

Abstract
Phosphorylation of the myosin regulatory light chain (RLC) by Ca2+-calmodulin–activated myosin light chain kinase (MLCK) is known to be essential for the inotropic function of the heart. In this study, we have examined the effects of MLCK-phosphorylation of transgenic (Tg) mouse cardiac muscle preparations expressing the D166V (aspartic acid to valine)–RLC mutation, identified to cause familial hypertrophic cardiomyopathy with malignant outcomes. Our previous work with Tg-D166V mice demonstrated a large increase in the Ca2+ sensitivity of contraction, reduced maximal ATPase and force and a decreased level of endogenous RLC phosphorylation. Based on studies demonstrating the beneficial and/or protective effects of cardiac myosin phosphorylation for heart function, we hypothesized that an ex vivo phosphorylation of Tg-D166V cardiac muscle may rescue the detrimental contractile phenotypes observed earlier at the level of single myosin molecules and in Tg-D166V papillary muscle fibres. We showed that MLCK-induced phosphorylation of Tg-D166V cardiac myofibrils and muscle fibres was able to increase the reduced myofibrillar ATPase and reverse an abnormally increased Ca2+ sensitivity of force to the level observed for Tg-wild-type (WT) muscle. However, in contrast to Tg-WT, which displayed a phosphorylation-induced increase in steady-state force, the maximal tension in Tg-D166V papillary muscle fibres decreased upon phosphorylation. With the exception of force generation data, our results support the notion that RLC phosphorylation works as a rescue mechanism alleviating detrimental functional effects of a disease causing mutation. Further studies are necessary to elucidate the mechanism of this unexpected phosphorylation-induced decrease in maximal tension in Tg-D166V–skinned muscle fibres.
Keywords: cardiac hypertrophy, mutation, myosin regulatory light chain, muscle fibres, myosin light chain kinase, phosphorylation, transgenic mice
Introduction
Familial hypertrophic cardiomyopathy (FHC) is an autosomal dominant disorder of the heart characterized by increased growth of myocytes (hypertrophy), myofilament disarray, fibrosis and a high incidence of sudden cardiac death (SCD) [1]. FHC was found to originate from mutations in cardiac sarcomeric proteins, such as β-myosin heavy chain, myosin essential (ELC) and regulatory (RLC) light chains, myosin-binding protein-C, actin, α-tropomyosin, troponin T, troponin I, troponin C and titin [1]. Mutations in the MYL2 gene encoding the human ventricular RLC constitute about 2% of the total sarcomeric protein mutations and are associated with a wide spectrum of morphological and functional abnormalities in the affected patients [1, 2]. To date, 10 RLC mutations have been identified by population studies to cause hypertrophic cardiomyopathy, eight of which are single amino acid substitutions and two are intronic splice site mutations [3-7].
In this report, we focus on one of the most malignant RLC FHC mutations, which is brought about by an aspartic acid to valine substitution at the last amino acid residue of RLC (D166V) [4, 8]. The resulting valine residue of the RLC is positioned in the hinge region (‘hook’) of the myosin heavy chain (MHC) that serves as a major source of cross-bridge compliance during the contractile cycle [9, 10]. Given the critical position of the D166V mutation in the RLC molecule and its location in relation to the myosin cross-bridge, it is evident why this one amino acid substitution could affect myosin function and transmission of force leading to abnormal cardiac contractility and FHC. Our previous work with transgenic (Tg) D166V mouse cardiac muscle preparations suggested that the manifestation of the mutation-induced phenotype could be correlated with a significantly decreased myosin RLC phosphorylation [11, 12]. Tg-D166V papillary muscle fibres demonstrated an increased Ca2+ sensitivity of contractile force, reduced maximal ATPase and force; functional changes that coincided with the significantly reduced in situ phosphorylation of RLC observed in rapidly frozen Tg-D166V samples compared to Tg-wild type (WT) hearts [11]. Similarly, single molecule detection studies with fluorescently labelled cardiac myofibrils from Tg-D166V mice showed decreased rates of cross-bridge cycling and a much lower RLC phosphorylation compared with Tg-WT myofibrils [12]. Based on these data, we hypothesized that the phenotype associated with the D166V mutation could be alleviated or partially reversed by an ex vivo Ca2+-calmodulin (CaM)–activated myosin light chain kinase (MLCK)-phosphorylation of the mutated myocardium. The hypothesis was supported by a large body of basic science and clinical findings demonstrating the beneficial role of RLC phosphorylation on cardiac muscle performance [13-15].
Here, we show that MLCK-induced phosphorylation of cardiac muscle preparation from Tg-D166V mice was able to increase the mutation-induced decreased levels of myofibrillar ATPase activity. In addition, a D166V-mediated increase in Ca2+ sensitivity of force was fully restored to the level of Tg-WT muscle fibres. Surprisingly, in contrast to Tg-WT muscle, which demonstrated a phosphorylation-induced increase in steady-state force, the maximal level of tension in phosphorylated Tg-D166V muscle fibres was decreased. Our results suggest that RLC phosphorylation may operate through different mechanisms in healthy and diseased hearts.
Materials and methods
Animals
All animal studies were conducted in accordance with institutional guidelines. The University of Miami has an Animal Welfare Assurance (A-3224-01, effective July 11, 2007) on file with the Office of Laboratory Animal Welfare (OLAW), National Institutes of Health. Tg mouse models expressing the WT or D166V mutant of the human ventricular myosin RLC were generated and characterized as described earlier [11]. As reported earlier, Tg-WT mice expressed ∼100% of human ventricular RLC protein (i.e. totally replaced the endogenous mouse ventricular RLC) and were compared to Tg-D166V mice that expressed ∼95% of the human RLC carrying the D166V mutation [11]. The animals used in this study were ∼6 months of age.
Phosphorylation of RLC in mouse cardiac myofibrils
Skinned cardiac myofibrils (CMF) from the right and left ventricles of Tg-WT and Tg-D166V mice were prepared as described earlier [16]. MLC kinase (MLCK) from rabbit skeletal muscle, isolated and purified as described in [17], was used to phosphorylate RLC. CMF were phosphorylated with Ca2+-CaM–activated MLCK in a solution containing: 250 μg myofibrils suspended in 20 mM phosphate buffer (pH 8), 30 mM KCl and 0.5 μM MLCK, 5.0 μM CaM, 0.1 mM CaCl2, 12.5 mM MgCl2 and 5 mM ATP. The phosphorylation reaction was carried out overnight on ice. These conditions were determined to result in fully phosphorylated CMF (Fig. 1, inset). After phosphorylation, CMF were washed thoroughly with buffer containing 30 mM MOPS, 60 mM KCl, 1 mM DTT, pH 7.0. The levels of RLC phosphorylation in Tg-WT and Tg-D166V CMF were determined using Urea/SDS-PAGE as described later.
Fig 1.
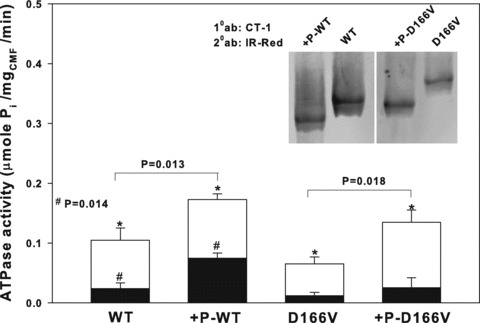
Myofibrillar ATPase activity measured in Tg-WT and Tg-D166V preparations before and after Ca2+-CaM MLCK treatment. Maximal ATPase activity was measured in pCa 4 (white bars), whereas basal ATPase activity was measured in pCa 8 (black bars). Inset: Urea/SDS-PAGE of phosphorylated and non-phosphorylated cardiac myofibrils (CMF). CMF were run on 8% polyacrylamide gel in the presence of 8 M urea and the RLC protein was visualized with CT-1 antibodies recognizing the total RLC protein content (Materials and Methods). Under these conditions, the phosphorylated form of RLC in +P-WT or +P-D166V CMF migrated faster than the non-phosphorylated WT or D166V. Note, that due to the D166V mutation, the non-phosphorylated D166V migrates slower than the non-phosphorylated WT. 10 ab: Primary CT-1 antibodies; 20 ab: secondary IR-Red antibodies.
ATPase assays
Myofibrillar ATPase activity assays using Tg-WT and Tg-D166V CMF were performed in a solution of 20 mM MOPS (pH 7.0), 40 mM KCl, 2.5 mM MgCl2, 2 mM EGTA and calculated CaCl2 concentration to produce 10−8 M (basal activity) or 10−4 M (maximal activity) of free [Ca2+][18]. After 5 min. of incubation at 30°C, the reaction was initiated with 2.5 mM ATP and terminated after 10 min. with 5% trichloroacetic acid. Released inorganic phosphate was measured according to the method of Fiske and Subbarow [19].
Phosphorylation of RLC in mouse cardiac papillary muscle fibres
Glycerinated-skinned papillary muscle fibres from Tg-WT and Tg-D166V mice were isolated from mouse left ventricles and prepared using established protocols [20]. Fibre bundles of three to five single muscle fibres were phosphorylated with Ca2+-CaM–activated MLCK in a solution containing 0.5 μM MLCK, 5 μM CaM in pCa 6 buffer [18]: 10−6 M free [Ca2+] (total calcium propionate, CaPr = 4.997 mM), 1 mM free [Mg2+] (total MgPr = 3.88 mM), 7 mM EGTA, 20 mM MOPS, 2.5 mM [Mg-ATP2−] (total Na2ATP = 3.056 mM), 15 mM creatine phosphate, 15 units/ml phosphocreatine kinase, pH 7.0 (adjusted with KOH), ionic strength 150 mM (adjusted with KPr = 38.41 mM), for 1 hr at room temperature. Alternatively, phosphorylation of fibre bundles was performed while mounted on a Guth force transducer. The fibres were skinned before phosphorylation with pCa 8 relaxing solution (same composition as pCa 6 except 10−8 M free [Ca2+]) containing 1% Triton X-100 for 30 min. Phosphorylation of the latter was performed in the solution of 0.5 μM MLCK, 5 μM CaM in pCa 6 for 30 min. at room temperature. Both treatments were equally efficient in achieving the same level of RLC phosphorylation (Fig. 3, insets). After phosphorylation, the fibres were washed in pCa 8 buffer and subjected to force measurements.
Fig 3.
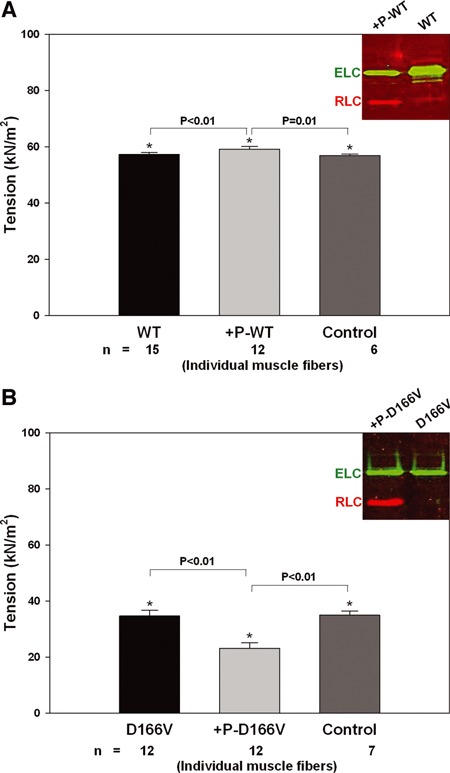
The effect of RLC phosphorylation on maximal force development in Tg-WT (A) and Tg-D166V (B) papillary muscle fibres. Maximal force was measured in pCa 4 buffer before and after CaM–MLCK treatment. Control WT and control D166V fibres were incubated in pCa 6 buffer with no CaM–MLCK complex. Insets: Western blot of experimental WT and +P-WT fibers (A); D166V and +P-D166V fibres (B). Phosphorylation of RLC in +P-WT or +P-D166V muscle fibres was detected with the +P-RLC antibodies specific for the phosphorylated form of cardiac RLC followed by a secondary goat anti-rabbit antibody conjugated with the fluorescent dye, IR red 800 (red bands). Total ELC, which served as a loading control, was detected with the monoclonal ab680 antibody followed by a secondary goat anti-mouse antibody conjugated with the fluorescent dye, Cy 5.5 (green bands). RLC: regulatory light chain of myosin (red); ELC: essential light chain of myosin (green).
Steady-state force measurements and myofilament Ca2+ sensitivity in phosphorylated and non-phosphorylated fibres
The fibres were tested for steady-state force development in a pCa 4 solution (composition is the same as pCa 8 buffer except the [Ca2+]= 10−4 M) and relaxed in pCa 8 solution. Steady-state force development was monitored for phosphorylated Tg-D166V and Tg-WT papillary muscle fibres as well as for their non-phosphorylated counterparts. To account for potential decreases in force due to fibre exposure to sub-maximal calcium concentrations during phosphorylation, control Tg-WT and Tg-D166V fibres were tested in parallel to those being phosphorylated after their 30-min. incubation in the pCa 6 solution with no added CaM–MLCK complex. Maximal force (in Newtons) was calculated per cross-section of muscle fibre and expressed in kN/m2. The cross-sectional area was calculated based on measurement of the fibre width using an SZ6045 Olympus microscope (zoom ratio of 6.3:1, up to 189 χ maximum magnification) and the assumption that the fibre is circular in diameter. The measurement was taken at ∼3 points along the bundles and averaged. The average length and diameter of small fibre bundles selected for the experiments were ∼1.2–1.4 and 90–120 μm, respectively. To determine the force–pCa dependence, the fibres were exposed to solutions of increasing Ca2+ concentrations (from pCa 8 to pCa 4) and force development was monitored. The data were analysed using the Hill equation yielding the pCa50 and nH (Hill coefficient) [21].
Analysis of RLC phosphorylation
The levels of RLC phosphorylation in rapidly frozen ventricular samples, CMF and papillary muscle fibres were determined using Urea/SDS-PAGE and phospho-RLC–specific antibodies (generously provided by Dr. Neal Epstein, National Institutes of Health). Specifically, to determine RLC phosphorylation in situ, after euthanasia the hearts from Tg-D166V and Tg-WT mice were excised and the ventricles were isolated and flash frozen in liquid nitrogen. The tissue was thawed in a buffer consisting of 8M Urea, 10 mM Tris-HCl, pH 7.0, 1% SDS, 1%β-mercaptoethanol (β-ME), 1 mM EDTA, 1 mM PMSF, 5 nM microcystin and protease inhibitor cocktail, and then homogenized in the same buffer for 2 min. at 25 Hz. This step was repeated until the tissue was completely homogeneous. The homogenates were combined with equal amounts of SDS-PAGE sample buffer and run on 15% polyacrylamide-SDS gel as described in [11]. To determine the level of RLC phosphorylation in CMF, ∼100 μl CMF suspended in 30 mM MOPS, 60 mM KCl, 1 mM DTT, pH 7.0 were combined with 70 mg of crystalline urea, 10 μl β-ME and 5 μl bromophenol blue and run on 8% polyacrylamide gel in the presence of 8M urea [22]. Under these conditions, the phosphorylated form of RLC in +P-WT and +P-D166V CMF migrated faster than the non-phosphorylated counterpart (Fig. 1, inset) [23]. The extent of RLC phosphorylation in papillary muscle fibres was tested after the fibres were sonicated in a solution of 1% SDS and 10%β-ME for 30 min. on ice, mixed with SDS sample buffer and run on 15% polyacrylamide-SDS gel. All gels were transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). Phosphorylated Tg RLC in +P-WT or +P-D166V muscle fibres was detected with the +P-RLC antibodies specific for the phosphorylated form of cardiac RLC [24], followed by a secondary goat anti-rabbit antibody conjugated with the fluorescent dye, IR red 800 (Rockland, Gilbertsville, PA, USA). The total RLC protein was determined with rabbit polyclonal CT-1 antibodies produced in this laboratory [20, 25]. Total ELC (ELC of myosin), which served as a loading control, was detected with the monoclonal ab680 antibody (Abcam, Cambridge, MA, USA) followed by a secondary goat anti-mouse antibody conjugated with the fluorescent dye, Cy 5.5. Blots were scanned and respective bands were quantified using the Odyssey Infrared Imaging System (LI-COR Inc., Lincoln, NE, USA).
Histopathology
After euthanasia, hearts from Tg-WT and Tg-D166V mice were excised and immersed in 10% buffered formalin [11, 20]. Slides of whole mouse hearts were prepared by American Histolabs, Inc. (Gaithersburg, MD, USA). The paraffin-embedded longitudinal sections of whole mouse hearts stained with haematoxylin and eosin (H&E) and Masson's trichrome were examined for overall morphology and fibrosis using a Dialux 20 microscope, 40χ/0.65 Leitz Wetzlar objective and AxioCam HRc (Zeiss, Thornwood, NY, USA) [11, 20, 25].
Statistical analysis
Data are expressed as the average of n experiments ± S.E. (standard error). Student's t-test was used for simple comparisons of two groups (e.g. non-phosphorylated versus phosphorylated). For multiple comparisons between groups, a one-way ANOVA and the Holm-Sidak method (Sigma Plot 11; Systat Software, Inc., San Jose, CA, USA) were used. Statistically significant differences were defined as P < 0.05.
Results
The effect of RLC phosphorylation on myofibrillar ATPase activity in Tg-WT and Tg-D166V mice
ATPase activity assays were performed on phosphorylated (+P) or non-phosphorylated–skinned cardiac myofibrils from Tg-WT and Tg-D166V mice. As shown in Figure 1, the maximal ATPase activity (μmole Pi/mg CMF/min) determined at pCa 4 was 1.6-fold lower in non-phosphorylated D166V CMF (0.065 ± 0.009, n= 5) compared to non-phosphorylated WT CMF (0.105 ± 0.021, n= 8). Following phosphorylation, the myofibrillar ATPase activity increased in both +P-WT (0.173 ± 0.009, n= 7) and +P-D166V (0.135 ± 0.020, n= 5) CMF; however, the phosphorylation-induced increase in Tg-D166V myofibrils was larger than that in Tg-WT (Fig. 1, white bars). Therefore, phosphorylation of Tg-D166V CMF was able to recover low levels of maximal myofibrillar ATPase activity observed for non-phosphorylated–mutated CMF and bring it to an intermediate level, between WT and +P-WT. Phosphorylation-induced changes were also observed in the basal levels of myofibrillar ATPase activity measured at pCa 8.0 (Fig. 1, black bars), but the differences were only significant in WT myofibrils (P= 0.014).
The effect of ex vivo RLC phosphorylation on Ca2+sensitivity of force development in Tg-WT and Tg-D166V–skinned muscle fibres
The effect of Ca2+-CaM MLCK-induced phosphorylation of the RLC on the force–pCa relationship was studied in skinned glycerinated fibres from Tg-D166V and Tg-WT mice. As seen in Figure 2, phosphorylation of Tg-WT fibres resulted in a small leftward shift in the force–pCa dependence with ΔpCa50= 0.06, for n= 7 individual muscle fibres (P= 0.038) demonstrating an increase in Ca2+ sensitivity upon phosphorylation. In contrast, phosphorylation of Tg-D166V fibres resulted in a large decrease in myofilament Ca2+ sensitivity with ΔpCa50=−0.20, n= 8 fibres (P < 0.01). Therefore, phosphorylation of D166V-mutated papillary muscle fibres reversed the increased Ca2+ sensitivity of force of the diseased D166V muscle and brought it back to the level observed for WT. No significant phosphorylation induced changes in myofilament co-operativity (nH) were observed in Tg-WT or Tg-D166V papillary muscle fibres (Fig. 2).
Fig 2.
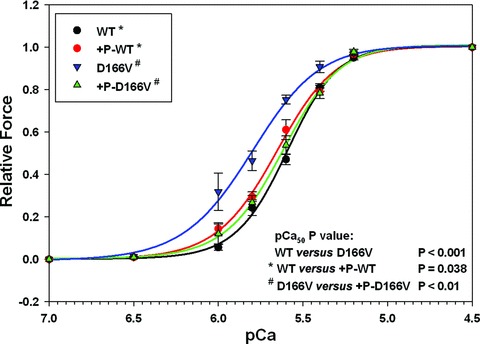
The effect of RLC phosphorylation on the force–pCa relationship in skinned muscle fibers from Tg-WT mice and Tg-D166V. The pCa50 and nH (Hill coefficient) values were: WT (black): pCa50= 5.60 ± 0.01, nH= 2.89 ± 0.15; +P-WT (red): pCa50= 5.66 ± 0.02, nH= 2.47 ± 0.16; D166V (blue): pCa50= 5.83 ± 0.04, nH= 2.16 ± 0.23; +P-D166V (green): pCa50= 5.63 ± 0.04, nH= 2.57 ± 0.19.
The effect of Ca2+-CaM MLCK-induced phosphorylation of RLC on maximal force in Tg-WT and Tg-D166V–skinned muscle fibres
We further studied the effect of RLC phosphorylation on the maximal steady-state force in skinned muscle fibres from Tg-WT and Tg-D166V mice. Phosphorylation of RLC in Tg-WT papillary muscle fibres with Ca2+-CaM MLCK resulted in a small but significant increase in maximal force; from Fmax= 57.30 ± 0.17 to Fmax= 59.19 ± 0.26 kN/m2 (Fig. 3A). Tg-WT fibres that were incubated in pCa 6 solution in the absence of CaM–MLCK and used as a control were indistinguishable from WT muscles (Fmax= 56.88 ± 0.20 kN/m2). The P values determined with one-way ANOVA and the Holm–Sidak method for pairwise multiple comparison procedures were: P < 0.01 for WT versus+P-WT, P= 0.01 for +P-WT versus control fibres and P > 0.05 for WT versus control fibres.
The gel shown in Figure 3A (inset: red bands) illustrates the level of RLC phosphorylation in +P-WT and WT muscles determined by SDS-PAGE and +P-RLC antibodies specific to the phosphorylated form of cardiac RLC (see Materials and Methods). As depicted in Figure 3B, phosphorylation of D166V papillary muscle fibres resulted in a large (∼35%) decrease in the maximal force from Fmax= 34.64 ± 0.56 to Fmax= 23.12 ± 0.55 kN/m2 (P < 0.01). Control Tg-D166V fibres treated in parallel in pCa 6 solution in the absence of CaM–MLCK (see Materials and Methods, for details) were indistinguishable from non-phosphorylated D166V preparations (P > 0.05) and showed no change in maximal force (Fmax= 34.92 ± 0.55 kN/m2; Fig. 3B). This result indicated that the decrease in force observed in MLCK-treated D166V fibres was clearly associated with RLC phosphorylation of Tg-D166V fibres. The gel shown in Figure 3B (inset) depicts RLC phosphorylation in +P-D166V and D166V fibres.
The effect of the D166V mutation on endogenous RLC phosphorylation in Tg-D166V versus Tg-WT mouse hearts
Left ventricles of mice tested in functional assays were rapidly frozen in liquid nitrogen and examined for endogenous levels of RLC phosphorylation by SDS-PAGE and immunoblotting with +P-RLC antibody. Similar to what we observed in our earlier study [11, 12], a significant (approx. threefold) decrease in RLC phosphorylation was detected in left ventricular extracts from Tg-D166V compared with Tg-WT mice (Fig. 4, left). The lower level of RLC phosphorylation in Tg-D166V was not due to a lower RLC content, as verified by the total RLC protein detected with CT-1 antibody in Tg-D166V compared to Tg-WT mice (Fig. 4, right).
Fig 4.
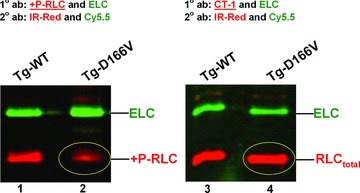
The effect of D166V mutation on RLC phosphorylation in situ. Left ventricles of Tg-WT and Tg-D166V mice were flash frozen in liquid N2 and subsequently analysed for level of RLC phosphorylation (Materials and Methods). Lanes 1 and 3: Tg-WT; lanes 2 and 4: Tg-D166V. Left: Immunoblotted with +P-RLC (antibody recognizing phosphorylated form of RLC: red). Right: Immunoblotted with CT-1 (antibody recognizing total RLC: red). Circled: D166V protein: phosphorylated (left), and total (right). IR-Red 800 and Cy5.5: secondary fluorescent antibodies.
The effect of low D166V phosphorylation on myofilament organization in Tg-D166V mice
To determine whether the decreased levels of RLC phosphorylation in Tg-D166V mouse hearts led to or were caused by the D166V-induced structural defects, the myocardium of Tg-D166V mice was compared with Tg-WT (Fig. 5). No major difference in heart morphology was observed between Tg-D166V and Tg-WT mice, as evidenced by haematoxylin and eosin tissue staining. Similarly, Masson's trichrome staining demonstrated no fibrosis in the hearts of Tg-D166V or Tg-WT mice (Fig. 5). Therefore, lower levels of endogenous RLC phosphorylation observed in left ventricles of Tg-D166V mice did not induce or were not caused by any abnormal tissue morphology (Fig. 5).
Fig 5.
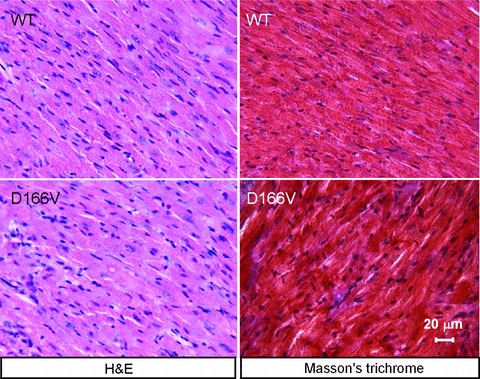
Representative longitudinal sections from left ventricles of ∼6-month-old Tg-WT and of ∼6-month-old Tg-D166V mice stained with haematoxylin and eosin (H&E) for overall morphology and Masson's trichrome for signs of fibrosis. Scale bar = 20 μm.
Discussion
This report is the first to demonstrate the physiological effects of RLC phosphorylation in cardiomyopathic Tg mouse hearts. Hypertrophic cardiomyopathy is one of the most common genetic cardiovascular disorders caused by mutations in genes encoding cardiac sarcomeric proteins but the molecular mechanism by which these single amino acid substitutions lead to specific disease phenotypes is yet to be elucidated. The pathways of target-specific interventions that could result in rescue of the pathological phenotype also remain undiscovered. In this study, we focus on one such mechanism acting through phosphorylation of myosin RLC and show that it can reverse or partially recover the detrimental consequences of the D166V mutation.
Effect of RLC phosphorylation in cardiac myofibrils and under load in skinned papillary muscle fibres
Based on studies demonstrating the effect of myosin phosphorylation on cardiac function and the observation that Tg FHC-mutated mouse hearts display low levels of RLC phosphorylation [11, 12, 16], we hypothesized that an ex vivo phosphorylation of Tg-D166V cardiac muscle may rescue detrimental contractile phenotypes associated with this malignant FHC mutation.
Experiments performed in cardiac muscle myofibrils, under unloaded conditions, clearly showed the beneficial effect of RLC phosphorylation on the ATPase activity in Tg-D166V muscle. Low levels of maximal ATPase activity observed in non-phosphorylated Tg-D166V myofibrils could be recovered by RLC phosphorylation (Fig. 1). Because the heart operates against a load of mean arterial blood pressure and peripheral resistance, the physiologically relevant effects of RLC phosphorylation are those observed under loaded conditions, that is when myosin performs work in isometrically contracting skinned papillary muscle fibres. Consistent with other reports (reviewed in Refs. 13 and 14), a phosphorylation-induced increase in maximal force production and Ca2+ sensitivity of tension were observed in Tg-WT muscle preparations. However, phosphorylation of the muscle carrying the malignant D166V-RLC mutation showed a much more complex pattern of changes that in some cases were strikingly different than those observed in the WT myocardium.
As reported previously [11], we observed an abnormally large increase in the calcium sensitivity of force in non-phosphorylated Tg-D166V muscle fibres compared to non-phosphorylated Tg-WT counterparts (Fig. 2). The MLCK-induced phosphorylation of RLC in Tg-D166V muscle shifted the force–pCa dependence back to that observed in Tg-WT muscle. Therefore, phosphorylation of RLC worked as a rescue mechanism in the mutation-compromised myocardium, reversing its overly sensitive responsiveness to calcium. Unexpectedly, the maximal level of force in phosphorylated Tg-D166V muscles was decreased (Fig. 3B). This effect was in contrast to Tg-WT muscle (Fig. 3A), which demonstrated a phosphorylation-induced increase in steady-state force. No such effect on maximal force was observed previously during cardiac [24, 26, 27] or skeletal [23, 28–31] muscle contraction. A plausible mechanism of this phosphorylation-induced decrease in maximal force is that the substitution of the bulky valine for the negatively charged aspartic acid in Tg-D166V muscle may lead to abnormal alignment of D166V myosin cross-bridges along actin filaments. This in turn would cause weaker binding and decreased force generation in Tg-D166V muscle fibres. The question remains as to whether these phosphorylation-induced changes in force generation in Tg-D166V muscle are beneficial or detrimental to the workings of the mutated hearts.
Attenuation of RLC phosphorylation in vivo: relation to heart disease
Our previous and current studies with Tg mouse models of FHC demonstrate a direct relationship between severity of hypertrophic phenotype and the level of RLC phosphorylation. Mouse hearts carrying the malignant R58Q-RLC mutation exhibited significantly lower levels of RLC phosphorylation compared to WT hearts or those of benign disease phenotypes [16, 32, 33]. The recent report from the Stull laboratory also showed that attenuation of RLC phosphorylation in cardiac MLCK knockout mice resulted in cardiomyocyte hypertrophy with histological evidence of necrosis and fibrosis [26]. These findings suggest that the D166V mutation-mediated decrease of RLC phosphorylation in Tg-D166V myocardium (Fig. 4) may initiate hypertrophic remodelling of the heart and stimulate the development of cardiomyopathic phenotype. It is interesting to note that whereas tissue abnormalities are clearly manifested in older Tg-D166V mice [11], they are not present in younger animals (Fig. 5). Most likely, pathological changes follow the functional abnormalities (inability to become phosphorylated, changes in ATPase, force, calcium sensitivity) observed in the mutated myocardium.
As shown by van der Velden et al., a largely reduced RLC phosphorylation was observed in end-stage heart failure patients [34-36]. The authors speculated that this could reflect an adaptive response of the myocardium to pathophysiological injury. Similar to what we think of phosphorylation-induced reduction in force and decreased Ca2+-sensitivity in Tg-D166V muscle fibres, it was proposed that dephosphorylation of RLC in end-stage failing hearts was to compensate for the potentially detrimental effects of an abnormally increased Ca2+ responsiveness [34-36].
Molecular mechanisms of RLC phosphorylation in Tg-D166V hearts
The unique position of the D166V RLC mutation, located near the junction between the myosin head and filamentous rod, makes this region of myosin especially sensitive to charge-induced conformational changes that ultimately may affect the myosin–actin interaction and force generation. At the molecular level, it is postulated that phosphorylated myosin cross-bridges move away from the thick-filament backbone to be closer to the actin filaments increasing the probability of myosin–actin interactions [13]. This hypothesis was recently confirmed by low-angle X-ray diffraction studies showing a phosphorylation-induced 3.4% decrease in inter-thick filament spacing in permeabilized cardiac muscle fibres [37]. The authors also showed that following RLC phosphorylation, the myosin cross-bridges assume a new position, which is farther from the surface of the thick filament backbone and closer to the thin filament [37]. Based on our force data, we predict that phosphorylation of RLC in Tg-D166V muscle fibres would position the D166V myosin cross-bridges at a greater distance from actin than in Tg-WT fibres.
Studies in skinned muscle fibres further demonstrated that the ordered array of myosin heads, characteristic of relaxed thick filaments becomes reversibly disordered by RLC phosphorylation, thus increasing myosin head mobility and accessibility to actin [38]. Recent studies from the Cooke laboratory using epifluorescence of mant-nucleotides in the presence of blebbistatin suggest that the non-phosphorylated myosin heads are highly ordered in relaxed skinned skeletal [39] and cardiac [40] muscle fibres, and become vastly disordered upon phosphorylation. We hypothesize that the D166V mutation may stabilize a disordered conformation of myosin cross-bridges in Tg-D166V muscle fibres, and that their lack of ordered orientation could be further magnified by phosphorylation.
The importance of RLC phosphorylation was suggested by Davis et al. and a distinct balance of RLC phosphorylation was proposed to underlie the stretch activation response in the heart [24, 41]. Results from the Moss group supported this concept and showed that RLC phosphorylation affects cardiac muscle contractility by accelerating the stretch activation response [42]. These findings suggest that by decreasing RLC phosphorylation in the hearts of Tg-D166V mice, the D166V mutation may interfere with the proper ratio of phosphorylated to non-phosphorylated RLC that is required to maintain the stretch activation response and cardiac function.
Closing remarks
The results of this study indicate that RLC phosphorylation plays an important role not only in the physiological performance of the heart, but also helps to maintain normal cardiac function in the diseased myocardium. It seems to be especially important in the adaptive responses of the heart to pathophysiological injury. Our work leads us to the conclusion that phosphorylation of myosin RLC works as a regulator of the acto–myosin interaction in both normal and cardiomyopathic hearts. However, a phosphorylation-induced tuning of cardiac function in the diseased heart could be different from the healthy heart and may vary depending on the type and the level of cardiac insult.
Acknowledgments
This work was supported by National Institutes of Health Grants HL071778 and HL090786 (D.S.C.) and American Heart Association Grant 10POST3420009 (P.M.). P.M., K.K., M.J. and D.S.C. designed the research; P.M., K.K. and M.J. performed the experiments; P.M., K.K., M.J. and D.S.C. analysed the data and wrote the paper.
Conflict of interest
No conflict of interest declared.
References
- 1.Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol. 2008;19:104–10. doi: 10.1111/j.1540-8167.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- 2.Szczesna D. Regulatory light chains of striated muscle myosin. Structure, function and malfunction. Curr Drug Targets Cardiovasc Haematol Disord. 2003;3:187–97. doi: 10.2174/1568006033481474. [DOI] [PubMed] [Google Scholar]
- 3.Poetter K, Jiang H, Hassanzadeh S, et al. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–9. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- 4.Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–32. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 5.Andersen PS, Havndrup O, Bundgaard H, et al. Myosin light chain mutations in familial hypertrophic cardiomyopathy: phenotypic presentation and frequency in Danish and South African populations. J Med Genet. 2001;38:E43. doi: 10.1136/jmg.38.12.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flavigny J, Richard P, Isnard R, et al. Identification of two novel mutations in the ventricular regulatory myosin light chain gene (MYL2) associated with familial and classical forms of hypertrophic cardiomyopathy. J Mol Med. 1998;76:208–14. doi: 10.1007/s001090050210. [DOI] [PubMed] [Google Scholar]
- 7.Morner S, Richard P, Kazzam E, et al. Identification of the genotypes causing hypertrophic cardiomyopathy in northern Sweden. J Mol Cell Card. 2003;35:841–9. doi: 10.1016/s0022-2828(03)00146-9. [DOI] [PubMed] [Google Scholar]
- 8.Richard P, Charron P, Carrier L, et al. Correction to: “Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy”. Circulation. 2004;109:3258. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 9.Brown JH, Kumar VS, O'Neall-Hennessey E, et al. Visualizing key hinges and a potential major source of compliance in the lever arm of myosin. Proc Natl Acad Sci USA. 2011;108:114–9. doi: 10.1073/pnas.1016288107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rayment I, Rypniewski WR, Schmidt-Base K, et al. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993;261:50–8. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 11.Kerrick WGL, Kazmierczak K, Xu Y, et al. Malignant familial hypertrophic cardiomyopathy D166V mutation in the ventricular myosin regulatory light chain causes profound effects in skinned and intact papillary muscle fibers from transgenic mice. FASEB J. 2009;23:855–65. doi: 10.1096/fj.08-118182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muthu P, Mettikolla P, Calander N, et al. Single molecule kinetics in the familial hypertrophic cardiomyopathy D166V mutant mouse heart. J Mol Cell Cardiol. 2010;48:989–98. doi: 10.1016/j.yjmcc.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sweeney HL, Bowman BF, Stull JT. Myosin light chain phosphorylation in vertebrate striated muscle: regulation and function. Am J Physiol. 1993;264:C1085–95. doi: 10.1152/ajpcell.1993.264.5.C1085. [DOI] [PubMed] [Google Scholar]
- 14.Morano I. Tuning the human heart molecular motors by myosin light chains. J Mol Med. 1999;77:544–55. doi: 10.1007/s001099900031. [DOI] [PubMed] [Google Scholar]
- 15.Huang J, Shelton JM, Richardson JA, et al. Myosin regulatory light chain phosphorylation attenuates cardiac hypertrophy. J Biol Chem. 2008;283:19748–56. doi: 10.1074/jbc.M802605200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abraham TP, Jones M, Kazmierczak K, et al. Diastolic dysfunction in familial hypertrophic cardiomyopathy transgenic model mice. Cardiovasc Res. 2009;82:84–92. doi: 10.1093/cvr/cvp016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenberg MJ, Mealy TR, Watt JD, et al. The molecular effects of skeletal muscle myosin regulatory light chain phosphorylation. Am J Physiol Regul Integr Comp Physiol. 2009;297:R265–74. doi: 10.1152/ajpregu.00171.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dweck D, Reyes-Alfonso A, Jr, Potter JD. Expanding the range of free calcium regulation in biological solutions. Anal Biochem. 2005;347:303–15. doi: 10.1016/j.ab.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 19.Fiske CH, Subbarow Y. The colorimetric determination of phosphorus. J Biol Chem. 1925;66:375–400. [Google Scholar]
- 20.Szczesna-Cordary D, Guzman G, Zhao J, et al. The E22K mutation of myosin RLC that causes familial hypertrophic cardiomyopathy increases calcium sensitivity of force and ATPase in transgenic mice. J Cell Sci. 2005;118:3675–83. doi: 10.1242/jcs.02492. [DOI] [PubMed] [Google Scholar]
- 21.Hill TL, Einsenberg E, Greene LE. Theoretical model for the cooperative equilibrium binding of myosin subfragment-1 to the actin-troponin-tropomyosin complex. Proc Natl Acad Sci. 1980;77:3186–90. doi: 10.1073/pnas.77.6.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szczesna D, Ghosh D, Li Q, et al. Familial hypertrophic cardiomyopathy mutations in the regulatory light chains of myosin affect their structure, Ca2+ binding, and phosphorylation. J Biol Chem. 2001;276:7086–92. doi: 10.1074/jbc.M009823200. [DOI] [PubMed] [Google Scholar]
- 23.Szczesna D, Zhao J, Jones M, et al. Phosphorylation of the regulatory light chains of myosin affects Ca2+ sensitivity of skeletal muscle contraction. J Appl Physiol. 2002;92:1661–70. doi: 10.1152/japplphysiol.00858.2001. [DOI] [PubMed] [Google Scholar]
- 24.Davis JS, Hassanzadeh S, Winitsky S, et al. The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell. 2001;107:631–41. doi: 10.1016/s0092-8674(01)00586-4. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Xu Y, Kerrick WGL, et al. Prolonged Ca2+ and force transients in myosin RLC transgenic mouse fibers expressing malignant and benign FHC mutations. J Mol Biol. 2006;361:286–99. doi: 10.1016/j.jmb.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 26.Ding P, Huang J, Battiprolu PK, et al. Cardiac myosin light chain kinase is necessary for myosin regulatory light chain phosphorylation and cardiac performance in vivo. J Biol Chem. 2010;285:40819–29. doi: 10.1074/jbc.M110.160499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olsson MC, Patel JR, Fitzsimons DP, et al. Basal myosin light chain phosphorylation is a determinant of Ca2+ sensitivity of force and activation dependence of the kinetics of myocardial force development. Am J Physiol Heart Circ Physiol. 2004;287:H2712–8. doi: 10.1152/ajpheart.01067.2003. [DOI] [PubMed] [Google Scholar]
- 28.Palmer BM, Moore RL. Myosin light chain phosphorylation and tension potentiation in mouse skeletal muscle. Am J Physiol. 1989;257:C1012–9. doi: 10.1152/ajpcell.1989.257.5.C1012. [DOI] [PubMed] [Google Scholar]
- 29.Davis JS, Satorius CL, Epstein ND. Kinetic effects of myosin regulatory light chain phosphorylation on skeletal muscle contraction. Biophys J. 2002;83:359–70. doi: 10.1016/S0006-3495(02)75175-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryder JW, Lau KS, Kamm KE, et al. Enhanced skeletal muscle contraction with myosin light chain phosphorylation by a calmodulin-sensing kinase. J Biol Chem. 2007;282:20447–54. doi: 10.1074/jbc.M702927200. [DOI] [PubMed] [Google Scholar]
- 31.Zhi G, Ryder JW, Huang J, et al. Myosin light chain kinase and myosin phosphorylation effect frequency-dependent potentiation of skeletal muscle contraction. PNAS. 2005;102:17519–24. doi: 10.1073/pnas.0506846102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greenberg MJ, Kazmierczak K, Szczesna-Cordary D, et al. Cardiomyopathy-linked myosin regulatory light chain mutations disrupt myosin strain-dependent biochemistry. Proc Natl Acad Sci U S A. 2010;107:17403–8. doi: 10.1073/pnas.1009619107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenberg MJ, Watt JD, Jones M, et al. Regulatory light chain mutations associated with cardiomyopathy affect myosin mechanics and kinetics. J Mol Cell Cardiol. 2009;46:108–15. doi: 10.1016/j.yjmcc.2008.09.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Velden J, Papp Z, Boontje NM, et al. Myosin light chain composition in non-failing donor and end-stage failing human ventricular myocardium. Adv Exp Med Biol. 2003;538:3–15. doi: 10.1007/978-1-4419-9029-7_1. [DOI] [PubMed] [Google Scholar]
- 35.van der Velden J, Papp Z, Boontje NM, et al. The effect of myosin light chain 2 dephosphorylation on Ca2+-sensitivity of force is enhanced in failing human hearts. Cardiovascular Research. 2003;57:505–14. doi: 10.1016/s0008-6363(02)00662-4. [DOI] [PubMed] [Google Scholar]
- 36.van der Velden J, Papp Z, Zaremba R, et al. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res. 2003;57:37–47. doi: 10.1016/s0008-6363(02)00606-5. [DOI] [PubMed] [Google Scholar]
- 37.Colson BA, Locher MR, Bekyarova T, et al. Differential roles of regulatory light chain and myosin binding protein-C phosphorylations in the modulation of cardiac force development. J Physiol. 2010;588:981–93. doi: 10.1113/jphysiol.2009.183897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levine RJ, Yang Z, Epstein ND, et al. Structural and functional responses of mammalian thick filaments to alterations in myosin regulatory light chains. J Struct Biol. 1998;122:149–61. doi: 10.1006/jsbi.1998.3980. [DOI] [PubMed] [Google Scholar]
- 39.Stewart MA, Franks-Skiba K, Chen S, et al. Myosin ATP turnover rate is a mechanism involved in thermogenesis in resting skeletal muscle fibers. Proc Natl Acad Sci U S A. 2010;107:430–5. doi: 10.1073/pnas.0909468107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hooijman P, Stewart MA, Cooke R. A new state of cardiac myosin with very slow ATP turnover: a potential cardioprotective mechanism in the heart. Biophys J. 2011;100:1969–76. doi: 10.1016/j.bpj.2011.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vemuri R, Lankford EB, Poetter K, et al. The stretch-activation response may be critical to the proper functioning of the mammalian heart. Proc Natl Acad Sci U S A. 1999;96:1048–53. doi: 10.1073/pnas.96.3.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stelzer JE, Patel JR, Moss RL. Acceleration of stretch activation in murine myocardium due to phosphorylation of myosin regulatory light chain. J Gen Physiol. 2006;128:261–72. doi: 10.1085/jgp.200609547. [DOI] [PMC free article] [PubMed] [Google Scholar]