in the large clade Ditrysia ( > 150,000 species) of the insect order Lepidoptera (butterflies and moths). Seeking to remedy the overall weak support for deeper divergences in an initial study based on five nuclear genes (6.6 kb) in 123 exemplars, we nearly tripled the total gene sample (to 26 genes, 18.4 kb) but only in a third (41) of the taxa. The resulting partially augmented data matrix (45% intentionally missing data) consistently increased bootstrap support for groupings previously identified in the five-gene (nearly) complete matrix, while introducing no contradictory groupings of the kind that missing data have been predicted to produce. Our results add to growing evidence that data sets differing substantially in gene and taxon sampling can often be safely and profitably combined. The strongest overall support for nodes above the family level came from including all nucleotide changes, while partitioning sites into sets undergoing mostly nonsynonymous versus mostly synonymous change. In contrast, support for the deepest node for which any persuasive molecular evidence has yet emerged (78–85% bootstrap) was weak or nonexistent unless synonymous change was entirely excluded, a result plausibly attributed to compositional heterogeneity. This node (Gelechioidea + Apoditrysia), tentatively proposed by previous authors on the basis of four morphological synapomorphies, is the first major subset of ditrysian superfamilies to receive strong statistical support in any phylogenetic study. A “more-genes-only” data set (41 taxa×26 genes) also gave strong signal for a second deep grouping (Macrolepidoptera) that was obscured, but not strongly contradicted, in more taxon-rich analyses.
Keywords: Ditrysia, gene sampling, Hexapoda, Lepidoptera, missing data, molecular phylogenetics, nuclear genes, taxon sampling
Nearly every large molecular phylogenetic study has faced the issue of what to do when the evidence to date fails to provide compelling resolution (as judged, e.g., by bootstrap support) of one or more deeper nodes of special interest. As resources are always limiting, there has been keen interest in the question of what design of additional gene and/or taxon sampling can confidently resolve those additional nodes with maximal efficiency. The discussion was initially framed as a search for the optimal dimensions of a complete taxon×gene matrix (e.g., Graybeal 1998). More recently, spurred by the provocative simulations of Wiens and colleagues (Wiens 2003, Wiens 2006; Wiens and Moen 2008), interest has grown in the possibility of achieving stronger deep node resolution by increased gene sampling for only a subset of taxa, which, in turn, might free resources for other project objectives. In general, the deliberate use of incomplete matrices, if effective, could reduce the total cost of convincingly resolving a given phylogenetic problem and/or increase the total number of nodes so resolvable with a given resource allotment (Driskell et al. 2004; de Queiroz and Gatesy 2007).
How often the postulated advantages of deliberately unbalanced sampling designs will be achieved in practice, however, is not clear. As shown by other simulation studies, the inclusion of taxa or genes with large blocks of missing data, even if generally beneficial, can sometimes result in obscured or even misleading phylogenetic signal (Huelsenbeck 1991; Wiens and Reeder 1995; Wiens 1998; Hartmann and Vision 2008). An especially pessimistic view of missing data was advanced by Lemmon et al. (2009), who argued that previous simulations have confounded the effects of adding incomplete data rows or columns per se with those of adding or subtracting phylogenetic information. From simulations and manipulated real data examples in which the only characters with missing data are parsimony uninformative, they concluded that addition of such characters can result in both strong support for false groupings and loss of support for true groupings. It is not clear that researchers would ever add completely uninformative characters, but one can imagine other circumstances, in which adding incompletely scored characters could result in misleading phylogenetic inference. Suppose, for example, that the incompletely scored characters were 1) more numerous than the completely scored ones, 2) more rapid, and variable among lineages, in evolutionary rate, and 3) scored only in a minority of taxa, scattered across the true phylogeny. Then the partially augmented matrix should be more prone to long-branch attraction than was the original complete matrix (Wiens 1998). One indication that an added set of incompletely scored characters had in fact introduced artifacts of phylogeny inference would be groupings from the partially augmented matrix that conflicted markedly with those from the original complete matrix.
The varying results from simulations highlight the importance of empirical examples in determining when and how often deliberately incomplete sampling designs will be efficient at improving deep node resolution, as opposed to ineffective or misleading. Relatively, few studies have been directed specifically at this issue. In several cases, incomplete augmentation of gene sampling has been shown to markedly increase node support without inducing evident artifacts due to missing data. Philippe et al. (2004) analyzed 36 species spread across the eukaryote phyla, each sequenced for up to 129 genes, with an average of 25% missing data. Nearly all nodes were strongly supported, and relationships among the phyla agreed well with most previous studies. Essentially identical results were obtained in separate analyses of partitions consisting of genes scored in 31 or more taxa versus those scored in fewer taxa and of matrices with additional cells randomly deleted to yield 50% missing data overall. Wiens et al. (2005) compared phylogenetic relationships across the frog family Hylidae inferred from 1) a complete matrix of 193 taxa scored for a single gene (mitochondrial 12S) versus 2) an incomplete matrix in which 81 of the 193 taxa were additionally scored for a second mitochondrial gene, 2 nuclear genes and 144 morphological characters. The incompletely augmented matrix yielded substantially higher support levels overall, particularly for deeper nodes, while strongly supported differences between the two analyses were lacking. Burleigh et al. (2009) compared relationships among angiosperm families inferred from a 97.1% complete matrix of 567 species scored for three genes versus a partially augmented matrix (27.5% incomplete) in which 378 of the 567 species had been sequenced for the additional gene matK, while a partially overlapping set of 240 species had been sequenced for the additional gene 26S rDNA. The five-gene and three-gene matrices yielded very similar topologies, but the deliberately incomplete five-gene matrix gave substantially higher bootstrap support on average. The single point of strong disagreement was ascribed to conflicting signal specifically in one gene, rather than to missing data per se. Somewhat different conclusions, however, emerged from a study (Driskell et al. 2004) that used sequence databases to assemble two very large but highly incomplete matrices, one for animals plus fungi (70 taxa×131 genes, 92% incomplete) and one for green plants (69 taxa×254 genes, 84% incomplete). Phylogenetic analysis of these yielded extensive agreement with previous evidence but also some anomalous groupings possibly arising from missing data. As the contrast among these findings illustrates, additional work is needed to delimit the conditions under which creating larger but more incomplete gene samples will be an effective strategy.
In this paper, we seek to further characterize the effectiveness of partially augmenting gene sampling by examining its ability to strengthen resolution of deeper divergences within the insect order Lepidoptera (moths and butterflies). Relationships among the 126 families and 46 superfamilies of this megadiverse group ( > 150,000 species) are still poorly understood. Regier et al. (2009) described an initial attempt to resolve higher-level relationships within the Ditrysia, a well-established clade that includes over 98% of all Lepidoptera, including the so-called “higher” moths and the butterflies. In that study, 123 species representing 55 families and 27 superfamilies were sampled for five protein-coding nuclear gene regions totaling 6.6 kb. Although bootstrap support for many intrafamily and some interfamily relationships was strong, robust support at deeper levels was mostly lacking. Intergene conflict was rare and weak resolution was accompanied by short branch lengths, suggesting that additional sequence might be the best way to achieve strong support.
Encouraged by the results of Wiens (2003), we sought to achieve stronger deep node support with high efficiency by sequencing just one-third (41) of the 123 species from Regier et al. (2009) for 21 additional genes not previously used in lepidopteran systematics. For these 41 taxa, we nearly tripled (to 18.4 kb) the total sequence length. The additional sequencing focused on the problematic “lower” ditrysian lineages, that is, those outside the huge postulated clade Obtectomera ( > 100,000 species; Minet 1991). Divergences of these lineages may date to the mid Cretaceous (90–70 Ma; Grimaldi and Engel 2005). We compared results from this deliberately unbalanced augmented data set to those from two nearly complete matrices, the previous 123 taxon×5-gene matrix and the new 41 taxon×26-gene data set. We first looked for groupings derived from the deliberately incomplete matrix that strongly contradicted trees from one or both complete matrices, as these would suggest artifacts caused by missing data. We then asked whether deep node support was increased or decreased, on average, and for which nodes, by partially increased gene sampling.
MATERIALS AND METHODS
Taxon Sampling and Specimen Acquisition
The taxon set for this study (listed in Table S1 available from http://www.sysbio.oxfordjournals.org), identical to that of Regier et al. (2009; see additional file 1 of that paper), consists of 123 species of ditrysian Lepidoptera. These are spread across the three major nested clades of Ditrysia recognized by Minet (1991), which in order from least to most inclusive are Macrolepidoptera < [tnqit]Obtectomera[/tnqit] < [tnqit]Apoditrysia[/tnqit] < Ditrysia. The sampling is most dense in Macrolepidoptera (66 species, 11 of 11 superfamilies) and nonmacrolepidopteran Obtectomera (17 species, 4 of 6 superfamilies), which together contain about two-thirds of lepidopteran species diversity. Thirty species of nonobtectomeran Apoditrysia are included, representing 8 of 11 superfamilies, and 7 species of nonapoditrysian Ditrysia, representing 4 of 5 superfamilies. One of the latter, Tineoidea (two species included), was used to root the tree, as tineoids are generally agreed to be the oldest ditrysian superfamily (Minet 1991; Kristensen and Skalski 1998). Altogether, the sample includes 27 of 33 superfamilies and 55 of 100 families of Ditrysia. The classification system in Table S1 generally follows the authorities in Kristensen (1998) with some exceptions (Pyraloidea: Solis and Maes 2002; Geometroidea: Holloway 1997; Scoble 1999; Hausmann 2003; Young 2006).
Specimens for this study, obtained with the help of numerous collectors (see Acknowledgments section), are stored at − 85оC in 100% ethanol as part of the AToLep collection at the University of Maryland (details at http://www.leptree.net/). Nucleic acid extractions were performed on the head and thorax for most species, leaving the rest of the body, including the genitalia, as a voucher. Wing voucher images for all adult exemplars are posted at http://www.leptree.net/. Mitochondrial CO-I “barcodes” for all specimens have been generated by the All-Leps Barcode of Life project (http://www.lepbarcoding.org/), allowing check of their identifications against the Barcode of Life Data system reference library (Ratnasingham and Hebert 2007), as well as future identifications of specimens whose identity is still pending (i.e., species listed as “sp.” or “unidentified” in this report).
All 123 taxa had previously been sequenced for five genes, as described in Regier et al. (2009). For this study, 21 additional genes were sequenced for 30 taxa (listed in Table S1) of “lower Ditrysia,” specifically, nonapoditrysian Ditrysia (7 exemplars, 4 of 5 superfamilies) and nonobtectomeran Apoditrysia (23 exemplars, 8 of 11 superfamilies). To represent the Obtectomera in our analyses of increased gene sampling, we also sequenced the 21 additional genes in 11 species of Macrolepidoptera, representing 6 of the 11 superfamilies. Exemplar sampling for the additional genes in this taxon-directed study was strongly heterogeneous, concentrated on the deeper nodes that we were especially keen to resolve.
Gene Sampling
The five nuclear genes sequenced by Regier et al. (2009) totaled 6633 base pairs (bp), not including 333 with uncertain alignments. They are: CAD (2928 bp; Moulton and Wiegmann 2003), DDC (1281 bp; Fang et al. 1997), enolase (1134 bp; Farrell et al. 2001), period (888 bp; Regier et al. 1998), and wingless (402 bp; Brower and DeSalle 1998). The percentage completeness of the sequence obtained for each gene in each species is shown in Table S2. Overall, the 123-species×5-gene data set is 90.4% complete.
The additional 21 gene regions comprise a total of 12,159 bp, not including 54 bp for which alignment was uncertain. They are among 68 gene regions for which primers were successfully developed and phylogenetic informativeness tested, across all the classes of Arthropoda (Regier, Shultz, et al. 2008). That screen included three diverse Lepidoptera, namely, Prodoxus quinquepunctellus (Prodoxidae, a nonditrysian family), Cydia pomonella (Tortricidae, nonobtectomeran Apoditrysia), and Antheraea paukstadtorum (Saturniidae, Macrolepidoptera). Given that the primers work across the subphyla and classes of Arthropoda, many of which originated over 500 Ma, it is not surprising that these genes are quite conservative. To maximize likely information content of the data set to be generated within the much younger lepidopteran radiation (probable origin < 200 Ma), we chose 21 gene regions, which amplified and sequenced well in all three test lepidopterans and had relatively high average rates of nonsynonymous change among the 68 total (see table 2 in Regier, Shultz, et al. 2008 for relative rate estimates across Arthropoda). The code names, gene names/functions, and lengths of the individual gene regions are given in Table 1. GenBank numbers for these sequences are listed in Table S1. The percentage completeness of the sequence obtained for each gene region in each species is shown in Table S2. Overall, the 41 species×26 gene matrix is 89.9% complete.
TABLE 2.
Comparison of bootstrap support (nodes above the family level) between five-gene complete and partially augmented matrices

|
TABLE 1.
Gene regions sequenced
| A. 21 gene segments adopted from arthropod study of (Regier, Shultz, et al.(2008)) | |||
| PCR amplicon name | Gene name/function | Fragment length(bp) | Average number of substitutions per nt2 sitea |
| 36fin1_3 | Syntaxin | 471 | 0.39 |
| 44fin2_3 | Glucosamine phosphate isomerase | 528 | 0.85 |
| 3007fin1_2 | Glucose phosphosphate dehydrogenase | 621 | 1.22 |
| 8091fin1_2 | Glucose phosphate isomerase | 666 | 1.22 |
| 3006fin1_2 | Dynamin | 222 | 1.24 |
| 113fin1_2 | Glycogen synthase | 975 | 1.27 |
| acc2_4 | Acetyl-coA carboxylase | 501 | 1.31 |
| 69fin2_3 | Clathrin coat assembly protein | 627 | 1.36 |
| 109fin1_2 | Gelsolin | 594 | 1.40 |
| 3070fin4_5 | Alanyl-tRNA synthetase | 705 | 1.43 |
| 262fin1_2 | Proteasome subunit | 501 | 1.48 |
| 268fin1_2 | AMP deaminase | 768 | 1.68 |
| 270fin2_3 | (Hypothetical protein) | 447 | 1.70 |
| 3017fin1_2 | Tetrahydrofolate synthase | 594 | 1.74 |
| 40fin2_3 | Phosphogluconate dehydrogenase | 750 | 1.77 |
| 8028fin1_2 | Nucleolar cysteine-rich protein | 324 | 1.81 |
| 42fin1_2 | Putative GTP-binding protein | 840 | 1.95 |
| 3059fin1_3 | Arginine methyltransferase | 732 | 2.23 |
| 197fin1_2 | Triosephosphate isomerase | 444 | 2.30 |
| 192fin1_2 | Glutamyl- & prolyl-tRNA synthetase | 402 | 2.78 |
| 265fin2_3 | Histidyl-tRNA synthetase | 447 | 3.86 |
| B. Gene segments from (Regier et al.(2009)), with estimated substitution rates | |||
| nt3b/noLR1 + nt2c | Gene name | Fragment length | “nt2 est.”d |
| 85.0 | CAD | 2928 | 1.60 |
| 18.4 | DDC | 1281 | 1.62 |
| 22.4 | Enolase | 1134 | 1.17 |
| 10.8 | Period | 888 | 6.00 |
| 42.4 | Wingless | 402 | 0.34 |
Notes: Provided are PCR amplicon names, gene names/functions and fragment lengths (excluding nucleotide characters of uncertain alignment) of the 21 additional gene regions sequenced for 41 taxa, ordered by evolutionary rate at nt2 on a phylogeny for 13 arthropod exemplars (Regier2008c).
Average number of nucleotide changes per second codon position site, estimated by ML on a constrained tree of 13 divergent arthropod species, from table 2 of (Regier, Shultz, et al.(2008)).
Average number of nucleotide changes per third codon position site, estimated by ML on a constrained tree of 32 species of Bombycoidea (Lepidoptera), from table 4 of (Regier, Cook, et al.(2008)).
Average number of nucleotide changes per site in a character set consisting of nt2 plus all nt1 sites at which no leucine or arginine occurs in any taxon, estimated by ML on a constrained tree of 32 species of Bombycoidea (Lepidoptera), from table 4 of (Regier, Cook, et al.(2008)). This is an estimate of the rate of nonsynonymous change. The ratio of rates at nt3 to rates in this character set is an estimate of the relative rate of synonymous to nonsynonymous substitution.
Approximation of nonsynonymous substitution rate, for comparison to the 21 additional gene fragments above. Gene 19 in table 2 of (Regier, Shultz, et al.(2008)), not included in the 21 additional genes of this study, is a 600 base pair piece of CAD. Estimates of nonsynonymous rates (noLR1 + nt2, above) for the five genes genes used by (Regier et al.(2009)) were first converted to proportions of the rate for CAD, then rescaled to reflect the ranking of CAD among the nt2 rates for the 21 additional gene fragments. The result is an approximate scale of comparison for rates of nonsynonymous substitution across all 26 genes, assuming that rates of substitution at nt2 and at nt1 sites undergoing only nonsynonymous substitutions are comparable.
Generation of DNA Sequence Data and Matrices
A detailed protocol of all laboratory procedures is provided by Regier, Shultz, et al. (2008). Further descriptions, including gene amplification strategies, polymerase chain reaction (PCR) primer sequences, and sequence assembly and alignment methods, can be found in Regier, Cook, et al. (2008), Regier, Grant, et al. (2008), Regier, Shultz, et al. (2008), and Regier et al. (2009). To summarize, total nucleic acids were isolated and specific regions of the cognate mRNAs were amplified by reverse transcriptase PCR. Specific bands were gel isolated and reamplified by PCR using heminested primers, when available. Visible bands that were too faint to sequence were reamplified using the M13 sequences at the 5' ends of all primers. PCR amplicons were sequenced directly on a 3730 DNA Analyzer (Applied Biosystems). Sequences were edited and assembled using the TREV, PREGAP4, and GAP4 programs in the STADEN package (Staden 1999). Multiple sequence alignments were made manually in Genetic Data Environment (Smith et al. 1994), and these were generally straightforward, given the overall conservation of the protein-coding nuclear gene sequences. A data exclusion mask of 387 bp of 18,792 total aligned sequences ( = 2.1% of total) for all 123 species was applied.
Taxon×Gene Data Set Design and Assessing the Effects of Incomplete Gene Sampling.—
To assess the effects of deliberately incomplete gene sampling, we compared the results of separate analyses on three taxon×gene data sets, depicted in Figure 1. The “five-gene complete matrix” (Fig. 1, left) is the 123-species×5-gene (6.6 kb) data set of Regier et al. (2009). The “partially augmented matrix” (Fig. 1, center), deliberately incomplete, is constructed by adding to the five-gene complete matrix the sequences of 21 additional genes (12.2 kb) for 41 (1/3) of the taxa. The block of data it is missing by design is about 45% of the total amount of sequence that would be present in a complete 123-species×26-gene data set. The more-genes-only matrix (Fig. 1, right) is a complete matrix containing just the 41 species sequenced for all 26 genes. Although for convenience, we will refer to the first and third matrices as “complete,” they are more accurately described as mostly complete. Despite our best efforts, each contains about 10% missing data due to occasional amplification or sequencing failures spread haphazardly across gene fragments and taxa (see Table S2). They nonetheless differ sharply from the partially augmented matrix, which, in addition to 10% haphazardly missing data, has 45% missing in a single 82 taxon×21 gene block.
FIGURE 1.
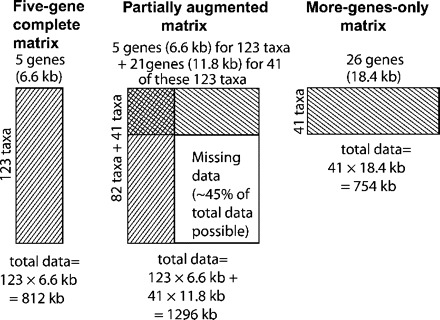
Diagram of gene and taxon sampling design, showing relationships among the three data sets analyzed. a) Five-gene complete matrix (123 taxa; from Regier et al. 2009). b) Partially augmented matrix, deliberately incomplete, created by adding 21 genes for just 41 of the 123 taxa in the five-gene complete matrix. c) More-genes-only matrix, consisting of just the 41 species sequenced for all 26 genes.
We used comparisons among these data sets to address two questions about the effects of incompletely augmented gene sampling. First, do the large blocks of missing data created by partial augmentation result in artifactual groupings? If so, then we might expect to see strong support, in trees from the partially augmented matrix, for groups which do not occur in trees from the five-gene complete matrix. Conversely, finding the same topology from the two matrices would imply that large missing data blocks in the partially augmented matrix do not themselves mislead phylogenetic inference.
Even if it did not induce artifacts, deliberately incomplete augmentation of gene sampling would be an ineffective strategy if failed to strengthen phylogenetic signal, or worse, obscured it. Therefore, we also asked whether bootstrap support was increased or decreased, on average and for which and how many nodes, by the partially augmented matrix as compared with the five-gene complete matrix. Our interest is in deeper divergences, so our comparisons included only nodes above the family level. Many such nodes are weakly supported in all trees from both matrices. To focus on aspects of the phylogeny on which our gene sample is most likely to provide substantial evidence, we initially restricted comparisons to nodes which were 1) shared between trees from the two matrices or found in one and not strongly contradicted by the other and 2) supported by at least 50% bootstrap in at least one tree. Each tree comparison was then repeated using a subset of these nodes that appeared especially sensitive to differences between the two data matrices as reflected in absolute differences of bootstrap values between trees of ≥ 10%. The somewhat arbitrary 10% cutoff for a “noteworthy” difference in bootstrap value excluded about 70% of the initial comparisons.
Even if the only effect of partial sequence augmentation were an increase in support for clades already present in an initial tree, one could doubt that the phylogeny estimate had been improved unless there were independent evidence that the groupings thus reinforced were real rather than artifactual. If, as in the case of ditrysian Lepidoptera, the phylogenetic problem is difficult and little studied, such evidence will necessarily be scarce. However, there is at least some prior morphological and/or molecular support for most of the groupings identified in this study. Matrix-based morphological phylogenetic studies are almost completely lacking, but synapomorphy-based arguments (of varying strength) for monophyly exist for a majority of the groups for which we find strong molecular support (Kristensen 1998), mainly within or among closely related superfamilies. In no case do our results strongly contradict groupings that appear to be strongly supported by morphology. In addition, our trees are similar in topology and in overall levels of support to, and in no case strongly conflict with, those found in a parallel molecular study by Mutanen et al. (2010). The genes used in the two studies are largely distinct; overlap, restricted to fragments of two genes (CAD and wingless; 1250 bp total), amounts to 19.8% of the gene sample (6303 bp) of Mutanen et al. (2010). The groupings above the family level that are supported by bootstrap of ≥ 50% in at least one analysis in our study and also supported by previous morphological or molecular evidence (Kristensen 1998; Mutanen et al. 2010), are noted in Table 2.
Data Partitions, Character Sets/Coding, Compositional Heterogeneity, and Model Selection.—
Our analyses placed special emphasis on the distinction between synonymous and nonsynonymous substitution. Previous studies using these same genes (Regier, Shultz, et al. 2008, Regier et al. 2009, Regier et al. 2010) showed that in addition to evolving faster overall, sites undergoing synonymous substitutions are especially prone to among-lineage base compositional heterogeneity, thereby obscuring and sometimes misleading phylogeny inference. To avoid this potential artifact, we partitioned nucleotides into sets undergoing mostly synonymous versus mostly nonsynonymous change, as follows (see Regier et al. 2009). We first isolated the subset of sites at the first codon position (nt1) which encodes no more than one leucine or arginine residue across all species in the data set, using a Perl script available in online appendix 4 of Regier, Cook, et al. (2008). Because only leucine and arginine codons can undergo synonymous change at nt1, synonymous change is not directly detectable in any pairwise comparisons of extant taxa for such characters. We combined these sites with nt2 to produce a partition, here termed “nonsynonymous nt1 + nt2,” which should reflect almost entirely nonsynonymous change (identical to the “noLRall2 + nt2” of Regier et al. 2009). The excluded sites, comprising on average 22.9% of all nt1 sites for the three data sets, were then combined with nt3 to create a partition here termed “possibly synonymous nt1 + nt3.” The great majority of changes in this partition should be synonymous, though there will also be a few nonsynonymous substitutions at both nt1 and nt3. This partitioning scheme was intended to improve separation of nonsynonymous from synonymous change over that achieved by partitions based solely on codon position. In preliminary studies (data not shown), it markedly improved support for nodes above the family level as compared with analyses with no partitioning.
If compositional heterogeneity is strong, even partitioning may not overcome its deleterious effects on phylogenetic inference. For this reason, we also used the “degen-1” coding of Regier et al. (2010), which in effect excludes synonymous change entirely. Degen-1 is an extension of the purine, pyrimidine-coding scheme (Philippe et al. 2004). Nucleotide sites at any codon position that have the potential of directly undergoing synonymous change, by virtue of the specific codon, they are part of, are fully degenerated, using standard International Union of Pure and Applied Chemistry codenames. For example, CAC and CAT (His) are both coded CAY, whereas TTA, TTG, CTT, CTC, CTA, and CTG (Leu) are all coded YTN. As a result, synonymous pairwise differences between species are entirely eliminated. Synonymous change becomes largely invisible to phylogenetic inference methods, and any compositional heterogeneity it produces is eliminated. Analysis under degen-1 coding (or of the “nonsynonymous nt1 + nt2” character subset) can be viewed as a computationally efficient approximation to a purely “mechanistic” amino acid model (one based on the genetic code but not incorporating empirical transition frequencies between amino acids; Yang et al. 2000; Seo and Kishino 2009). In the remainder of this paper, we will refer to degen-1 as “all nonsynonymous” coding.
We checked the level of compositional heterogeneity in the different character sets just described using the more-genes-only matrix (41 taxa×26 genes), for which we conducted separate chi-square tests of among-taxon heterogeneity using PAUP* 4.0b10 (Swofford 2003), for all nucleotides, for just nt1 plus nt2, and for nonsynonymous nt1 + nt2. We used jModeltest (Posada 2008) to select an appropriate substitution model for each nucleotide character set (nonsynonymous nt1 + nt2, possibly synonymous nt1 + nt3, all nonsynonymous) in each taxon×gene data set. In all cases, the model favored under all selection criteria was general time reversible + gamma + I. This model was applied separately to both character subsets in the partitioned analysis.
Prior to the main analyses, we performed single gene analyses to characterize the degree of conflicting signal among genes. These analyses were carried out, separately on the nonsynonymous nt1 + nt2 and all nonsynonymous character sets, for each of the 26 genes in the 41-taxon more-genes-only matrix. We looked for groupings that conflicted at least moderately ( ≥ 70% bootstrap) with other individual genes, with the all-gene result, or with conventional understanding of relationships. As such conflict proved to be rare (see Results section), we concatenated all 26 genes for subsequent analyses.
Phylogenetic Analyses
All of our phylogenetic analyses were based on the maximum likelihood (ML) criterion as implemented in Genetic Algorithm for Rapid Likelihood Inference (GARLI; version 0.961; Zwickl 2006). We used the program default settings, including random stepwise addition starting trees, except that we halved the number of successive generations yielding no improvement in likelihood score that prompts termination (genthreshfortopoterm = 10,000), as suggested for bootstrapping in the GARLI manual. Each search for an optimal tree consisted of 500 GARLI runs, whereas bootstrap analyses consisted of 1000–2000 replicates, each based on a single heuristic search replicate. Optimal tree searches and bootstrap analyses were parallelized using grid computing (Cummings and Huskamp 2005) through The Lattice Project (Bazinet and Cummings 2009). For consistency in the characterization of results, we will refer to bootstrap support of 70–79% as “moderate” and support ≥ 80% as “strong.” In the main body of this report, we show mostly trees whose terminal nodes have been collapsed to the level of family or subfamily. We do this because higher-level relationships are the focus of this study and because the intrafamily relationships found here are very similar to those described in detail by Regier et al. (2009). We do however provide the full trees, showing all terminal taxa, as Figures S1–S6. Except for Lasiocampidae (3 species), individual families are represented by 2 or fewer species in the 41-species analyses; in the 123-species analyses, the number of representatives ranges up to 12 (Geometridae). The aligned data matrices used and best trees found in our ML tree searches are available at TreeBASE v.2 (http://www.treebase.org/; study accession number 11299).
RESULTS
Gene Agreement and Conflict
The results of the bootstrap analyses for each individual gene and for all genes in the more-genes-only data set (41 taxa×26 genes), under the all nonsynonymous and nonsynonymous nt1 + t2 character sets (both reflecting nonsynonymous change only), are shown in Table S3. Overall, very few nodes above the family level were moderately or strongly supported by any individual gene. Grouping of the two Gelechioidea was the only interfamily relationship strongly supported by any individual gene (bootstrap percentage [BP] = 88% and 86% for all nonsynonymous and nonsynonymous nt1 + nt2, respectively, for syntaxin). Four relationships among superfamilies received bootstrap support ≥ 70% from at least one gene, but only one of these (Choreutoidea + Alucitoidea, BP = 75% and 71%) was present in any of the combined gene results, and none received support ≥ 80%. In only five instances were conflicting groupings moderately or strongly supported by two individual genes or by an individual gene versus the 26 genes combined (see details in Table S3). Two of these disagreements affect only relationships within a single family, and all concern differing placements of just a single exemplar. Because the intergene conflicts involve only a small fraction of genes and taxa, we judged gene concatenation to be a reasonable approach to estimating lepidopteran phylogeny above the family level.
Effects of Deliberately Incomplete Augmentation of Gene Sampling
Figure 2 contrasts the among-family relationships and corresponding bootstrap supports inferred from the five-gene complete matrix with those from the partially augmented matrix, in which 41 of 123 species have been sequenced for 21 additional genes. For both the partitioned analysis of all nucleotides and the all nonsynonymous analysis, the two matrices yield very similar trees, differing only by rearrangements of a few weakly supported groupings. For a given character treatment, no groups are strongly supported by the partially augmented matrix that do not also occur in the ML tree from the five-gene complete matrix, whereas all groups supported strongly by the five-gene complete matrix are also strongly supported in trees from the partially augmented matrix.
FIGURE 2.
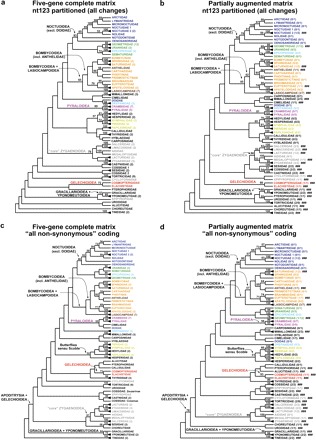
Comparison of ML trees of family relationships inferred from the five-gene complete matrix (left column) to those from the partially augmented matrix (123 taxa×5 or 26 genes; right column), simplified from full 123-taxon trees shown in Figures S1–S4. Black triangles denote families with multiple exemplars. Numbers in parentheses after family name represent number of exemplars for the five-gene complete matrix, number with 26 genes/total number for partially augmented matrix). ### denotes families with one or more exemplars scored for 26 genes for partially augmented matrix. BPs > 50% are shown above branches; number of replicates is 1000 for a) and b), 2000 for c) and d). a) nt123 partitioned, five-gene complete matrix; b) nt123 partitioned, partially augmented matrix; c) all nonsynonymous coding, five-gene complete matrix; d) all nonsynonymous coding, partially augmented matrix.
Although the topology remains essentially unchanged under increased gene sampling, the partially augmented matrix provides consistently greater support for deeper nodes. Table 2 lists the 31 nodes subtending two or more families that receive bootstrap support of > 50% under at least one of the two character treatments for one or both data matrices and are not contradicted by other groupings with > 50% bootstrap support. As shown in the table summary, under both character treatments, a substantial majority of the relevant nodes (those for which the two data matrices yield different bootstrap values) gets higher support from the partially augmented matrix. The strongest effect occurs in the nt123 partitioned analyses, for which 19 of 24 contrasts overall (79%), and 8 of 8 (100%) showing a difference of 10% or more, show higher bootstraps for the partially augmented matrix. For the all nonsynonymous coding, which captures nonsynonymous changes only, the partially augmented matrix has the higher bootstrap value for 15 of 21 comparisons overall (71%) and 5 of 5 (100%) with 10% or more difference. Some of these increases in support are substantial. For example, support for Apoditrysia + Gelechioidea, always < < 50% for the five-gene complete matrix, increases to 78% for the partially augmented matrix under all nonsynonymous coding (Fig. 2d). Support for Gelechioidea increases by 20% (from 73% to 93%) and that for monophyly of the “core” Zygaenoidea (Regier et al. 2009) by 18% (from 81% to 99%), under that same coding. Support for Lasiocampoidea + Bombycoidea increases by 20% (65% to 85%) for nt123 partitioned. Overall, pronounced increase ( ≥ 10%) in bootstrap support under partial augmentation of gene sampling occurs most often for nodes subtending at least two species sequenced for 26 genes (summarized in Table 2, bottom). However, the association is not absolute, and support was also increased for several groups (e.g., Pyraloidea) sequenced only for five genes.
Overall, for the partially augmented matrix, nt123 partitioned analysis (all changes included) yields higher bootstraps than all nonsynonymous coding (only nonsynonymous changes included) for 23 of the 30 nodes at which the 2 differ, including 20 of 22 for which the difference is ≥ 10%. Partitioned analysis supports 18 nodes overall by 70% or greater bootstrap, as compared with 10 for all nonsynonymous, but this differential mostly reflects relatively shallow nodes, that is, groupings of families within superfamilies. In marked contrast, the only instance of substantial support for a deep “backbone” node is the 78% bootstrap for Gelechioidea + Apoditrysia under all nonsynonymous coding; this grouping does not even occur in the ML tree for the nt123 partitioned analysis.
Analysis of the more-genes-only matrix (41 species×26 genes) yields among-family relationships very similar to those found for the two 123-taxon data sets (cf. Figs. 2 and 3; see also Figs. S1–S13). For each character treatment, no strongly supported groups are found that do not also occur in trees from the five-gene complete and partially augmented data sets. Moreover, all groupings of two or more families supported strongly by either of the two 123-taxon data sets also occur in trees from the more-genes-only matrix, provided those families are represented among the 41 species in that matrix. Bootstrap support is higher from the more-genes-only data set than from the five-gene complete matrix for all or nearly all nodes for which the two data sets yield different bootstrap values (8/9 under all nonsynonymous coding, 8/8 for nt123 partitioned). The differences are often substantial, exceeding 30% in three cases. The differential support could in theory reflect either greater gene sampling or lesser taxon sampling in the more-genes-only matrix because bootstrap support is known to be inversely correlated with taxon number, other things being equal (Zharkikh and Li 1995; Susko 2009). As seen in Table 2, however, bootstrap support is only sometimes and only slightly greater in the more-genes-only matrix than in the partially augmented matrix, which has the same number of taxa (123) as the five-gene complete matrix. This observation points to greater gene sampling as the main cause of higher support in the more-genes-only data set.
FIGURE 3.
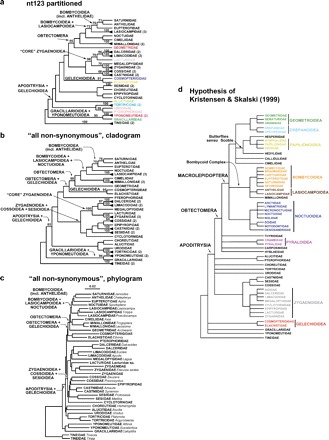
a)–c) ML trees of family relationships inferred from more-genes-only data set (41 taxa×26 genes), simplified (except in c) from full 41-taxon trees shown in Figures S5 and S6. Black triangles denote families with multiple exemplars; number of exemplars shown in parentheses after family name. a) all nonsynonymous coding, phylogram; b) all nonsynonymous coding, cladogram; c) nt123 partitioned. BPs > 50% are shown above branches; number of replicates is 1000 for a), 2000 for b). d) Relationships among the sampled families (only) according to the morphology-based working hypothesis of Kristensen and Skalski (1998).
Nucleotide Compositional Heterogeneity
Chi-square tests on the more-genes-only matrix (41 taxa×26 genes), with or without prior removal of invariant characters, strongly rejected compositional homogeneity for all character sets that include synonymous differences (nt123 and nt12; P < 0.001). The one exception is marginal nonsignificance of heterogeneity in nt12 with invariant characters included (P = 0.09). By contrast, homogeneity is not rejected for the nonsynonymous nt1 + nt2 character set, either with (P = 0.966) or without (P > 0.999) invariant characters.
DISCUSSION
Effects of Deliberately Incomplete Augmentation of Gene Sampling
Our results provide clear support for the efficacy of partially augmented gene sampling in improving an estimate of phylogeny above the family level for ditrysian Lepidoptera. The partially augmented and five-gene complete matrices yield nearly identical topologies and similar rank orders of support among nodes, and all nodes with ≥ 70% bootstrap support from the partially augmented matrix are also present in the ML tree for the five-gene complete matrix. Thus, the major block of missing data in the partially augmented matrix (Fig. 1), amounting to 45% of the total possible sequence for a complete matrix of these dimensions, appears not to induce artifactual groupings. Moreover, partial augmentation results in consistently higher bootstrap support; for example, for all nodes at which support differs by ±10% or more between matrices, support is greater from the partially augmented matrix than from the five-gene complete matrix (Table 2). Finally, all 19 nodes that have moderate to strong support from the partially augmented matrix also have prior support from morphology (Kristensen 1998), molecular data (Mutanen et al. 2010), or both, strengthening the evidence that they are not artifacts.
Our results add to the growing evidence that under a wide range of circumstances, data sets with substantially different gene, and taxon sampling can be safely combined to improve estimates of deeper-level phylogeny, even if a majority of the genes are sampled in only a minority of taxa in the combined matrix. Earlier, we cited three case studies supporting this conclusion, ranging broadly across organismal groups and taxonomic levels, from species and higher taxa of hylid frogs (Wiens et al. 2005) to families of angiosperm (Burleigh et al. 2009) to phyla of eukaryotes (Philippe et al. 2004). To this list can be added another recent study in Lepidoptera that used most of the genes analyzed in the current report. Zwick et al. (2011) sequenced an additional 20 genes (11.7 kb) for 24 of 50 species spread across the families of Bombycoidea + Lasiocampoidea, previously sequenced for five genes (6.6 kb) by Regier, Grant, et al. (2008). The partially augmented matrix, 33% deliberately incomplete, yielded essentially the same topology as the five-gene complete matrix, while bootstrap support was substantially increased, especially for deeper nodes. These cases all support the generalization (Wiens 2003) that missing data are not problematic if there is substantial signal in the data that are present. In contrast, however, probable artifacts of incomplete sampling were evident in the much sparser, 84–92% incomplete matrices of Driskell et al. (2004). Thus, a greater variety of empirical studies is needed to fully demarcate the regions of taxon×gene sampling space, in which incomplete sampling is an effective strategy.
It might be argued that the effects of incomplete gene sampling will soon become irrelevant, as next-generation sequencing technologies (NGST) will lead to economical scoring of very large numbers of genes in all taxa. Although NGSTs hold tremendous promise, we doubt that they will soon eliminate the motivation for unbalanced sampling designs. Many phylogenetic problems may prove to be solved most cost-effectively by combining the broad taxon sampling of existing databases with expanded NGST gene sampling for only subsets of taxa. Moreover, varying future applications of NGST, for example, complete genome sequencing versus RNA-seq of highly expressed genes only (e.g., Hittinger et al. 2010), will yield very different numbers of genes. The question will remain of whether these data sets can safely be combined.
Lepidopteran Phylogeny: Problems and Progress
Thus far, we have emphasized the prevailing increase in node support conferred by deliberately incomplete augmentation of the gene sample. An additional, more somber conclusion to emerge from our results, however, is that deep-level ditrysian phylogeny is a difficult problem. The hypothesis of deep-level relationships that Regier et al. (2009) set out to test (Kristensen and Skalski 1998) is depicted in Figure 3d. Most of the increased node support from increased gene sampling in the present study applies to interfamily relationships and a few pairings of related superfamilies, not the deeper divisions hypothesized by Kristensen and Skalski (1998). Only one node subtending three or more superfamilies is strongly supported by the expanded data set, even in a tree region in which most of the exemplars had 18.4 kb of sequence (Fig. 2). Mutanen et al. (2010) report a similar paucity of strong support for deep nodes. These observations, coupled with the very short branches evident along the backbone of the phylogram in Figure 3c, suggest that lower ditrysian relationships might be conclusively resolvable only by very large amounts, and possibly new kinds, of data (e.g., Jian et al. 2008).
Although progress is thus likely to remain incremental, our current results provide several significant steps beyond those reported by Regier et al. (2009), who review current understanding of ditrysian phylogeny in detail. The 78–85% bootstraps we find for Apoditrysia + Gelechioidea under all nonsynonymous analysis (Figs. 2d and 3b) constitute the first substantial statistical support for monophyly of any major subset of ditrysian superfamilies. This grouping is the more plausible because it was previously proposed by Kristensen and Skalski (1998) based on two putative synapomorphies in male genital structures (Robinson and Nielsen 1993) and two in proboscis morphology (Rammert 1994). Why then does the association of Gelechioidea with Apoditrysia, to the exclusion of Gracillarioidea and Yponomeutoidea, emerge in molecular analyses only weakly (41 taxon analyses) or not at all (123 taxon analyses) unless synonymous change is entirely excluded? Assuming this morphology supported grouping to be valid, it appears that, at this depth, synonymous change obscures the signal from nonsynonymous change even when it is modeled separately. A plausible explanation for that observation is nonstationarity of synonymous substitution. Highly significant heterogeneity of base composition is seen when all nucleotides are considered but disappears when we consider only sites undergoing nonsynonymous change.
Monophyly for Apoditrysia as currently defined, although a long-standing hypothesis, is not supported by any of our trees (Figs. 2 and 3). Instead, the supposedly nonapoditrysian superfamily Gelechioidea always falls among the putative apoditrysians. Morphological support for Apoditrysia, however, is limited to a single proposed synapomorphy, namely, relatively short and stout apodemes on abdominal sternum II (Minet 1983). No exact position for Gelechioidea is strongly supported, but in our analyses, they always group with some or all members of the putative clade Obtectomera (Figs. 2 and 3). Within Obtectomera, we find further evidence beyond Regier et al. (2009) in favor of a sister group relationship between Bombycoidea and Lasiocampidae (BP = 85% under partitioned nt123 for the partially augmented matrix). We also find much stronger support than previous molecular studies (Regier et al. 2009; Mutanen et al. 2010) for several superfamilies defended by morphological synapomorphies, including Pyraloidea, Gelechioidea, and core Zygaenoidea.
Finally, we take encouragement from the strong bootstrap support (85% under all nonsynonymous coding; Fig. 3b) provided by the more-genes-only matrix (41 taxa×26 genes) for a clade containing all five sampled superfamilies of Obtectomera: Macrolepidoptera (no nonmacrolepidopteran Obtectomera were included in this data set). The two 123-taxon data sets (Fig. 2), which include many more representatives of Macrolepidoptera and other Obtectomera, provide almost no bootstrap support for Macrolepidoptera or any substantial subset thereof (excluding the butterflies, which this and two previous studies show not to group with other macrolepidopterans; Regier et al. 2009, Mutanen et al. 2010). Inspection of Figure 2 shows, however, that both 123-taxon matrices do support monophyly for Macrolepidoptera when pruned to exclude taxa not included in the more-genes-only data set, arguing that the presence of this grouping in the 41-taxon matrix is not an artifact of under-sampling. Thus, the more-genes-only result can be taken as evidence for the existence of strong signal for some version of this large putative clade ( > 80,000 species) in the 123-taxon data sets, particularly the partially augmented matrix, despite the lack of convincing support for any individual node in that region of the tree. Why does the additional taxon sampling obscure this signal? Part of the reason, probably, is the stringency of the conventional measure of bootstrap support, namely bootstrap majority rule consensus trees. Because such consensus trees do not take partial agreement on a monophyletic group into account, they can greatly underestimate the degree of structure in a large data set (Sanderson 1989). Robust inference of very large phylogenies is likely to require multiple approaches to separating underlying large-scale signal from smaller-scale anomalies, such as “rogue” taxa (Thomson and Shaffer 2010), which obscure it. Deliberate undersampling of taxa, coupled with expanded gene sampling, may be one useful tool in this quest.
SUPPLEMENTARY MATERIAL
Supplementary material, including data files and/or online-only appendices, can be found at http://www.sysbio.oxfordjournals.org/.
FUNDING
Financial support was provided by the US National Science Foundation's Assembling the Tree of Life program, award numbers 0531626 and 0531769; the Spanish Government (Ministerio de Ciencia e Innovación) (CGL2008-00605 to J.B.); US National Science Foundation (DEB 0515699 to D. H. Janzen).
Supplementary Material
Acknowledgments
We are greatly indebted to generous colleagues for supplying specimens for this study, including J. K. Adams, D. Adamski, K. Nishida, V. Becker, N. Bloomfield, T. Burbidge, J. Farr, G. Clarke, R. F. Denno, E. Edwards, M. Epstein, M. Fibiger, T. Friedlander, J. Giebultowicz, W. Hallwachs, A. Hausmann, R. J. B. Hoare, K. R. Horst, R. Hutchings, N. Hyde'n, D. H. Janzen, W. Kelly, I. J. Kitching, R. LeClerc, M. J. Matthews, N. McFarland, D. Messersmith, A. Mitchell, R. Poole, K.-T. Park, J. O. Nelson, E. S. Nielsen, R. S. Peigler, K. Pullen, R. Robertson, M. A. Solis, G. Tremewan, A. Venables, A. Willis, K. Wolfe, and S.-H.Yen. S. Zhao and K. Jiang provided technical assistance, and K. Mitter made essential contributions as specimen collection and database manager. The manuscript was greatly improved by comments from Editor J. Sullivan, Associate Editor K. Kjer, J. Wiens, and two anonymous reviewers, but they are not to blame for its remaining faults.
References
- Bazinet AL, Cummings MP. The lattice project: a grid research and production environment combining multiple grid computing models. In: Weber MHW, editor. Distributed and grid computing—science made transparent for everyone. Principles, applications and supporting communities. Marburg (Germany): Tectum Publishing House; 2009. pp. 2–13. [Google Scholar]
- Brower AVZ, DeSalle R. Mitochondrial vs. nuclear DNA sequence evolution among nymphalid butterflies: the utility of wingless as a source of characters for phylogenetic inference. Insect Mol. Biol. 1998;7:1–10. doi: 10.1046/j.1365-2583.1998.71052.x. [DOI] [PubMed] [Google Scholar]
- Burleigh JG, Hilu KW, Soltis DE. Inferring phylogenies with incomplete data sets: a 5-gene, 567-taxon analysis of angiosperms. BMC Evol. Biol. 2009;9:61. doi: 10.1186/1471-2148-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings MP, Huskamp JC. Grid computing. EDUCAUSE Rev. 2005;40:116–117. [Google Scholar]
- de Queiroz A, Gatesy J. The supermatrix approach to systematics. Trends Ecol. Evol. 2007;22:34–41. doi: 10.1016/j.tree.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Driskell AC, Ané C, Burleigh JG, McMahon MM, O'Meara BC, Sanderson MJ. Prospects for building the tree of life from large sequence databases. Science. 2004;306:1172–1174. doi: 10.1126/science.1102036. [DOI] [PubMed] [Google Scholar]
- Fang Q, Cho S, Regier J, Mitter C, Matthews M, Poole R, Friedlander T, Zhao S. A new nuclear gene for insect phylogenetics: dopa decarboxylase is informative of relationships within Heliothinae (Lepidoptera: Noctuidae) Syst. Biol. 1997;46:269–283. doi: 10.1093/sysbio/46.2.269. [DOI] [PubMed] [Google Scholar]
- Farrell BD, Sequeira AS, O'Meara B, Normark BB, Chung JH, Jordal BH. The evolution of agriculture in beetles (Curculionidae: Scolytinae and Platypodinae) Evolution. 2001;55:2011–2027. doi: 10.1111/j.0014-3820.2001.tb01318.x. [DOI] [PubMed] [Google Scholar]
- Graybeal A. Is it better to add taxa or characters to a difficult phylogenetic problem? Syst. Biol. 1998;47:9–17. doi: 10.1080/106351598260996. [DOI] [PubMed] [Google Scholar]
- Grimaldi D, Engel MS. Evolution of the insects. Cambridge (UK): Cambridge University Press; 2005. [Google Scholar]
- Hartmann S, Vision TJ. Using ESTs for phylogenomics: can one accurately infer a phylogenetic tree from a gappy alignment? BMC Evol. Biol. 2008;8:95. doi: 10.1186/1471-2148-8-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann A, editor. The Forum Herbulot world list of family group names in Geometridae. 2003. Available from: http://www.herbulot.de/ [Google Scholar]
- Hittinger CT, Johnston M, Tossberg JT, Rokas A. Leveraging skewed transcript abundance by RNA-sequencing to increase the genomic depth of the tree of life. Proc. Natl. Acad. Sci. U S A. 2010;107:1476–1481. doi: 10.1073/pnas.0910449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway JD. The moths of Borneo, pt. 10, Geometridae: Sterrhinae, Larentiinae. Malay Nat. J. 1997;51:1–242. [Google Scholar]
- Huelsenbeck JP. When are fossils better than extant taxa in phylogenetic analysis? Syst. Zool. 1991;40:458–469. [Google Scholar]
- Jian S, Soltis PS, Gitzendanner MA, Moore MJ, Li R, Hendry TA, Qiu Y-L, Dhingra A, Bell CD, Soltis DE. Resolving an ancient, rapid radiation in Saxifragales. Syst. Biol. 2008;57:38–57. doi: 10.1080/10635150801888871. [DOI] [PubMed] [Google Scholar]
- Kristensen NP. Lepidoptera, moths and butterflies, volume 1: evolution, systematics, and biogeography. In: Fischer M., editor. Handbook of zoology: a natural history of the phyla of the animal kingdom, volume IV, Arthropoda: Insecta, part 35, Lepidoptera, moths and butterflies. Berlin (Germany): Walter de Gruyter, Inc; 1998. [Google Scholar]
- Kristensen NP, Skalski AW. Phylogeny and palaeontology. In: Kristensen NP, editor. Handbook of zoology: a natural history of the phyla of the animal kingdom, Volume IV, Arthropoda: Insecta, part 35, Lepidoptera, moths and butterflies, Volume 1: evolution, systematics, and biogeography. Berlin (Germany): Walter de Gruyter, Inc.; 1998. pp. 7–25. [Google Scholar]
- Lemmon AR, Brown JM, Stanger-Hall K, Lemmon EM. The effect of ambiguous data on phylogenetic estimates obtained by maximum likelihood and Bayesian inference. Syst. Biol. 2009;58:130–145. doi: 10.1093/sysbio/syp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minet J. Étude morphologique et phylogénétique des organes tympaniques des Pyraloidea. Généralités et homologies. (Lep. Glossata) Ann. Soc. Entomol. Fr. (N.S.) 1983;19:175–207. [Google Scholar]
- Minet J. Tentative reconstruction of the ditrysian phylogeny (Lepidoptera: Glossata) Ent. Scand. 1991;22:69–95. [Google Scholar]
- Moulton JK, Wiegmann BM. Evolution and phylogenetic utility of CAD (rudimentary) among Mesozoic-aged eremoneuran Diptera (Insecta) Mol. Phylogenet. Evol. 2003;31:363–378. doi: 10.1016/S1055-7903(03)00284-7. [DOI] [PubMed] [Google Scholar]
- Mutanen M, Wahlberg N, Kaila L. Comprehensive gene and taxon coverage elucidates radiation patterns in moths and butterflies. Proc. R. Soc. Lond. B. Biol. Sci. 2010;277:2839–2848. doi: 10.1098/rspb.2010.0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H, Snell EA, Bapteste E, Lopez P, Holland PW, Casane D. Phylogenomics of eukaryotes: impact of missing data on large alignments. Mol. Biol. Evol. 2004;21:1740–1752. doi: 10.1093/molbev/msh182. [DOI] [PubMed] [Google Scholar]
- Posada D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- Rammert U. Morphologische Untersuchungen zur Aufdeckung der stammesgeschichtliche Verhältnisse der basalen Gruppen der ditrysen Lepidopteren (Lepidoptera: Ditrysia) [dissertation] Flintbek (Germany): Universität Bielefeld; 1994. 193 pp., 25 pls. [Google Scholar]
- Ratnasingham S, Hebert PD. Bold: the barcode of life data system. Mol. Ecol. Notes. 2007;7:355–364. doi: 10.1111/j.1471-8286.2007.01678.x. Available from: http://www.barcodinglife.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regier JC, Cook CP, Mitter C, Hussey A. A phylogenetic study of the "bombycoid complex" (Lepidoptera) using five protein-coding nuclear genes, with comments on the problem of macrolepidopteran phylogeny. Syst. Ent. 2008;33:175–189. [Google Scholar]
- Regier JC, Fang QQ, Mitter C, Peigler RS, Friedlander TP, Solis MA. Evolution and phylogenetic utility of the period gene in Lepidoptera. Mol. Biol. Evol. 1998;15:1172–1182. doi: 10.1093/oxfordjournals.molbev.a026024. [DOI] [PubMed] [Google Scholar]
- Regier JC, Grant MC, Peigler RS, Mitter C, Cook CP, Rougerie R. Phylogenetic relationships of wild silkmoths (Lepidoptera: Saturniidae) inferred from four protein-coding nuclear genes. Syst. Ent. 2008;33:219–228. [Google Scholar]
- Regier JC, Shultz JW, Ganley ARD, Hussey A, Shi D, Ball B, Zwick A, Stajich JE, Cummings MP, Martin JW, Cunningham CW. Resolving arthropod phylogeny: exploring phylogenetic signal within 41 kb of protein-coding nuclear gene sequence. Syst. Biol. 2008;57:920–938. doi: 10.1080/10635150802570791. [DOI] [PubMed] [Google Scholar]
- Regier JC, Shultz JW, Zwick A, Hussey A, Ball B, Wetzer R, Martin JW, Cunningham CW. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature. 2010;463:1079–1083. doi: 10.1038/nature08742. [DOI] [PubMed] [Google Scholar]
- Regier JC, Zwick A, Cummings MP, Kawahara AY, Cho S, Weller S, Roe A, Baixeras J, Brown JW, Parr C, Davis DR, Epstein M, Hallwachs W, Hausmann A, Janzen DH, Kitching IJ, Solis MA, Yen S-H, Bazinet AL, Mitter C. Toward reconstructing the evolution of advanced moths and butterflies (Lepidoptera: Ditrysia): an initial molecular study. BMC Evol. Biol. 2009;9:280. doi: 10.1186/1471-2148-9-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson GS, Nielsen ES. Tineid genera of Australia. Monographs on Australian Lepidoptera. 1993;2:1–344. [Google Scholar]
- Sanderson MJ. Confidence limits on phylogenies: the bootstrap revisited. Cladistics. 1989;5:113–129. doi: 10.1111/j.1096-0031.1989.tb00559.x. [DOI] [PubMed] [Google Scholar]
- Scoble MJ. The Lepidoptera, form, function and diversity. Oxford: Oxford University Press; 1992. [Google Scholar]
- Scoble MJ. Geometrid moths of the world: a catalogue (Lepidoptera, Geometridae) Volume 1 and 2. Stenstrup (Denmark): CSIRO Publishing and Apollo Books; 1999. [Google Scholar]
- Seo T-K, Kishino H. Statistical comparison of nucleotide-, amino-acid-, and codon-substitution models for the evolutionary analysis of protein-coding sequences. Syst. Biol. 2009;58:199–210. doi: 10.1093/sysbio/syp015. [DOI] [PubMed] [Google Scholar]
- Smith SW, Overbeck R, Woese CR, Gilbert W, Gillevet PM. The genetic data environment and expandable GUI for multiple sequence analysis. Comput. Appl. Biosci. 1994;10:671–675. doi: 10.1093/bioinformatics/10.6.671. [DOI] [PubMed] [Google Scholar]
- Solis MA, Maes KVN. Preliminary phylogenetic analysis of the subfamilies of Crambidae (Pyraloidea: Lepidoptera) Belgian J. Entom. 2002;4:53–95. [Google Scholar]
- Susko E. Bootstrap support is not first-order correct. Syst. Biol. 2009;58:211–223. doi: 10.1093/sysbio/syp016. [DOI] [PubMed] [Google Scholar]
- Staden R. Staden package. Cambridge (UK): MRC Laboratory of Molecular Biology; 1999. Available from: http://www.mrc-lmb.cam.ac.uk/pubseq/ [Google Scholar]
- Swofford DL. PAUP*: phylogenetic analysis using parsimony (* and other methods), version 4.0b 10. Sunderland (MA): Sinauer Associates; 2003. [Google Scholar]
- Thomson RC, Shaffer HB. Sparse supermatrices for phylogenetic inference: taxonomy, alignment, rogue taxa, and the phylogeny of living turtles. Syst. Biol. 2010;59:42–58. doi: 10.1093/sysbio/syp075. [DOI] [PubMed] [Google Scholar]
- Wiens JJ. Does adding characters with missing data increase or decrease phylogenetic accuracy? Syst. Biol. 1998;47:625–640. doi: 10.1080/106351598260635. [DOI] [PubMed] [Google Scholar]
- Wiens JJ. Missing data, incomplete taxa, and phylogenetic accuracy. Syst. Biol. 2003;52:528–538. doi: 10.1080/10635150390218330. [DOI] [PubMed] [Google Scholar]
- Wiens JJ. Missing data and the design of phylogenetic analyses. J. Biomed. Inform. 2006;39:34–42. doi: 10.1016/j.jbi.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Wiens JJ, Fetzner JW, Parkinson CL, Reeder TW. Hylid frog phylogeny and sampling strategies for speciose clades. Syst. Biol. 2005;54:719–748. doi: 10.1080/10635150500234625. [DOI] [PubMed] [Google Scholar]
- Wiens JJ, Moen DS. Missing data and the accuracy of Bayesian phylogenetics. J. Syst. Evol. 2008;46:307–314. [Google Scholar]
- Wiens JJ, Reeder TW. Combining data sets with different numbers of taxa for phylogenetic analysis. Syst. Biol. 1995;44:548–558. [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen A-MK. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CJ. Molecular relationships of the Australian Ennominae (Lepidoptera: Geometridae) and implications for the phylogeny of the Geometridae from molecular and morphological data. Zootaxa. 2006;1264:1–147. [Google Scholar]
- Zharkikh A, Li W.- H. Estimation of confidence in phylogeny: the complete-and-partial bootstrap technique. Mol. Phylogenet. Evol. 1995;4:44–63. doi: 10.1006/mpev.1995.1005. [DOI] [PubMed] [Google Scholar]
- Zwick A, Regier JC, Mitter C, Cummings MP. Increased gene sampling yields robust support for higher-level clades within Bombycoidea (Lepidoptera) Syst. Ent. 2011;36:31–43. [Google Scholar]
- Zwickl DJ. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion[PhD dissertation] Austin (TX): The University of Texas; 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.