

Abstract
Much evidence indicates that soluble amyloid beta (Aβ) oligomers are key mediators of early cognitive loss, but the localization and key peptide species remain unclear. We have used flow cytometry analysis to demonstrate that surviving Alzheimer's disease (AD) synapses accumulate both Aβ and p-tau. The present experiments use peptide-specific xMAP assays and Western blotting to identify the Aβ peptide species in synaptosome-enriched samples from normal human subjects, neurologic controls, and AD cases. Aβ40 peptide levels did not vary, but both Aβ42 and Aβ oligomers were increased in soluble AD extracts, with oligomer levels 20-fold higher in aqueous compared to detergent extracts. In Western blots, a ladder of SDS-stable oligomers was observed in AD cases, varying in size from monomer, the major peptide observed, to larger assemblies up to about 200 kD and larger. Multiple oligomers, including monomer, small oligomers, a 56 kD assembly, and APP were correlated with the Aβ level measured in flow cytomety-purified synaptosomes. These results suggest that multiple APP processing pathways are active in AD synapses and multiple soluble oligomeric assemblies may contribute to synaptic dysfunction.
Keywords: synaptosome, flow cytometry, Parkinson's disease, synaptophysin, PSD-95, A11 antibody, OC antibody
1. Introduction
A number of studies have documented that levels of soluble amyloid beta (Aβ) peptides are superior to amyloid deposits as correlates of cognitive decline in Alzheimer's disease (AD; McLean et al., 1999; Naslund et al., 2000). Accordingly, the original amyloid cascade hypothesis has evolved to propose that soluble oligomeric Aβ assemblies precede deposition and are the proximal cause of synaptic dysfunction and early impairment in AD (see Walsh and Selkoe, 2007 for review). However, the size of the key assembly state and relevant downstream pathways remain the subject of intense study. Among natural low-n assemblies, dimers and/or trimers in particular have been isolated from AD brain and shown to impair cognition in vitro (Cleary et al., 2005; Townsend et al., 2007; Klyubin et al., 2008; Shankar et al., 2008). In the Tg2576 mouse model a larger assembly, (Aβ*56), possibly a multimer of smaller oligomers, was associated with cognitive decline in Tg2576 mice (Lesne et al., 2006).
Soluble Aβ peptides are associated with synaptic loss (Lue et al., 1999), and multiple studies have shown that soluble oligomers bind to dendritic spines in primary cultures (Lacor et al., 2004, 2007). Recent evidence also suggests that brief passive immunotherapy has acute and extended benefits on synaptic density and plasticity (Rozkalne et al., 2009; Spires-Jones et al., 2009). Consistent with synaptic Aβ release, interstitial Aβ levels are increased by synaptic activity (Cirrito et al., 2005, 2006), and have been shown to correlate with neurological status in patients with brain injury (Brody et al., 2008). Reasoning that study of surviving synaptic terminals is critical for understanding the sources for synaptic Aβ production and release as well as pathways leading to loss of synapses, we have analyzed human synaptosomal preparations by flow cytometry analysis and have shown that Aβ accumulates in synaptic terminals in multiple regions of AD brain. P-tau also accumulates in Aβ-bearing synapses, and the co-localization of Aβ and p-tau is accompanied by increased synaptosome size, modest losses of PSD-95, and increased cholesterol and GM1 ganglioside (Gylys et al., 2004, 2007, 2008). With flow cytometry, the synaptosomal Aβ signal is best detected by an N-terminal antibody (10G4) that does not discriminate between peptides; the present study correlates the flow cytometry signal with a series of peptide and conformation-specific antibodies along with a series of Aβ peptide-specific assays on the luminex platform. We report here that monomeric Aβ is prominent among multiple SDS-stable soluble Aß species, including a 56 kDa assembly, in synaptic terminals from AD cortex.
2. Materials and Methods
2.1 Materials
The monoclonal anti-Aβ antibody 10G4 has been described previously (Mak et al., 1994). Polystyrene microsphere size standards were purchased from Polysciences, Inc. (Warrington, PA), and rhodamine-conjugated anti-mouse antibody from Chemicon (San Diego, CA). The following monoclonal antibodies were purchased: anti-SNAP-25 (Sternberger Monoclonals Inc., Lutherville, MD), anti-PSD 95 (Upstate Biotechnology, Lake Placid, NY), 6E10 antibody (Signet Labs, Dedham, MA), anti-synaptophysin from Abcam (Cambridge, MA), 4G8 antibody (Covance, Denver, PA), and anti-APP 3E9 (MBL, Naka-ku Nagoya, Japan). A11 was the kind gift of C. Glabe (UC Irvine, CA), and OC antibody was received from R. Kayed (UTMB, Galveston, TX). The rabbit anti-Aβ42 and anti-Aβ-40 antibodies were from T. Golde (Mayo Clinic, Jacksonville, FL).
2.2 Human brain specimens
Brain samples, primarily superior parietal (A7) cortex were obtained at autopsy from the Alzheimer's Disease Research Centers at USC and UCLA; for some experiments frontal (A9) or parietal (A39) samples were substituted. Samples were obtained from a total of 14 cases (10 females, 4 males); 7 were diagnosed clinically and histopathologically with AD, and 3 were neurological control cases. The control cases included 2 Parkinson's disease (PD) and 1 tauopathy case. The 4 cognitively normal aged controls were confirmed histopathologically. The mean age of AD cases was 86.3, and 84.6 for normal and control cases. The mean postmortem interval for AD cases was 8.2 h, and for normal and control cases was 7.0 h.
2.3 P-2 preparation
Samples (∼0.3-5g), were minced and slowly frozen on the day of autopsy in 10% DMSO and 0.32M sucrose and stored at −70°C until homogenization. The P-2 (crude synaptosome) fraction was prepared as described previously (Gylys et al., 2003), briefly, the homogenate was first centrifuged at 1000 g for ten minutes; the resulting supernatant was centrifuged at 10,000 g for 20 minutes to obtain the crude synaptosomal pellet. Aliquots of P-2 are routinely cryopreserved in 0.32M sucrose and banked at -70°C until the day of the experiment.
2.4 Immunolabeling of P-2 fraction
P-2 aliquots were immunolabeled for flow cytometry analysis according to a method for staining of intracellular antigens (Schmid et al., 1991). Pellets were fixed in 0.25% buffered paraformaldehyde (1 hr, 4°C) and permeabilized in 0.2% Tween20/PBS (15 min., 37°C). Antibodies were labeled directly with Alexa Fluor 488 or 647 reagents according to kit directions (Zenon Alexa Fluor Labeling Kit, Invitrogen, Carlsbad CA). The labeled antibody mixture was added to P-2 aliquots (5 μl of P-2 pellet/sample, ∼5-8 μg/μl) and incubated at RT for 30 min. Pellets were washed 2 times with 1 ml 0.2% Tween20/PBS, resuspended in PBS buffer (0.75 ml) for flow cytometry analysis. The synaptosomal pellet was dispersed for all washes and for incubations with fixative, detergent, and antibody, then collected by centrifugation (1310 × g at 4°C).
2.5 Flow cytometry
Data was acquired using an BD-FACSCalibur analytic flow cytometer (Becton-Dickinson, San Jose, CA) equipped with argon 488 nm, helium-neon 635 nm, and helium-cadmium 325 nm lasers. 5,000 particles were collected and analyzed for each sample. Debris was excluded by establishing a size threshold set on forward light scatter. Alexa 488 and Alexa 647 fluorochromes were detected by the LSR's FL1, Ssc-W, photomultiplier tube detectors, respectively. Analysis was performed using FCS Express software (DeNovo Software, Ontario, CAN).
2.6 Synaptosome extracts
Crude P2 fractions (x μl P-2 pellet, ∼ μg protein) were first extracted by sonication in a detergent-free buffer (10 mM Tris, 1 mM EGTA, 10% sucrose, pH 7.5) and then spun at 25,000 g. The supernatant of this detergent-free extraction was used to quantify the levels of aqueous soluble Aβ species. The remaining pellet was extracted by sonication in the same buffer containing 1% N- lauroylsarkosyl (NLS) and spun at 300,000 g. This supernatant was analyzed as the detergent soluble fraction.
3.7 Luminex assay
The aqueous and detergent soluble fractions were quantified using specific bead-based kits for aggregated Aβ, Aβ40, and Aβ42 (Invitrogen, CA). Assays were performed on a Luminex instrument using X-map Technology (Austin, TX) and xPONENT software. Standard curves were constructed from authentic standards included with each kit; the lower limit of quantification was 52 pg/mL for aggregated Aβ; 50 pg/mL for Aβ40 and Aβ42. Each extract was analyzed in duplicate for each analyte.
2.8 Statistics
Student's t tests were calculated using the Vassarstat interactive statistical website (http://faculty.vassar.edu/lowry/VassarStats.html; Richard Lowry, Poughkeepsie, NY, USA). Correlation coefficients were calculated with the Spearman correlation coefficient procedure.
2.9 Western Blotting
Samples (20 μg P-2 sample/lane) were boiled in Laemmli loading buffer (2%SDS, Invitrogen) and electrophoresed on 4-20% Tris-Tricine gradient gels, then transferred to PVDF membranes. Because actin and similar genes may be affected in AD samples, membranes were stained with Ponceau (immediately after transfer) and Coomassie blue (after immunolabeling) to ensure equal protein loading. Only blots with equal loading were quantified and included in figures. Membranes were blocked for 1 hr. at room temperature in 10% non-fat dried milk in PBS, followed by incubation overnight at 4°C with primary antibodies (1:1000) in PBS containing 0.05% Tween 20 (PBS-T) and 1.5% (W/V) albumin. After rinsing in PBS-T, the membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-mouse IgG (1:50,000) or anti-rabbit IgG (1:100,000) in PBS-T with 1.5% albumin for 1 hr. Immunolabeled proteins were visualized by enhanced chemiluminescence (ECL) detection reagents (SuperSignal West Femto, Thermo Scientific, Rockford, IL). Resulting films were scanned and quantified on a UVP BioSpectrum 600 imaging system (Upland, CA) using VisionWorks software.
3. Results
3.1 Elevated Aβ in intact AD synaptosomes is accompanied by high levels of APP
Synaptosomes are resealed synaptic boutons that form when fresh tissue is gently homogenized with the appropriate shear force in isotonic sucrose. The preparation has been thoroughly characterized over several decades; typical synaptosomes contain presynaptic cytoplasm, synaptic vesicles, mitochondria and cytoskeleton proteins enclosed by a spherical membrane, along with attached fragments of postsynaptic membrane and postsynaptic densities (see Dunkley et al. 2008 for review). Flow cytometry analysis of synaptosomes enables focus on a large and highly pure population (3000-5000 analyzed/case) of human synaptic terminals; by using an analysis gate to include only particles between 0.75 and 1.5 microns, we have previously shown the synaptosomal population in flow cytometry studies to be ∼95% pure. This compares to ∼ 80% for a gradient-purified synaptosomal preparation (Gylys et al., 2004b; 2007, Fein et al., 2008; Wolfe and Kapatos al 1989). Synaptosomal purity for the present experiments is illustrated by SNAP-25 immunolabeling of a representative parietal cortex sample (Fig.1A), compared to background (Fig.1B).
Fig 1. Flow cytometry analysis of Aβ and APP in AD synaptosome.
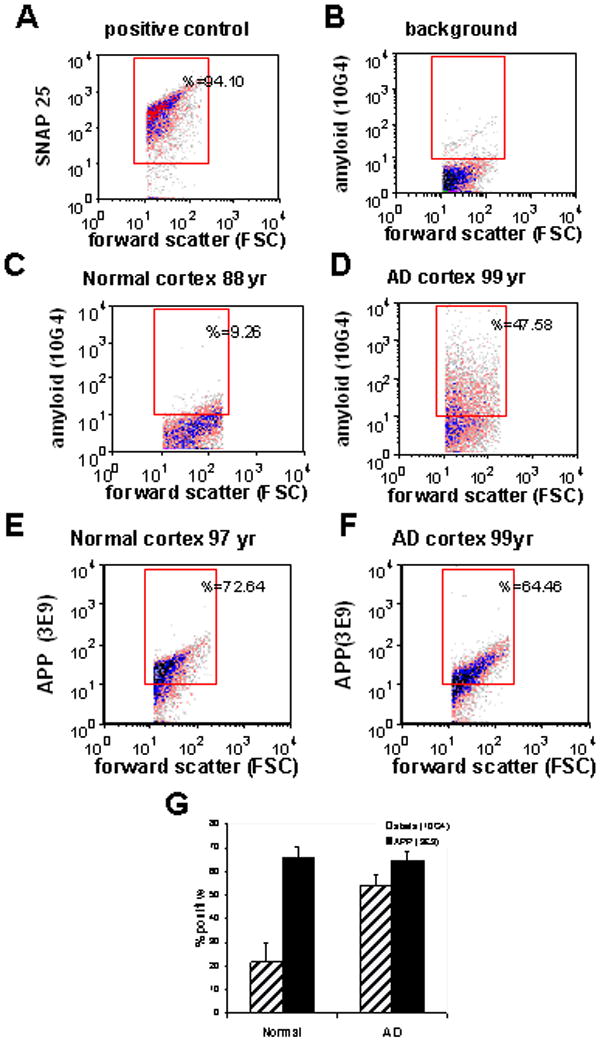
Representative flow cytometry controls are shown for: (A) an AD parietal cortex sample labeled for SNAP-25 as a positive control and indicator of synaptosomal purity, and (B) background labeling in the presence of a non-specific isotype control. Representative synaptosomal amyloid beta (Aβ) labeling is shown for a parietal cortex sample from an aged normal control case (C), and an AD case (D). Synaptosomal APP labeling is shown for a parietal cortex sample from an aged normal control case (E), and from an AD case (F). Size of positive fraction for APP and Aβ in aged cognitively normal (n=5) and AD cases (n=19); data was collected from 5000 terminals for each sample (p< 0.001, Student's t test).
Flow cytometry analysis of parietal cortex synaptosomes was used to compare Aβ labeling (with the antibody 10G4, an N-terminal antibody directed against residues 5-17), with labeling for amyloid precursor protein (APP, 3E9 antibody; Fig. 1C,D). Marked synaptic Aβ accumulation is observed in AD parietal cortex, with little Aβ detected in cognitively normal aged control cases. APP is present at high levels in synapses, with ∼65-70% of synaptosomes positive for APP in AD and in controls (Fig.1E,F). In aggregate, the fraction of Aβ-positive synaptosomes increases from 22% (±8.2, n=5) in age-matched control subjects to 55% in AD parietal cortex (±4.6, n=19; p< 0.001); however, the fraction of synaptosomes positive for APP in controls (65%; (±4.9, n=5) was unchanged in surviving terminals in AD (65%;±3.9, n=19).
Sixty-four percent of synaptic terminals were labeled for Aβ with the 10G4 antibody in a representative Parkinson's disease (PD) case (Fig. 2A), and an even larger fraction (78%) was Aβ-positive in a PD case with 2 years of dementia, suggesting that synaptic Aβ elevations are not limited to AD.
Fig. 2. Flow cytometry analysis of N-terminal and C-terminal antibodies; Aβ in Parkinson's disease (PD) and mixed dementia.
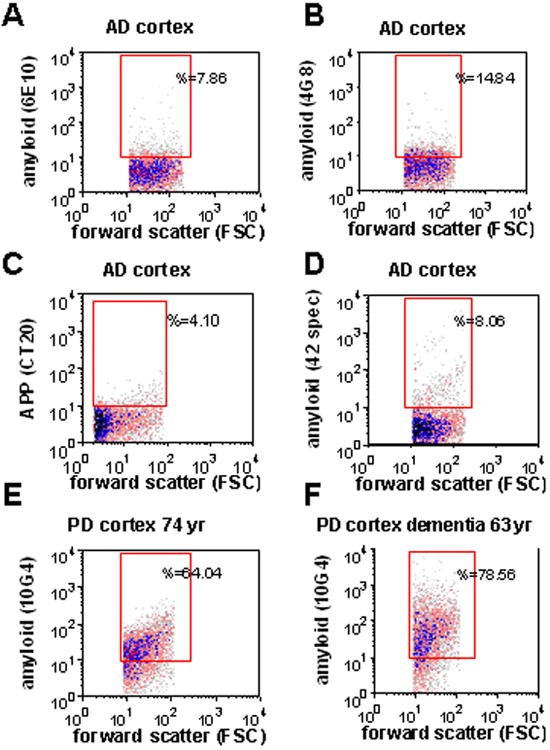
(A-D), Representative samples of AD cortex showing minimal immunolabeling in intact synaptosomes for the N-terminal antibodies 6E10 (A), 4G8 (B), CT20, a C-terminal APP antibody (C), and an Aβ 42-specific antibody (D). Synaptosomal Aβ immunolabeling for a parietal cortex sample from a Parkinson's disease case without dementia (E; case information in Table 1, lane 5), and for a dementia case with a 2 year history of dementia caused by Parkinson's disease (F; case information in Table 1, lane 6).
With flow cytometry, bright specific immunolabeling is obtained with the antibody 10G4 in intact AD synaptosomes that are fixed and permeabilized for labeling of intracellular antigens (Fig.1D,G). However, a Aβ42-specific C-terminal antibody labels a positive fraction just above background, indicating that the C-terminal epitope is not exposed in situ (Fig. 2A). The N-terminal antibody 6E10, which is directed against a similar epitope to 10G4, likewise labels a much smaller fraction of synaptosomes in the same case (Fig. 2B). No immunolabeling is observed in intact synaptosomes for the N-terminthe conformation-specific antibodies A11 and OC, which detect prefibrillar and fibrillar oligomers respectively (not shown). Taken together, these results indicate limited flow cytometric detection of Aβ peptide-specific epitopes in intact synaptosomes. This conclusion is in line with the observation that antigen masking has historically limited Aβ detection in situ, and is a factor contributing to false-negative results for intraneuronal localization of Aβ (Gouras et al., 2005).
3.2 Soluble oligomers and Aβ42 are the primary peptides in aqueous extractions
We next measured Aβ with a number of biochemical assays to confirm and extend the flow cytometry observations of synaptic Aβ accumulation. The goal was to compare Aβ pools and peptides in a series of AD and normal cases; synaptosome-enriched P-2 fractions (crude synaptosomes; SEF) were used in order to obtain sufficient volume for the analyses. Western blots were used to demonstrate the enrichment of pre- and post-synaptic elements in the P-2 compared to the initial sucrose homogenate and the initial low-speed pellet (P-1), which contains nuclei and large membrane fragments (Fig. 3A,B). Synaptophysin, a synaptic vesicle protein involved in docking of presynaptic vesicles prior to exocytosis (Sudhof, 1995), was increased in the P-2 compared to the initial homogenate by 2.3-fold (p<0.01); P-2 enrichment for the post-synaptic NMDA scaffold protein PSD-95 (Kornau et al., 1995) was 1.92-fold (Fig. 3B; p<0.05). Differential distribution of SDS-stable Aβ peptide assemblies between the P-2 and initial homogenate is shown in Fig. 3 (C,D). Higher levels of two Aβ oligomers (43 kDa and tetramer) in the initial homogenate compared to the P-2 fraction (Aβ-42-specific antibody), with Aβ monomer levels higher in the P-2 (6E10 antibody). Increased monomer may result from breakdown of higher order oligomers during the Western analysis procedure, and seems likely to indicate impaired Aβ clearance in synaptic terminals.
Fig. 3. Biochemical analysis of Aβ42, Aβ42 and oligomeric amyloid beta in synaptosome-enriched fractions (P-2).
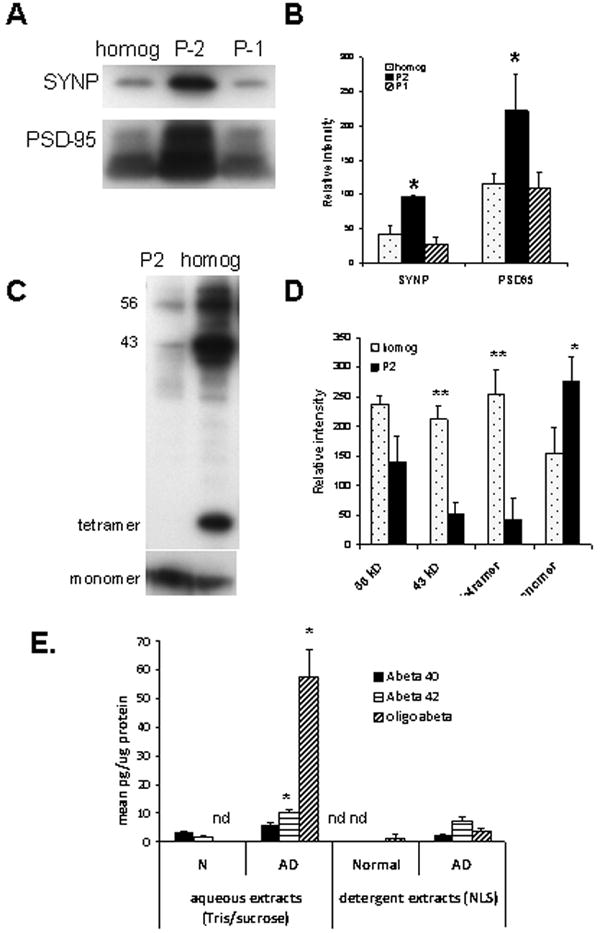
(A) Western blots comparing levels of pre-synaptic (synaptophysin, SYNP) and post-synaptic (PSD-95) markers in the initial homogenate, initial nuclear fraction (P-1) and synaptosome-enriched (crude synaptosome; P-2), with quantification illustrated in (B; n=4, **p<0.01, *p< 0.02 compared to initial homogenate). (C) Western blot showing Aβ level in P-2 compared to homogenate, with quantification in (D; **p<0.01, *p< 0.05 compared to initial homogenate). (E) Bead-based luminex biochemical assay for aqueous and detergent (1% N-laurylsarcosyl) extracts of synaptosome-enriched fractions from normal (n=2) and AD cases (n=7); nd, not detectable, *p<0.05 for AD vs. N and for aqueous vs. detergent-soluble extracts.
To determine the specific Aβ peptide(s) in synaptosomes, we used specific bead-based immunoassays using the XMAP technology (Luminex, Austin, Texas) to quantify three Aβ species (Aβ 40, Aβ42, and Aβ oligomers of 40 or 42 amino acids) in sequential extractions of P-2 fractions prepared from fresh unfixed AD samples. To measure Aβ in soluble and detergent fractions, P-2 pellets were first extracted by sonication in detergent-free buffer (Tris/EGTA/sucrose); the supernatant after centrifugation was used for the soluble extracts. The pellet was sonicated in the same buffer plus 1% N-laurylsarcosyl, centrifuged, and the supernatant used for the detergent extracts. Figure 3E shows that Aβ40 levels did not differ between groups, but Aβ42 was higher in aqueous vs. detergent extracts in AD samples (10.35±0.74 vs. 7.38±1.23 pg/μg; p<0.05; n=7 aqueous samples and 4 detergent samples). Aβ42 was also elevated in AD compared to control samples in aqueous (10.35±0.74 vs. 1.69±0.15 pg/μg; p<0.05) extracts; Aβ42 was undetectable in detergent extracts from control cases (n=2). The small number of controls was prompted by the large tissue volume required for extraction experiments, together with the limited availability of aged control cases and previous results showing relatively little synaptic Aβ pathology in control cases (Gylys et al., 2004, 2007). The aggregated Aβ assay is a bead-based sandwich assay that uses the same antibody for capture and detection, therefore only proteins with multiple copies of the Aβ N-terminus are quantified. Aggregated Aβ levels were undetectable in control samples, but were 20-fold higher in aqueous compared to detergent extracts in AD samples (57.57±9.51 vs 2.62±3.71 pg/μg; p<0.05). Taken together, the luminex assays indicate that soluble Aβ is primarily Aβ42 peptide, with the majority in oligomeric (n≥2) form. Because assays using the same antibody for capture and detection can not distinguish between multiple monomers bound to a carrier such as receptors for advance glycation endproducts (RAGE) or apolipoprotein E, the measure of oligomers may include carrier-bound Aβ.
3.3 Aβ Monomer and a 56 kD SDS-stable peptide assembly are prominent among multiple Aß species
In order to gain size information about SDS-stable Aβ assemblies in synaptosome-enriched fractions, western blotting experiments were performed with a series of anti-Aβ antibodies. For the present study we examined synaptosomes from a series of AD (n=7) and normal control cases (n=4). The series included three additional neurologic control cases: two with PD, and a third tauopathy dementia case that showed marked tau-immunoreactive neurofibrillary tangles and neuropil threads but no diffuse or neuritic plaques. Neurologic controls were included to better understand mixed pathology and the disease specificity of synaptic pathology in AD; for example, one of the PD cases displayed mixed pathology with a 2 year history of dementia, and a diagnosis of Lewy Body AD was considered for this case. Case information and synaptic Aβ levels measured by flow cytometry are presented for each case in Table 1 along with the lane number for each case in western analysis.
Table 1. Case information for Synaptosome Samples.
Cases used for Western analysis. Neuropil threads and plaques are reported for frontal cortex and refer to silver (Gallyas) stain. The plaque number includes plaques with and without cores; sparse (<5/field), mod (6 to 20/field), freq (21 to 30/field). RFU, mean relative fluorescence for Aβ and p-tau antibody fluorescence measured by flow cytometry (mean in superior parietal A7 cortex).
| Lane | Sex | Age | PMI (h) | Braak & Braak score | Frontal cortex atrophy | Neuritic plaques | Neurofib. changes (neuropil threads) | amyloid fluor. (RFU) | p-tau fluor (RFU) |
|---|---|---|---|---|---|---|---|---|---|
| Normal cases | |||||||||
| 1 | F | 97 | 5.5 | - | mild | 0 | 0 | 19.73 | 23.89 |
| 2 | M | 93 | 8.5 | - | mild | 0 | 0 | 45.16 | 25.27 |
| 3 | M | 82 | 14.9 | Normal with hippocampal sclerosis | mod | 0 | 0 | 27.36 | 26.24 |
| 4 | F | 105 | 9 | - | mod | 0 | 0 | 23.25 | 17.19 |
| PD Cases | |||||||||
| 5 | M | 74 | 7.8 | PD | mild | 0 | 0 | 50.35 | 33.80 |
| 6 | M | 63 | 3.5 | PD vs. Lewy body AD 2 yr dementia | mod | mod | sparse | 66.50 | 25.18 |
| 7 tauopathy | F | 72 | 5 | dementia | prom | 0 | marked | 43.99 | 140.96 |
| AD cases | |||||||||
| 8 | F | 89 | 12 | IV-V; 10 yr dementia | mild | mod | sparse | 79.23 | 43.07 |
| 9 | F | 87 | 10 | V-VI; 5 yr dementia | mild | freq | mild | 145.79 | 79.84 |
| 10 | F | 80 | 11 | VI; 7 yr dementia | mod | mod | prom | 77.11 | 23.99 |
| 11 | F | 86 | 5 | VI; 11 yr dementia | severe | mod | mod | 104.92 | 58.70 |
| 12 | M | 82 | 5 | VI; mixed AD/Lewy body AD | severe | sparse | sparse | 82.71 | 41.40 |
| 13 | M | 83 | 7 | V; 2 yr dementia | mild | freq | mod | 290.95 | 94.80 |
| 14 | F | 99 | 11.5 | V; 2 yr dementia | mod | sparse | 0 | 74.85 | 45.64 |
In most AD cases, a ladder of SDS-stable oligomers was detected with multiple antibodies, varying in size from monomer to larger assemblies up to about 200kD. The N- terminal antibodies 6E10 and 10G4 are directed against similar Aβ epitopes, (6E10 against residues 5-10 and 10G4 against 5-17), but they differentially detect oligomers and APP (Fig. 4A,B). The antibody 10G4 labels primarily tetramers, oligomers in the range of 40-48 kDa; 10G4 also preferentially detected large aggregates. The 6E10 antibody (Fig. 4B) preferentially detected APP and small to mid-range oligomers including a 56 kDa species. With both antibodies, strong monomer bands were detected in all AD cases. In the same cases, an antibody specific for Aβ42 peptide (Fig. 4C), prominently labeled a 56 kDa dodecamer (Aβ-star) among a ladder of oligomers. The 56 kDa assembly was not detected by an antibody directed against Aβ40, which, like 10G4, preferentially detected the 43 kDa assembly (Fig. 4D). Quantification of AD-selective Aβ peptides is shown in Fig. 4E; monomer and three Aβ assemblies (large aggregates, 56 kD and 36 kD), along with APP and the c-terminal fragment were significantly increased in AD vs. normal synaptosome-enriched samples. Increased APP in AD immunoblots but not flow labeling of intact synaptosomes suggests localization to an interior compartment, possibly endosomal/lysosomal.
Fig. 4. Anti-Aβ antibody Western blots in synaptosome-enriched fractions.
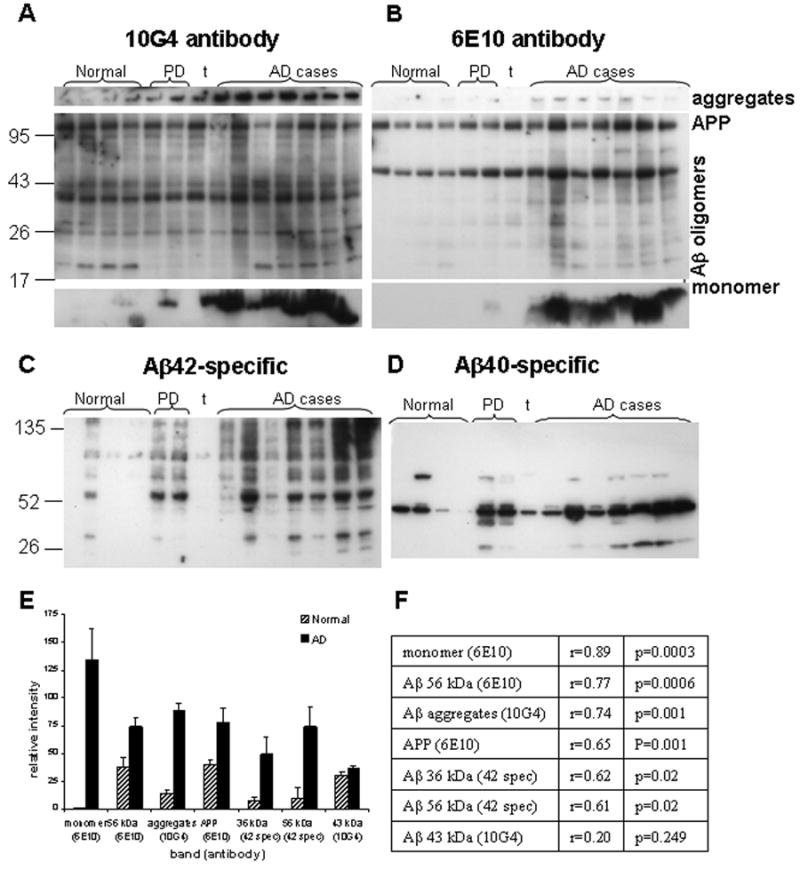
Western blots are shown for a series of normal (n=4) and AD cases (n=7). Controls include two PD cases and one tauopathy case (t); the right PD case (lane 6) had a 2 year history of dementia. Case information is presented in Table 1. Anti-Aβ antibodies illustrated are the N-terminal antibodies 10G4 (A), and 6E10 (B), and for C-terminal antibodies directed at Aβ42 (C), and at Aβ40 (D). (E) Quantification of AD-selective peptide assemblies from Western blots; p<0.05 for all comparisons except the 43 kDa band. (F) Correlation of Western blot Aβ peptides with flow cytometry Aβ fluorescence in size-purified synaptosomes (see Table 1, amyloid fluor. column).
The tauopathy case was positive for APP with several antibodies (22c11, 3E9, CT20, not shown), but Aβ assemblies were poorly detected in multiple experiments. In terms of Aβ, both PD cases demonstrated a ladder of mid-range to large oligomers with the 42-specific antibody. The PD case with dementia (720, lane 6) had a differential diagnosis of AD, Lewy body varient with a final diagnosis of PD; in most experiments this case demonstrated prominent Aβ monomer and aggregates that were similar to the pathology observed in the AD cases.
3.4 Monomers, APP, and multiple Aβ oligomers correlate with synaptic Aβ measured by flow cytometry in size-purified synaptosomes
In contrast to measures of Aβ by size-gated flow cytometry in a population of synaptosomes that is ∼95% pure, the P-2 fractions used for Western blots are enriched in synaptosomes, but also contain free mitochondria and vesicles, myelin, and membrane fragments in addition to intact synaptosomes. Therefore Western blot results were correlated with the synaptic Aβ level for each case measured by flow cytometry analysis of P2 fractions with the 10G4 antibody (Table 1). A number of Aβ-related peptides and assemblies from the Western blot series correlated significantly with the flow cytometry measure of Aβ fluorescence (Fig. 4F), with the highest correlation observed for Aβ monomer. Other assemblies with significant correlations included APP, aggregates, and SDS-stable assemblies at 36 kD and 56 kD.
The conformation dependent antibody A11, selective for pre-fibrillar oligomers, also labeled a major assembly at about 43 kDa, along with strong trimer/tetramer bands at 12 and 16 kDa in three of the seven AD cases (Fig. 5A). Interestingly, the trimer/tetramer assembly was present in both PD cases, and was most prominent in the non-demented PD case (711, left-most PD lane). However, these may also represent alpha synuclein or other oligomers, as A11 sensitivity to oligomers is not limited to Aβ oligomers (Kayed et al., 2003). Fibrillar oligomers were detected by the OC antibody in P-2 fractions as a series of SDS-stable oligomers between 26 and 56 kDa in size (Figure 5B); bands at ∼42, 45, and 51 kD were the most specific for AD. Fibrillar oligomers are conformationally distinct from pre-fibrillar oligomers (Kayed et al., 2007; Tomic et al., 2009) and were also prominent in the non-demented Parkinson's case. The widely used 4G8 antibody (directed against Aβ residues 17-24; Fig. 5C), in contrast to other N-terminal antibodies, did not sensitively detect oligomers and primarily labeled a major band at ∼43 kDa; this band was also observed with the 10G4 and 40-specific antibodies and was seen in normal as well as AD cases (Fig. 4A,D). A 43 kDa assembly is consistent with an Aβ nonomer as described for in vitro Aβ assemblies (Bitan et al., 2003); this assembly is also prominent in Westerns immunolabeled for apoE and may represent an Aβ dimer bound to apoE (Permanne et al., 1997).
Fig. 5. Aβ peptide species conformation.
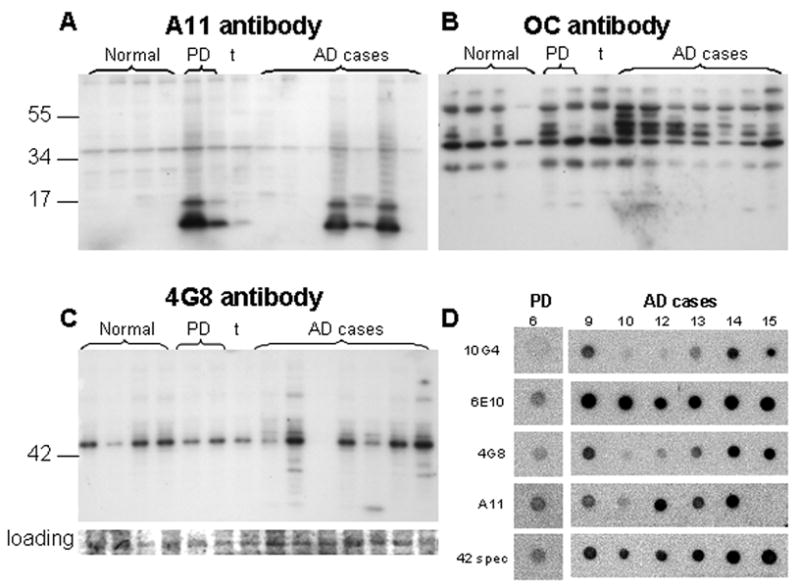
Western blots labeled with conformation-selective antibodies in same normal (n=4) and AD cases (n=7) used for Fig. 3. (A) Prefibrillar oligomers labeled with A11; (B) fibrillar oligomers labeled with the antibody OC. (C) Western blot for the N-terminal Aβ antibody 4G8 with Coomasie blue-stained membrane as loading control. (D) Dot blots using detergent-free sonicated synaptosome-enriched fractions; cases shown correspond to the lane number in (A) and (C), i.e., the PD case is the right-most case in lane 6 of Western blots.
Most protein complexes and oligomers dissociate when treated with SDS, but for amphipathic proteins such as Aβ, oligomers may be formed or stabilized in the presence of SDS micelles (Bitan et al., 2005). Therefore, dot blotting experiments were performed on P-2 fractions sonicated in buffer without detergent for one of the PD cases and 6 of the AD cases in the series. Due to the low Aβ signal for controls in Western blots and limited cryopreserved tissue volume, dot blot experiments did not include aged normal controls. Fig. 5D demonstrates results generally consistent with Western blotting results, with strong 6E10 and Aβ42-specific labeling in most AD cases, and strong conformation-dependent A11 immunolabeling of cases showing the strong trimer/tetramer band.
4. Discussion
Soluble oligomeric Aβ species are widely hypothesized to contribute to synaptic failure and early cognitive loss in AD, but the source, sequence of events, and key peptide species in pathogenesis have remained elusive, particularly with respect to human Aβ pathology. Using a series of aged normals, neurologic controls and AD cases, the present study demonstrates that synaptic terminals contain high levels of soluble oligomers, and Aβ42 rather than Aβ40 peptide. Aβ40 would be the expected dominant species if synaptic Aβ simply reflected production rates. In size-purified synaptosomes analyzed by flow cytometry, the Aβ signal correlates with the prominent monomer band on Western blots, along with multiple oligomeric Aβ assemblies, C-terminal fragments, and APP.
A role for synaptic Aβ in the progression of AD pathology is suggested by many lines of evidence, including the reduction of hippocampal pathology in APP transgenic mice by lesions of projection axons (Lazarov et al., 2002; Sheng et al., 2002) and the transsynaptic induction of neurofibrillary tangle pathology following Aβ fibril injection (Gotz et al., 2001). Activity-dependent synaptic release of Aβ has also been observed (Cirrito et al., 2005), and has been shown to require clathrin-mediated endocytosis (Cirrito et al., 2008). These authors suggest Aβ generation and release at or near the synapse following APP internalization associated with synaptic vesicle recycling. This hypothesis is supported by the present results showing increased APP in AD by Western blot but not in intact synaptosomes, suggesting endosomal APP location. The present results are also directly supported by the recent observation of Aβ release from TgCRND8 synaptoneurosomes. TgCRND8 mice overexpress two APP mutations; in these experiments blockade of release by a glutamate antagonist suggested a potential therapeutic target (Kim et al., 2010).
Dimeric Aβ has been inversely associated with MMSE in synaptoneurosomes (Williams et al., 2009), and recently both monomeric and dimeric Aβ from soluble and detergent extracts was strongly associated with dementia in AD cases (McDonald et al., 2010). The present observation of abundant monomer but little free dimer in synaptosome-enriched samples, even when probed with multiple antibodies, may result from preparation/exposure issues since Aβ immunoprecipitation was not performed in the present experiments and short exposure times are optimal for quantification. Alternatively, it is possible that dimeric Aβ in our samples is bound to apoE or other carrier proteins within pre-synaptic terminals and detected as a larger oligomer on immunoblots. The abundance of synapse-associated Aβ that runs as monomer on SDS gels is consistent with recent observations of high monomer levels in AD cortex (McDonald et al., 2010). As suggested by McDonald and colleagues, the high levels of synapse-associated Aβ monomer and small oligomers observed here may be membrane-associated and represent the active synaptotoxic peptide species. This hypothesis is consistent with extensive evidence that Aβ oligomers potently inhibit LTP (Selkoe 2008), control synaptic levels of the NMDA receptor (Snyder et al., 2005), and induce dendritic remodeling (Lacor et al., 2007; Shankar et al., 2007). However, the relatively high signal for Aβ monomer and soluble oligomers in disrupted synaptosomes and in Western blots relative to intact synaptosomes analyzed by flow cytometry seems more likely to indicate sequestration of Aβ species within intra-synaptic compartments, consistent with internalization and breakdown but not clearance of SDS-stable Aβ assemblies within surviving AD terminals. Aβ monomer may also be generated from higher order oligomers during Western analysis procedures. Alternatively, increased Aβ monomer in surviving terminals may also indicate active synaptic Aβ generation.
Gamma-secretase activity required for Aβ production has been localized to endosomes and synaptic membrane and vesicle fractions in rat brain and cell culture (Frykman et al., 2010), and processing of the γ-secretase substrate EphA4 is enhanced by synaptic activity (Inoue E., et al., 2009). The volume of early endosomes is markedly increased early in AD; early endosomes are close to the cell surface, prominent in processes, and have been suggested as a likely site for upregulated APP processing and Aβ generation in AD (Cataldo et al., 1997). Taken together with the Aβ generation observed in studies of released interstitial Aβ, the observation here of monomers and multiple oligomeric assemblies together with increased APP and AD-associated C-terminal fragments suggests that significant APP processing occurs within a large fraction of cortical synapses, within endosomes or along the synaptic cleft. However, given that considerable membrane recycling occurs at the synapse following vesicular release, a significant fraction of synaptosome-associated Aβ peptides might result from internalization of extracellular Aβ.
A good deal of evidence supports endosomal/lysosomal structures as a likely location for Aβ within AD synapses. Ultrastructural analysis in transgenic mouse models and human AD sections has previously shown association of Aβ with late endosomes near synapses (Takahashi et al., 2002; 2008). Moreover, like other components of the endosomal/lysosomal system, autophagic vacuoles are robustly upregulated in AD cortex (Cataldo et al., 1997; Nixon et al., 2005), which has been suggested to follow from impaired clearance of autophagic vacuoles (Bolund et al., 2008). Aβ monomer has been shown to have neuroprotective functions via antioxidant and insulin-like growth factor-mediated pathways (Zou et al., 2002; Giuffrida et al., 2009). Therefore, in the present study the high level of synaptic Aβ monomer may have played a role in the survival of Aβ-bearing terminals in the primarily late stage cases. Indeed, enhancement of lysosomal function has been associated with restoration of synaptic markers in mouse models of AD (Butler et al., 2006), suggesting that a compensatory lysosomal upregulation may slow synapse loss and cell death. Reduced synaptic Aβ monomer degrading enzymes in AD might account for the elevated Aβ monomer levels.
Both PD cases in the current series showed multiple synaptic Aβ oligomers with an Aβ42-specific antibody. Consistent with observations that mixed AD/PD pathology progresses more rapidly, the PD case with a 2 year history of dementia closely resembled pathology in the AD cases. A diagnosis of Lewy body variant AD was considered for this case because of the mild to moderate plaque pathology, but this case did not display neurofibrillary tangles, and the Lewy bodies were primarily in the brainstem rather than throughout the cortex. Neither dementia nor plaque/tangle pathology was observed in the other PD case, which showed prominent low molecular weight oligomers labeled by the conformation-dependent antibody A11. The synaptic Aβ pathology we observe in PD cases is in line with observations that Aβ and a-synuclein co-immunoprecipitate in patients with AD/PD and in transgenic mice (Tsigelny 2008), and with a hypothesis that interactions between misfolded proteins may contribute to augmentation of pathology in mixed neurodegenerative disease.
The polyclonal antibodies A11 and OC were raised against pre-fibrillar and fibrillar oligomers, respectively. However, the present results also demonstrate significant sensitivity to Aβ tertiary structure with monoclonal antibodies, since differential labeling of Aβ assemblies was observed in denatured samples in Western analysis. Conformational sensitivity is confounded to some degree with sequestration within intrasynaptosomal compartments such as lysosomes, as illustrated by differential labeling in situ (flow cytometry of intact synaptosomes), compared to dot blots of native proteins from mechanically disrupted synaptosomes. The previously unrecognized conformational sensitivity of monoclonal antibodies, along with the abundance of synaptic Aβ in our studies likely results from our extensive use of unfixed, short PMI tissue that is cryopreserved. This conclusion is in line with the observation that epitope masking has historically limited Aβ detection in situ, and is a factor contributing to false-negative results for intrneuronal localization of Aβ (Gouras et al., 2005).
In addition to serving as a possible seed for extracellular deposits, high levels of synaptic Aβ may be an upstream initiator of protein misfolding and neurodegeneration-related cascades, including p-tau pathology and disruption of axonal transport and synaptic transmission. These cascades are likely to be complex and interrelated, and signal a need for continued exploration with the ultimate goal of preventing early stages of synaptic dysfunction and cognitive decline.
Acknowledgments
This work was supported by NIH AG27465 to KHG, by NIH NS43946 to GMC, by NIA AG18879 to CAM. HVV is supported by the Daljit S. and Elaine Sarkaria Chair in Diagnostic Medicine. Tissue was obtained from the the Alzheimer's Disease Research Center Neuropathology Cores of USC (NIA 050 AG05142), UCLA (NIA P50 AG 16970), and UC Irvine (NIA P50 AG016573). Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility supported by NIH CA16042 and AI 28697, and by the JCCC, the UCLA AIDS Institute, the David Geffen School of Medicine and the Chancellor's Office at UCLA. We are grateful to Kevin Crespel for technical assistance.
Footnotes
Conflict of interest: There are no conflicts of interest to report for any of the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bitan G, Kirkitadze MD, E A, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Amyloid β-protein(Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. PNAS. 2003;100:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitan G, Fradinger EA, Spring SM, Teplow DB. Neurotoxic protein oligomers--what you see is not always what you get. Amyloid. 2005;12:88–95. doi: 10.1080/13506120500106958. [DOI] [PubMed] [Google Scholar]
- Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–37. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ, Holtzman DM. Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science. 2008;321:1221–4. doi: 10.1126/science.1161591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler D, Nixon RA, Bahr BA. Potential compensatory responses through autophagic/lysosomal pathways in neurodegenerative diseases. Autophagy. 2006;2:234–7. doi: 10.4161/auto.2729. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Pieroni C, Nixon RA. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer's disease: neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. J Neurosci. 1997;17:6142–51. doi: 10.1523/JNEUROSCI.17-16-06142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–22. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Dunkley PR, Jarvie PE, Robinson PJ. A rapid Percoll gradient procedure for preparation of synaptosomes. Nat Protoc. 2008;3:1718–28. doi: 10.1038/nprot.2008.171. [DOI] [PubMed] [Google Scholar]
- Falzone TL, Stokin GB, Lillo C, Rodrigues EM, Westerman EL, Williams DS, Goldstein LS. Axonal stress kinase activation and tau misbehavior induced by kinesin-1 transport defects. J Neurosci. 2009;29:5758–67. doi: 10.1523/JNEUROSCI.0780-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fein JA, Sokolow S, Miller CA, Vinters HV, Yang F, Cole GM, Gylys KH. Co-localization of amyloid beta and tau pathology in Alzheimer's disease synaptosomes. Am J Pathol. 2008;172:1683–92. doi: 10.2353/ajpath.2008.070829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frykman S, Hur JY, Franberg J, Aoki M, Winblad B, Nahalkova J, Behbahani H, Tjernberg LO. Synaptic and endosomal localization of active gamma-secretase in rat brain. PLoS ONE. 2010;5:e8948. doi: 10.1371/journal.pone.0008948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, Garozzo D, Nicoletti F, Rizzarelli E, Copani A. Beta-amyloid monomers are neuroprotective. J Neurosci. 2009;29:10582–7. doi: 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–5. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer's disease. Neurobiol Aging. 2005;26:1235–44. doi: 10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Tan AM, Cole GM. Apolipoprotein E enhances uptake of soluble but not aggregated amyloid-beta protein into synaptic terminals. J Neurochem. 2003;84:1442–51. doi: 10.1046/j.1471-4159.2003.01643.x. [DOI] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Wiley DJ, Cole GM. Rapid annexin-V labeling in synaptosomes. Neurochem Int. 2004;44:125–31. doi: 10.1016/s0197-0186(03)00146-3. [DOI] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Cole GM. Enrichment of presynaptic and postsynaptic markers by size-based gating analysis of synaptosome preparations from rat and human cortex. Cytometry A. 2004;60:90–6. doi: 10.1002/cyto.a.20031. [DOI] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer's disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–17. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Miller CA, Cole GM. Increased cholesterol in Abeta-positive nerve terminals from Alzheimer's disease cortex. Neurobiol Aging. 2007;28:8–17. doi: 10.1016/j.neurobiolaging.2005.10.018. [DOI] [PubMed] [Google Scholar]
- Inoue E, Deguchi-Tawarada M, Togawa A, Matsui C, Arita K, Katahira-Tayama S, Sato T, Yamauchi E, Oda Y, Takai Y. Synaptic activity prompts gamma-secretase-mediated cleavage of EphA4 and dendritic spine formation. J Cell Biol. 2009;185:551–64. doi: 10.1083/jcb.200809151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanisms of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007;2:18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Fraser PE, Westaway D, St George-Hyslop PH, Ehrlich ME, Gandy S. Group II metabotropic glutamate receptor stimulation triggers production and release of Alzheimer's amyloid {beta}42 from isolated intact nerve terminals. J Neurosci. 30:3870–5. doi: 10.1523/JNEUROSCI.4717-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–40. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–7. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106:4012–7. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004;24:10191–200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22:9785–93. doi: 10.1523/JNEUROSCI.22-22-09785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–62. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak K, Yang F, Vinters HV, Frautschy SA, Cole GM. Polyclonals to beta-amyloid(1-42) identify most plaque and vascular deposits in Alzheimer cortex, but not striatum. Brain Res. 1994;667:138–42. doi: 10.1016/0006-8993(94)91725-6. [DOI] [PubMed] [Google Scholar]
- Mc Donald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, Selkoe DJ, Ince PG, Walsh DM. The presence of sodium dodecyl sulphate-stable A{beta} dimers is strongly associated with Alzheimer-type dementia. Brain. 2010;133:1328–1341. doi: 10.1093/brain/awq065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–6. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Moreno H, Yu E, Pigino G, Hernandez AI, Kim N, Moreira JE, Sugimori M, Llinas RR. Synaptic transmission block by presynaptic injection of oligomeric amyloid beta. Proc Natl Acad Sci U S A. 2009;106:5901–6. doi: 10.1073/pnas.0900944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–7. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- Permanne B, Perez C, Soto C, Frangione B, Yu C, Wisniewski T. Detection of apolipoprotein E/dimeric soluble amyloid beta complexes in Alzheimer's disease brain supernatants. Biochem Biophys Res Commun. 1997;240:715–720. doi: 10.1006/bbrc.1997.7727. [DOI] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L, LaDu M, Busciglio J, Brady S. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc Natl Acad Sci U S A. 2009;106:5907–12. doi: 10.1073/pnas.0901229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozkalne A, Spires-Jones TL, Stern EA, Hyman BT. A single dose of passive immunotherapy has extended benefits on synapses and neurites in an Alzheimer's disease mouse model. Brain Res. 2009;1280:178–185. doi: 10.1016/j.brainres.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid I, Uittenbogaart CH, Giorgi JV. A gentle fixation and permeabilization method for combined cell surface and intracellular staining with improved precision in DNA quantification. Cytometry. 1991;12:279–85. doi: 10.1002/cyto.990120312. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–13. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Price DL, Koliatsos VE. Disruption of corticocortical connections ameliorates amyloid burden in terminal fields in a transgenic model of Abeta amyloidosis. J Neurosci. 2002;22:9794–9. doi: 10.1523/JNEUROSCI.22-22-09794.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–8. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Spires-Jones TL, Mielke ML, Rozkalne A, Meyer-Luehmann M, de Calignon A, Bacskai BJ, Schenk D, Hyman BT. Passive immunotherapy rapidly increases structural plasticity in a mouse model of Alzheimer disease. Neurobiol Dis. 2009;33:213–20. doi: 10.1016/j.nbd.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–8. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Stokin GB, Almenar-Queralt A, Gunawardena S, Rodrigues EM, Falzone T, Kim J, Lillo C, Mount SL, Roberts EA, McGowan E, Williams DS, Goldstein LS. Amyloid precursor protein-induced axonopathies are independent of amyloid-beta peptides. Hum Mol Genet. 2008;17:3474–86. doi: 10.1093/hmg/ddn240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–53. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–79. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK. Co-occurrence of Alzheimer's disease beta-amyloid and tau pathologies at synapses. Neurobiol Aging. 2008;31:1145–52. doi: 10.1016/j.neurobiolaging.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomic JL, Pensalfini A, Head E, Glabe CG. Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol Dis. 2009;35:352–8. doi: 10.1016/j.nbd.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–92. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsigelny IF, Crews L, Desplats P, Shaked GM, Sharikov Y, Mizuno H, Spencer B, Rockenstein E, Trejo M, Platoshyn O, Yuan JX, Masliah E. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer's and Parkinson's diseases. PLoS ONE. 2008;3:e3135. doi: 10.1371/journal.pone.0003135. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007;101:1172–84. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Williams C, Mehrian Shai R, Wu Y, Hsu YH, Sitzer T, Spann B, McCleary C, Mo Y, Miller CA. Transcriptome analysis of synaptoneurosomes identifies neuroplasticity genes overexpressed in incipient Alzheimer's disease. PLoS ONE. 2009;4:e4936. doi: 10.1371/journal.pone.0004936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, Kapatos G. Flow cytometric analysis of rat striatal nerve terminals. J Neurosci. 1989;9:94–105. doi: 10.1523/JNEUROSCI.09-01-00094.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou K, Gong JS, Yanagisawa K, Michikawa M. A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage. J Neurosci. 2002;22:4833–41. doi: 10.1523/JNEUROSCI.22-12-04833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]