

Summary
When DNA damage is detected, checkpoint signal networks are activated to stop the cell cycle, and DNA repair processes begin. Inhibitory compounds targeting components of DNA damage response pathways have been identified and are being used in clinical trials, in combination with chemotherapeutic agents, to enhance cancer therapy. Inhibitors of checkpoint kinases, Chk1 and Chk2, have been shown to sensitize tumor cells to DNA damaging agents, and treatment of BRCA1/2-deficient tumor cells, as well as triple negative breast cancers, with poly(ADP-ribose) polymerase (PARP) inhibitors has shown promise. But systematic studies to determine which tumor subtypes are likely to respond to these specific inhibitors have not been reported. The current study was designed to test sensitivity of specific breast cancer subtype-derived cells to two classes of these new inhibitory drugs, PARP and Chk1 inhibitors. Luminal, HER2 over-expressing and triple negative breast cancer-derived cells were tested for sensitivity to killing by PARP inhibitors, ABT-888 and BSI-201, and Chk1 inhibitor, PF-00477736, alone or in combination with gemcitabine or carboplatin. Each of the triple negative breast cancer cell lines showed strong sensitivity to the Chk1 inhibitor, but only the BRCA1-deficient breast cancer cell lines showed sensitivity to the PARP inhibitors, suggesting that in vitro testing of cancer cell lines of specific subtypes, with panels of the different PARP and Chk1 inhibitors, will contribute to stratification of patients for clinical trials using these classes of inhibitors.
Introduction
Several lines of investigation have focused our interest on expression of DNA damage response (DDR)-associated proteins in breast cancer, particularly triple negative (TN) breast cancers, lacking expression of hormone and HER2 receptors: 1) reports of activated DDR checkpoints in preneoplastic lesions (1–3); 2) findings that BRCA1/2-deficient breast cancer cell lines were exquisitely sensitive to cell killing by inhibitors of PARP activity (4,5), through a synthetic lethal mechanism involving loss of homologous recombination repair (HRR), due to BRCA1/2 mutation, and inhibition of other repair pathways, by PARP inhibition (6–8); 3) demonstration that BRCA1-deficient breast cancers are mostly TN, with basal-like phenotype, a subtype associated with defects in some types of DNA repair (9) and endowed with ‘BRCAness’ (for review, 10); 4) reports that loss of expression of Fhit, another tumor suppressor with DDR involvement, occurred in ~90% of BRCA1 and 2-mutated breast cancers (11–13) and coordinate loss of expression of Fhit and Wwox fragile tumor suppressors was significantly associated with the TN subtype (14,15); it was also reported previously that Chk1 is highly expressed in TN breast cancers (16). For these reasons, we were interested in how alterations in expression of DDR checkpoint and repair-associated proteins, might contribute to ‘BRCAness’ and to responses to drugs targeting activated DDR checkpoints or DNA repair pathways.
In normal cells, single or double-strand DNA breaks (SSBs, DSBs) lead to activation of checkpoint responses, through signal transduction cascades and post-translational modifications such as phosphorylation and ADP ribosylation, and result in cell cycle arrest or apoptosis. PARP enzymatic activity is essential for repair of DNA SSBs via the base excision repair pathway. PARP1, the best characterized of the PARP superfamily members, binds to SSB sites and catalyzes addition of ADP-ribose polymer (PAR) chains to itself and other effectors of base excision repair [cited in 7]. Small molecule inhibitors of PARP activity have shown promise for therapy of cancers, particularly BRCA1/2 mutated cancers, alone or combined with cytotoxic drugs (6–8). BRCA mutant cells are dependent on other DNA repair pathways, including base excision repair, that help prevent development of DSBs, to compensate for inability to repair DSBs by HRR. When PARP and therefore base excision repair are inhibited, the unrepaired SSBs cause collapse of replication forks, leading to DSBs and cell death. Such synthetic lethality represents a new strategy for development of anti-cancer drugs (7).
The serine/threonine protein kinases, ATM and ATR, are key proteins in DNA-damage checkpoint responses and their respective downstream targets, Chk2 and Chk1, have roles in regulation of G1/S and G2/M checkpoint responses. Inhibiting Chk1 also represents a “synthetic lethal” therapeutic strategy through inhibition of the defense of tumor cells against lethal damage induced by DNA-directed chemotherapeutic agents (17). Occurrence of DSBs is followed by ATM or ATR phosphorylation of histone H2AX. The phosphorylated form, γH2AX, recruits DNA repair proteins (18), including BRCA1 (19), to DNA breaks.
To determine if TN breast cancers are especially sensitive to a specific synthetic lethal therapeutic strategy, particularly since PARP1 inhibitors have been undergoing clinical trials for such breast cancers (20), we have tested sensitivity of breast cancer-derived cell lines of defined subtypes to PARP or Chk1 inhibitors, with or without combination treatment with cytotoxic drug.
Materials and Methods
Cell lines and culture conditions
Breast cancer cell lines of defined subtypes (21, 22) were cultured in RPMI1640 (Sigma-Aldrich, St. Louis, MO) (T47D, ZR-75-1, MDA-MB-231, MDA-MB-468) with 10% FBS and 100 μg/ml Gentamycin (Sigma), or Dulbecco Modified Eagle Medium with the same supplements (MCF-7, SK-BR-3, BT-20, MDA-MB-453, MDA-MB-436) at 37 C, in 5% CO2. All cell lines were obtained as frozen stocks from the laboratory of Tim H-M. Huang, of our department. Dr. Huang’s laboratory obtained the cells directly from the laboratory of Joe Gray, where they were characterized for subtype by expression profiling (22). In our laboratory, the cells were thawed and used within 10 tissue culture passages. SUM-149PT is an inflammatory breast cancer cell line (23) that carries a BRCA1 mutation.
Inhibitors and chemotherapy drugs
PF-00477736 (a gift of Pfizer Inc.) potently and specifically inhibits Chk1 with a K(i) of 0.49 nM, abrogates cell cycle arrest induced by DNA damage and enhances cytotoxicity of chemotherapeutic agents (17), including gemcitabine and carboplatin. Concentrations of gemcitabine (Tecoland Corporation, Edison, NJ) and carboplatin (MP Biomedicals, LLC. OH USA) used in our studies were as reported previously (24, 25). 10 mM Hydroxyurea (HU, Sigma) was used to induce DNA damage in breast cancer cells (24), to assess checkpoint protein expression after treatment with PF-00477736.
The PARP inhibitor, ABT-888 (Veliparib, provided through the Cancer Therapy Evaluation Program, National Cancer Institute), consists of benzimidazoles and displays excellent potency against PARP1/2 enzymes with a K(i) of 5 nM in whole cell assay, is aqueous soluble, orally bioavailable, and demonstrated good in vivo efficacy in xenograft models (26). We obtained the PARP inhibitor, BSI-201 (Iniparib) (7), through the Medicinal Chemistry Shared Facility at the OSU Comprehensive Cancer Center.
Cell survival assays
Methods for assessment of effect of PARP or Chk1 inhibitor activity on survival of breast cancer cells were adapted from published reports (4,5,27–29). MTS assays were performed for each drug or drug combination, as follows: 80% confluent cells were trypsinized and 7500 cells/well reseeded into 96 well plates in triplicate; 24 h later, medium was replaced with 100μL fresh medium, including increasing concentrations of PARP inhibitor in 25μLmedium added into 100μL medium. For PARP inhibition experiments, 72 h after adding drugs, MTS assay was performed after addition of 20μL CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega). For Chk1 inhibition experiments, 16 h after addition of inhibitor, medium was replaced with fresh medium, including Chk1 inhibitor, with or without gemcitabine or carboplatin; 48 h later, MTS assay was performed to estimate cell viability.
Clonogenic assay
Breast cancer cells were cultured in 6 well plates and treated with PF-00477736 or ABT-888 for 16 h for assessing clonogenicity after inhibitor alone. For clonogenicity assays after combination treatment with inhibitor plus chemotherapeutic drug, gemcitabine or carboplatin (10 ng/mL) was then added; 48 h later, cells were trypsinized and 2000 cells reseeded in inhibitor-free medium in 6 well plates in triplicate. After 14 days of incubation without inhibitor, cells were fixed in methanol:acetic acid (3:1), stained with crystal violet, and colonies counted.
Western blot analysis
Cells were scraped in lysis buffer, including protease inhibitors. Protein concentrations were determined by Bradford assay, using USA Bio-Rad protein assay reagent (Bio-Rad). Aliquots of 25 μg total protein were run on SDS-PAGE gels and transferred to Hybond ECL membrane (Amersham Pharmacia, USA). Membranes were immunoblotted with primary antisera: rabbit polyclonals anti-Chk1, anti-Chk2, and anti-ER-α and mouse monoclonal anti-Cdc25A, (Santa Cruz Biotechnology, Santa Cruz, CA); rabbit polyclonals anti-phospho-Chk1 (Ser317 and Ser345), anti-phospho-Chk2 (T68) and anti-phospho-Histone H2AX and rabbit monoclonal anti-HER2 (Cell Signaling Technology); rabbit polyclonal anti-PARP (Roche Applied Science); rabbit polyclonal anti-Brca1 (30); rabbit polyclonal anti-Fhit and anti-Wwox (14); mouse monoclonal anti-GAPDH (Calbiochem), followed by secondary goat-anti-rabbit or goat-anti-mouse serum (Invitrogen). For assessment of inhibition of PARP activity, cells treated with or without ABT-888 at 5 or 50 μM concentration for 72 h were analyzed for levels of PAR by Western blot (31). Anti-PAR rabbit polyclonal antibody was purchased from Trevigen.
Statistical analysis
Results of MTS assays were expressed as average values of three or four measurements. Results of clonogenic assays were represented as average values of three measurements in two different experiments. Error bars for all experiments represent one standard error around the mean. p values were determined by Student’s two-sided t test relative to untreated cells.
Results
Subtypes of Cell Lines
The cell lines used were characterized by Neve et al (22), who reported transcriptional expression profiling of 51 breast cancer cell lines, that allowed identification of specific breast cancer subtypes according to defined subtype specific profiles (21). MCF-7, T47D, ZR-75-1, SK-BR-3, MDA-MB-453 were identified as luminal subtype; BT-20, MDA-MB-468, MDA-MB-231 and MDA-MB-436 were identified as basal type. The SUM-149PT cell line, derived from an inflammatory breast cancer with BRCA1 mutation, has also been identified as basal subtype (32) and was used in a subset of experiments. Finn et al (33) reported HER2 status of 39 human breast cancer cell lines and found that SK-BR-3 and MDA-MB-453 showed amplification and expression of the HER2 gene, though expression was not high in the latter cell line, as we confirmed by western blot assay. Since these two cells are negative for expression of estrogen receptor (ER, shown later in results), we have considered these cells as HER2 over-expressing.
Effects of PARP inhibition on breast cancer-derived cells
First we examined viability of nine of the breast cancer cell lines representing each of the subtypes, by MTS assay in the presence of the PARP inhibitor ABT-888; see Fig. 1A for the dose response curve of the cell lines. Only one cell line, MDA-MB-436, with BRCA1 deficiency, showed sensitivity to ABT-888 at 10 to 50 μM concentration after 72 h treatment. To confirm the PARP inhibitory activity of ABT-888, we assessed the level of polymerized PAR in five breast cancer cell lines, as well as the transformed MCF10A cells, as shown in Supporting Fig S1. ABT-888 inhibits PARP activity in each of the cell lines tested, as shown by western blot assay. To determine if a second well-characterized PARP inhibitor would show differential effects on breast cancer cell lines, we examined the dose response of a subset of breast cancer cells to treatment with BSI-201 by MTS assay. Again, the MDA-MB-436 cells were most sensitive to BSI-201 (see Fig. 1B).
Fig. 1.

Dose response of breast cancer cell lines to treatment with PARP inhibitors. A. Nine breast cancer cell lines were assessed for ABT-888 sensitivity by MTS assay. The line graph shows results of averages (± SD) of at least triplicate assays as % change in cell number relative to untreated cells after 72 h treatment (1, 10, 25, 50 μM ABT-888); B. Similar dose response curve for treatment of a subset of breast cancer cell lines with BSI-201.
We also performed clonogenic assays after ABT-888 treatment of a subset of these cell lines. Results are shown for specific cell lines in Fig 2A, which illustrates that MDA-MB-436 cells are most sensitive, with almost no colony growth at 5 μM ABT-888; SUM-149PT cells were the next most sensitive with ~60% inhibition of colony growth at 50 μM. Moreover, combination treatment of ABT-888 and gemcitabine did not show an additive effect in MTS assay, though MCF-7 cells were sensitive to gemcitabine, as shown in Fig. 2B, and the TN cell, MDA-MB-468, showed some sensitivity to gemcitabine alone at 100 ng/ml (Fig 2B) in MTS assay. The BRCA1-deficient TN MDA-MB-436 cells exhibited significant sensitivity to ABT-888 alone in MTS assays (Fig. 2B and Fig. S2 for additional breast cancer cell lines) and addition of gemcitabine showed no effect on survival of these cells in MTS assay. Clonogenic assays with 5 μM ABT-888 plus 10 μg/ml carboplatin were attempted but with this drug combination no colonies were observed with the cell lines tested (MCF-7, T47D, MDA-MB-468, MDA-MB-231, MDA-MB-436). Effects of PARP inhibition on breast cancer cell lines of specific subtype are summarized in Table 1.
Fig. 2.
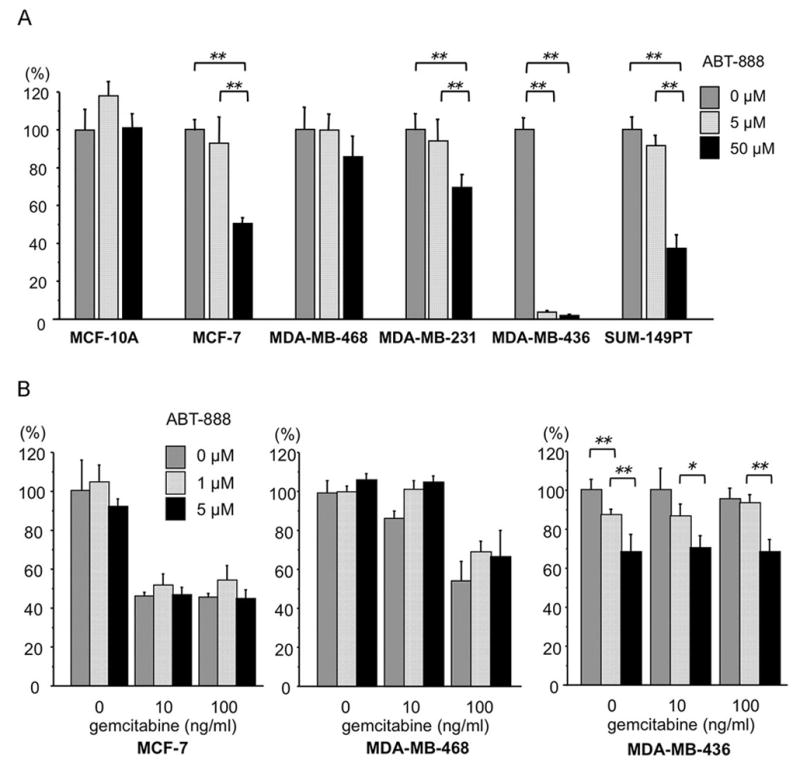
Effects of PARP inhibitor and/or gemcitabine on breast cancer cell lines. A. Clonogenic assay of breast cancer cell lines treated with ABT-888 concentrations shown; the bar graphs show results as averages (± SD) of at least triplicate assays; p values relative to non-treated cells were determined by Student’s t test; **, P<0.01; B. MCF-7, MDA-MB-468 and MDA-MB-436 cells were assessed for ABT-888 and/or gemcitabine sensitivity by MTS assay; the bar graphs show results as averages (± SD) of at least triplicate assays; p values relative to non-treated cells were determined by Student’s t test; *, P<0.05, **, P<0.01.
Table 1.
Summary of results of inhibition and protein expression experiments
| Cell line | MCF-7 | T47D | ZR-75-1 | SK-BR-3 | MDA-MB-453 | BT-20 | MDA-MB-468 | MDA-MB-231 | MDA-MB-436 |
|---|---|---|---|---|---|---|---|---|---|
| subtype | luminal | luminal | luminal | HER2+ | HER2+ | TN | TN | TN | TN |
| Protein expression | l. met | l. met | l. met | l. met | l. met | primary | PE | PE | PE |
|
| |||||||||
| Chk1 | + | +++ | + | + | +++ | ++ | +++ | +++ | + |
| pChk1(Ser317) | ± | − | − | − | ± | ± | ± | + | + |
| Chk2 | + | +++ | + | + | + | +/− | + | + | + |
| pChk2 (T68) | − | + | − | ± | − | ± | ± | − | +++ |
| γH2AX | + | − | − | ++ | ++ | ++ | + | ++ | ++ |
| PARP | ++ | + | +++ | + | ++ | ++ | ++ | ++ | +++ |
| Wwox | + | − | ++ | ++ | − | ++ | + | − | − |
| Fhit | + | ± | ++ | ++ | ++ | − | + | ++ | − |
| BRCA1 | + | + | + | + | + | + | + | + | +* |
| p53, reported | +/− | ++* | − | + | − | ++ | + | ++* | − |
| ER | ++ | +++ | + | − | − | − | − | − | − |
| HER2 | − | − | − | ++ | ± | − | − | − | − |
| GAPDH | ++ | + | +++ | ++ | ++ | +++ | ++ | ++ | +++ |
|
| |||||||||
| Effect of drug (MTS assay) | |||||||||
|
| |||||||||
| PF-00477736 | − | + | ++ | ++ | +++ | + | +++ | ||
| additive w gem | − | + | + | ++ | + | + | + | ||
| with carboplatin | − | + | + | ||||||
| ABT-888 | − | − | − | − | − | − | − | − | ++ |
| with gem | − | − | − | ||||||
| BSI-201 | − | − | − | ++ | |||||
|
| |||||||||
| Effect of drug (clonogenic assay) | |||||||||
|
| |||||||||
| PF-00477736 | + | ++ | ++ | + | |||||
| additive w gem | − | − | − | ||||||
| ABT-888 | − | − | − | +++ | |||||
expresses the mutant form of the protein
l. met, derived from lung metastasis; PE, derived from pleural effusion
Effects of Chk1 inhibition on breast cancer-derived cells
Next, we performed MTS assays of effects of PF-00477736 alone for: four basal subtype cell lines, BT-20, MDA-MB-468, MDA-MB-231, MDA-MB-436; two luminal, MCF-7, T47D; and a HER2 positive subtype cell, MDA-MB-453 (see Fig. 3A, Fig. S3A; left section of bar graphs for each cell line). PF-00477736 showed dose-related effects in MDA-MB-453, BT-20, MDA-MB-468 and MDA-MB-436 cells (results summarized in Table 1). Results showed that MCF-7 cells were unaffected by PF-00477736, and that the basal-like MDA-MB-231 and luminal T47D cells were not strongly responsive to PF-00477736 alone.
Fig. 3.

Effect of Chk1 inhibitor and chemotherapeutic drugs on survival of breast cancer cell lines of defined subtype. A. MCF-7, MDA-MB-468 and MDA-MB-436 cells were assessed for PF-00477736 and/or gemcitabine sensitivity by MTS assay; the bar graphs show results as averages (± SD) of at least triplicate assays; p values were determined by Student’s t test; *, P<0.05, **, P<0.01. B. The same cells were assessed for effects of PF-00477736 and/or gemcitabine by clonogenic assay; the bar graphs show results of triplicate experiments as % colony formation relative to untreated cells; % change in colonies formed after treatment (0.2 or 1 μM PF-00477736 and/or 10 ng/ml gemcitabine) of breast cancer cell lines; p values were determined by Student’s t test; *, P<0.05, ***, P<0.001; C. Effects of PF-00477736 and carboplatin treatment on survival of breast cancer cells. The bar graphs illustrate results of at least triplicate MTS assays as averages ± SD, relative to untreated cells; p values were determined by Student’s t test relative to the non-treated cells; *, P<0.05, **, P<0.01.
To further evaluate effects on breast cancer cells of PF-00477736 alone and combined with chemotherapeutic drugs, we performed cell toxicity tests by MTS and clonogenic assays. Results for selected cell lines are shown in the bar graphs in Fig. 3 (Fig. S3, for remaining cell lines). Luminal subtype MCF-7 cells were completely insensitive to PF-00477736 in MTS assay, showed slight sensitivity in clonogenic assay, exhibited high sensitivity to gemcitabine in both MTS and clonogenic assays (Fig. 3A, B) and were insensitive to carboplatin in MTS assay (Fig. 3C). MDA-MB-468 TN cells were sensitive to PF-00477736 in MTS assay and highly sensitive in clonogenic assay with no colony survival at 1 μM (Fig. 3A, B); these TN cells showed a small additive effect of gemcitabine plus PF-00477736, and carboplatin plus PF-00477736 in MTS assays (Fig. 3A, C). In MTS assays the BRCA1-deficient TN cell, MDA-MB-436, showed significant sensitivity to PF-00477736, little or no added sensitivity with gemcitabine and significant additive sensitivity with carboplatin (Fig. 3A, C). In clonogenic assays, the BRCA1-deficient MDA-MB-436 cells, like the HCC-1937 BRCA1-deficient cells (not shown), showed extreme sensitivity to PF-00477736 alone, such that surviving colonies were not detected (Fig. S3B). The BRCA1-mutant SUM-149PT cells were less sensitive than the TN cells to Chk1 inhibition, but more sensitive than MCF-7, with no colonies surviving 10 μM PF-00477736; the transformed but non-cancerous MCF10A cells were also highly sensitive to Chk1 inhibition in clonogenicity assays (Fig. S3B).
Expression of DDR-associated proteins
We examined expression of specific DDR-associated proteins in the breast cancer cell lines, to seek markers that might predict sensitivity to the PARP or Chk1 inhibitors in specific breast cancer subtypes. Fig. 4 illustrates expression of specific proteins in nine breast cancer cell lines after immunoblot analyses. Chk1 was expressed in all cell lines but most abundantly in T47D luminal cells, MDA-MB-453 HER2-expressing and in the TN cell lines; pChk1 was expressed at very low levels in several of the cell lines and higher levels in most of the Chk1 inhibitor-sensitive cells, MDA-MB-453, BT-20, MDA-MB-468, MDA-MB-231, MDA-MB-436; pChk2 was strongly expressed in T47D luminal cells and in BRCA1-deficient MDA-MB-436 cells. γH2AX was expressed at moderate levels in HER2+ and TN cells, possibly indicating presence of persistent DSBs. PARP was abundant in most of the cells, including MDA-MB-436 cells, the cell that was most responsive to PARP inhibition under conditions tested, confirming that expression of PARP protein did not correlate with response to ABT-888. Expression of the tumor suppressor proteins, Wwox, Fhit, and p53 did not show correlations among themselves, though Fhit loss correlated with expression of pChk2 for all cell types shown in Fig 4 (summarized in Table 1). ER and HER2 expression was assessed to confirm the reported subtypes of these cell lines (21, 22, 33–35).
Fig. 4.

Expression of DDR-associated proteins in breast cancer cell lines. Immunoblot analyses illustrate expression of subtype specific proteins (ER, HER2) and DDR-associated proteins. GAPDH levels serve as loading controls.
PF-00477736 affects levels of expression of pChk1, γH2AX and Cdc25A in sensitive cells
Next we examined expression of DDR and cell cycle-associated proteins in breast cancer cells after treatment with PF-00477736, with or without exposure to HU, a replication stress inducer. pChk2 expression level appears to be unaffected by treatment with one or both of the agents (Fig. 5). Chk1 level in MDA-MB-436 may be reduced in the presence of PF-00477736, especially after HU treatment, while pChk1 level is increased by PF-00477736 in the sensitive cells, MDA-MB-468 and 436, with or without HU treatment. Furthermore, pChk2 is expressed in the PF-00477736-sensitive cells shown, and induced in MCF-7 cells only after treatment with HU or the inhibitor. Collectively, these findings may reflect a greater dependency on the Chk1/Chk2 checkpoint pathways for survival of TN, compared to luminal cancer cells, and elevated expression of Chk proteins may represent markers for cancer cells susceptible to Chk1 inhibitors. γH2AX expression is induced by PF-00477736 exposure, only in the sensitive cells, MDA-MB-468 and 436, especially with the combination of PF-00477736 and HU treatment. The pChk1 downstream target, Cdc25A, is highly expressed in the PF-00477736-sensitive cells, is reduced in expression by HU-induced replication stress and this reduction is inhibited by the PF-00477736 inhibition of Chk1 activity.
Fig. 5.

Effects of PF-00477736, with or without HU, on expression of proteins involved in the DDR checkpoint pathway. MCF-7, MDA-MB-468 and MDA-MB-436 were treated with 0.2 μM PF-00477736 for 16 h. HU (10 μM) was then added to selected wells; 2 h later, cells were lysed and lysates analyzed by immunoblot.
Discussion
Several reviews of the synthetic lethality concept, involving inhibition of a branch of the DDR pathway in cancers with a different repair pathway crippled during cancer development, have summarized ongoing tests of this concept in clinical trials (6–8, 17). At least seven PARP inhibitors are being tested, including ABT-888, which is in Phase II trials for metastatic melanoma and breast cancer in combination with temozolomide (cited in 7). Several of these inhibitors are in phase II trials for treatment of BRCA-associated breast or ovarian cancer (reviewed in 6–8).
Inhibitors of Chk1/2 have also been shown to sensitize tumor cells to DNA damaging agents (8, 29). Several specific inhibitors of Chk1 or 2 kinase, including PF-00477736, exhibit Chk1 or 2 inhibition at various concentrations; PF-00477736 displays greatest specificity for Chk1 autophosphorylation and has been shown to sensitize cancer cells to anticancer agents (17). Up to four inhibitors of Chk1/2 are in Phase I-III trials for breast and other solid cancers in combination with various cytotoxic agents (cited in 8, 29).
Reviews of these new inhibitor studies have emphasized that success will depend on identification of markers that identify patients with checkpoint or DNA repair defects to stratify patients that should be treated with a specific novel targeted therapy (8). Nevertheless, few reports have described efforts to test specific cancer types for sensitivity to PARP or checkpoint inhibitors in advance of clinical trials. Asakawa et al (36) used an ingenious method to determine status of DDR proteins in vivo. These investigators obtained biopsies of breast cancers before and after first cycle neoadjuvant treatment and assessed nuclear focus formation of DDR proteins by immunocytochemical analysis. In this study, the presence of BRCA1, γH2AX or Rad51 foci before, or Rad51 foci after treatment, was inversely correlated with response to chemotherapy. Similar studies predict which cancers need synthetic lethality treatment, ie combination DDR inhibitor plus chemotherapy.
We have used breast cancer-derived cell lines of defined subtype to assess sensitivity of subtypes to specific DDR inhibitors and in parallel have examined expression levels of DNA checkpoint and repair associated proteins, in initial attempts to identify biomarkers to predict sensitivity to PARP or Chk1 inhibition.
The results suggest that neither TN status nor expression of PARP is a good predictor of sensitivity to PARP inhibition, at least not inhibition by ABT-888; triple negativity of breast cancers may be a good predictor of sensitivity to Chk1 inhibition. Additionally, the protein expression results suggest that expression of pChk1 may be a strong predictor of Chk1 inhibitor response, with near 100% sensitivity and high specificity; γH2AX expression also showed high sensitivity but less specificity for prediction of response to Chk1 inhibition, and absence of either Fhit or Wwox tumor suppressor protein expression was also correlated with PF-00477736 sensitivity in the breast cancer cells tested. It will be important to examine larger panels of cancer subtypes, and expanded panels of inhibitors and biomarkers, but results of this study suggest that future stratification of cancer patients based on biomarker expression in their cancers will lead to optimum design of synthetic lethal strategies for combination therapies.
The PARP inhibitor, BSI-201 or Iniparib, has been used in phase I and II trials for treatment of TN breast cancer (20). According to this report, Iniparib plus gemcitabine and carboplatin improved the rate of clinical benefit and the rate of overall response in TN cancers, though there was not a comparison with breast cancers of other subtypes. Our test of dose response of a subset of the breast cancer cell lines (Fig. 1B) showed a highly variable response to BSI-201 treatment, with the MDA-MB-436 cells the most sensitive. Very recently, Drew et al (37), have reported that another PARP inhibitor, AG014699 (≤10 μM), was cytotoxic to cells with mutated BRCA1/2 or epigenetically silenced BRCA1 but not to cells without BRCA1/2 mutations or that were heterozygous for BRCA2 mutation.
It is clear from all of the reported studies on inhibitors of DDR proteins, that these inhibitors will have impacts on cancer treatment; but it also seems clear that extensive in vitro studies of specific subtypes of breast cancers, and other cancer types, can refine sensitivity to specific inhibitors and drug combinations and stratify cancer subtypes for predicted responses to specific treatments.
Supplementary Material
PARP inhibition by ABT-888 treatment in breast cancer cell lines.
{kind=link}
Effect of PARP inhibitor and PARP inhibitor plus gemcitabine on breast cancer cell lines.
{kind=link}
Effects of PF-00477736, alone or combined with gemcitabine, on breast cancer cells.
{kind=link}
Acknowledgments
Supported by US Public Health Service/National Cancer Institute Grants CA120516, CA132453, CA115965 and training grants T32GM068412 and F31CA157150-01 (to JCS).
We thank our OSU CCC colleagues, Drs. Tim H-M. Huang, Tao Zuo and Jeffrey Parvin, for contribution of breast cancer cell lines. We gratefully acknowledge the gifts of ABT-888 by the CTEP program of the National Cancer Institute, and the PF-00477736 inhibitor by Pfizer, Inc.
Footnotes
All authors declare no potential or actual financial or other conflicts of interest
Disclosure Statement
The authors have no conflict of interest.
References
- 1.Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 2.Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 3.Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26:7773–79. doi: 10.1038/sj.onc.1210881. [DOI] [PubMed] [Google Scholar]
- 4.Farmer H, McCabe R, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 5.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–17. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 6.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 7.Rowe BP, Glazer PM. Emergence of rationally designed therapeutic strategies for breast cancer targeting DNA repair mechanisms. Breast Cancer Research. 2010;12:203–14. doi: 10.1186/bcr2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolderson E, Richard DJ, Zhou BS, Khanna KK. Recent advances in cancer therapy targeting proteins involved in DNA double strand break repair. Clin Cancer Res. 2009;15:6314–20. doi: 10.1158/1078-0432.CCR-09-0096. [DOI] [PubMed] [Google Scholar]
- 9.Alli E, Sharma VB, Sunderesakumar P, Ford JM. Defective repair of oxidative DNA damage in triple-negative breast cancer confers sensitivity to inhibition of poly(ADP-ribose) polymerase. Cancer Res. 2009;69:3589–96. doi: 10.1158/0008-5472.CAN-08-4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4:814–19. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 11.Ingvarsson S, Agnarsson BA, Sigbjornsdottir BI, et al. Reduced Fhit expression in sporadic and BRCA2-linked breast carcinomas. Cancer Res. 1999;59:2682–89. [PubMed] [Google Scholar]
- 12.Turner BC, Ottey M, Zimonjic DB, et al. The fragile histidine triad/common chromosome fragile site 3B locus and repair-deficient cancers. Cancer Res. 2002;62:4054–60. [PubMed] [Google Scholar]
- 13.Silva Soares EW, de Lima Santos SC, Bueno AG, et al. Concomitant loss of heterozygosity at the BRCA1 and FHIT genes as a prognostic factor in sporadic breast cancer. Cancer Genet Cytogenet. 2010;199:24–30. doi: 10.1016/j.cancergencyto.2010.01.019. [DOI] [PubMed] [Google Scholar]
- 14.Guler G, Uner A, Guler N, et al. The fragile genes, FHIT and WWOX, are coordinately inactivated in invasive breast cancer: correlations with clinical features. Cancer. 2004;100:1605–14. doi: 10.1002/cncr.20137. [DOI] [PubMed] [Google Scholar]
- 15.Guler G, Huebner K, Himmetoglu C, et al. Fragile histidine triad protein, WW domain-containing oxidoreductase protein, and activator protein 2 gamma expression levels correlate with basal phenotype in breast cancer. Cancer. 2009;115:899–908. doi: 10.1002/cncr.24103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verlinden L, Vanden Bempt I, Eelen G, et al. The E2F-regulated gene Chk1 is highly expressed in triple-negative estrogen receptor/progesterone receptor/HER-2 breast carcinomas. Cancer Res. 2007;67:6574–81. doi: 10.1158/0008-5472.CAN-06-3545. [DOI] [PubMed] [Google Scholar]
- 17.Blasina A, Hallin J, Chen E, et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol Cancer Ther. 2008;7:2394–404. doi: 10.1158/1535-7163.MCT-07-2391. [DOI] [PubMed] [Google Scholar]
- 18.Fillingham J, Keogh MC, Krogan NJ. GammaH2AX and its role in DNA double-strand break repair. Biochem Cell Biol. 2006;84:568–77. doi: 10.1139/o06-072. [DOI] [PubMed] [Google Scholar]
- 19.Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006;34:1416–26. doi: 10.1093/nar/gkl010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Shaughnessy J, Osborne C, Pippen JE, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med. 2011 Jan 5; doi: 10.1056/NEJMoa1011418. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 21.Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–27. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Golen KL, Davies S, Wu ZF, et al. A novel putative low-affinity insulin-like growth factor-binding protein, LIBC (lost in inflammatory breast cancer), and RhoC GTPase correlate with the inflammatory breast cancer phenotype. Clin Cancer Res. 1999;5:2511–2519. [PubMed] [Google Scholar]
- 24.Hoadley KA, Weigman VJ, Fan C, et al. EGFR associated expression profiles vary with breast tumor subtype. BMC Genomics. 2007;8:258. doi: 10.1186/1471-2164-8-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jordheim LP, Guittet O, Lepoivre M, Galmarini CM, Dumontet C. Increased expression of large subunit of ribonucleotide reductase is involved in resistance to gemcitabine in human mammary adenocarcinoma cells. Mol Cancer Ther. 2005;4:1268–76. doi: 10.1158/1535-7163.MCT-05-0121. [DOI] [PubMed] [Google Scholar]
- 26.Penning TD, Zhu GD, Gandhi VB, et al. Discovery of the Poly(ADP-ribose) polymerase (PARP) inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the treatment of cancer. J Med Chem. 2009;52:514–23. doi: 10.1021/jm801171j. [DOI] [PubMed] [Google Scholar]
- 27.Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;22:3785–90. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- 28.McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–15. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 29.O’Connor MJ, Martin NMB, Smith GC. Targeted cancer therapies based on the inhibition of DNA strand break repair. Oncogene. 2007;26:7816–24. doi: 10.1038/sj.onc.1210879. [DOI] [PubMed] [Google Scholar]
- 30.Schlegel BP, Starita LM, Parvin JD. Overexpression of a protein fragment of RNA helicase A causes inhibition of endogenous BRCA1 function and defects in ploidy and cytokinesis in mammary epithelial cells. Oncogene. 2003;22:983–91. doi: 10.1038/sj.onc.1206195. [DOI] [PubMed] [Google Scholar]
- 31.Donawho CK, Luo Y, Luo Y, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13:2728–37. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 32.Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011 Jun 1;:pii, 45014. doi: 10.1172/JCI45014. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finn RS, Dering J, Ginther C, et al. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/”triple-negative” breast cancer cell lines growing in vitro. Breast Cancer Res Treat. 2007;105:319–26. doi: 10.1007/s10549-006-9463-x. [DOI] [PubMed] [Google Scholar]
- 34.Nielson TO, Hsu FD, Jensen K, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10:5367–74. doi: 10.1158/1078-0432.CCR-04-0220. [DOI] [PubMed] [Google Scholar]
- 35.Cheang MC, Voduc D, Bajdik C, et al. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res. 2008;14:1368–76. doi: 10.1158/1078-0432.CCR-07-1658. [DOI] [PubMed] [Google Scholar]
- 36.Asakawa H, Koizumi H, Koike A, et al. Prediction of breast cancer sensitivity to neoadjuvant chemotherapy based on status of DNA damage repair proteins. Breast Cancer Res. 2010;12:R17. doi: 10.1186/bcr2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drew Y, Mulligan EA, Vong WT, et al. Therapeutic potential of Poly(ADP-ribose) Polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J Natl Cancer Inst. 2011;103:334–46. doi: 10.1093/jnci/djq509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PARP inhibition by ABT-888 treatment in breast cancer cell lines.
Effect of PARP inhibitor and PARP inhibitor plus gemcitabine on breast cancer cell lines.
Effects of PF-00477736, alone or combined with gemcitabine, on breast cancer cells.