

Abstract
The role of endogenously produced H2S in mediating varied physiological effects in mammals has spurred enormous recent interest in understanding its biology and in exploiting its pharmacological potential. In these early days in the field of H2S signaling, large gaps exist in our understanding of its biological targets, its mechanisms of action and the regulation of its biogenesis and its clearance. Two branches within the sulfur metabolic pathway contribute to H2S production: (i) the reverse transsulfuration pathway in which two pyridoxal 5′-phosphate-dependent (PLP) enzymes, cystathionine β-synthase and cystathionine γ-lyase convert homocysteine successively to cystathionine and cysteine and (ii) a branch of the cysteine catabolic pathway which converts cysteine to mercaptopyruvate via a PLP-dependent cysteine aminotransferase and subsequently, to mercaptopyruvate sulfur transferase-bound persulfide from which H2S can be liberated. In this review, we present an overview of the kinetics of the H2S-generating reactions, compare the structures of the PLP-enzymes involved in its biogenesis and discuss strategies for their regulation.
Keywords: PLP, H2S, cystathionine β-synthase, cystathionine γ-lyase, cysteine aminotransferase
Although known for its toxic effects for several centuries, the recognition that endogenously produced hydrogen sulfide (H2S) mediates a wide range of physiological effects, is relatively recent [1, 2]. The toxicity of H2S is comparable to that of cyanide and at concentrations ≥500 ppm (~15 mM), H2S causes rapid death [3, 4]. In contrast, most of the physiological effects of H2S appear to be elicited in the ten to several hundreds of micromolar concentration range (reviewed in [5, 6]). The potential effects of H2S range from its roles in neuromodulation, cardioprotection, smooth muscle relaxation and regulation of insulin release to being a pro- or anti-inflammatory mediator [5–9]. Interestingly, H2S can induce a state of suspended animation by decreasing body temperature and metabolic rate even in non-hibernating animals [10]. H2S has also been associated with increased lifespan in worm [11].
Although highly variable concentrations of H2S have been reported in mammalian tissues and plasma (reviewed in [12]), the emerging consensus is that the concentration of this gas is very low [13]: ~100 pM in blood to ~15 nM in tissues other than aorta, where the concentration is 20–100-fold higher than in other tissues [14]. H2S is a weak acid with pKa1 and pKa2 values of 6.9 and >12 [15] and at the physiological pH of 7.4, the ratio of HS−:H2S is 3:1. It is unclear whether the hudrosulfide ion or H2S is the biological mediator of the various physiological effects that have been reported. The steady-state concentration of H2S is governed by its rate of production and its rate of removal by the mitochondrial sulfide oxidation pathway, which couples to oxidative phosphorylation [7, 16]. H2S can also be removed or sequestered via reaction with disulfides or with hemeproteins. Despite the recent profusion of literature on the biological effects of H2S, major gaps exist in our understanding of the regulation of its biogenesis and its catabolism, the identity of its molecular targets, and the mechanisms of its action. This review focuses on the kinetics, structures and regulation of PLP-dependent enzymes involved in the evolution of H2S in mammals (Figure 1).
Figure 1.

Scheme showing the various H2S generating reactions catalyzed by enzymes in the reverse transsulfuration pathway (CBS and CGL) and the cysteine catabolic pathway (CAT/AAT and MST).
A. H2S Generation by PLP-dependent Enzymes
Two cytoplasmic PLP enzymes, cystathionine β-synthase (CBS) and cystathionine γ-lyase (CGL) are primarily responsible for H2S biogenesis in mammalian tissues [7, 17] (Figure 1). A third PLP-dependent pathway utilizes aspartate aminotransferase (AAT), which also has cysteine aminotransferase (CAT) activity, in combination with the PLP-independent 3-mercaptopyruvate sulfurtransferase (MST), to generate sulfane sulfur, which in the presence of a reductant, can liberate H2S [18, 19] (Figure 1). CAT/AAT and MST are present in both the cytoplasmic and mitochondrial compartments [20–22].
CAT/AAT is an aminotransferase, which catalyzes the reversible interconversion of dicarboxylic acids and keto acids in a reaction in which the PLP cofactor alternates between the aldehyde and amine forms as shown in equations 1–2 (and Scheme 1). The wealth of structural/mutagenesis and kinetic studies on CAT/AAT makes it one of the best-studied aminotransferases [23–30].
Scheme 1.

Reactions catalyzed in the CAT/AAT-MST Pathway
| [1] |
| [2] |
MST transfers sulfane sulfur from mercaptopyruvate to an active site cysteine (or cyanide) to form a persulfide (equation 3) or to generate thiocyanate. H2S can be liberated from the MST-persulfide by a reductant as shown in equation 4, where RSH is a thiol reductant.
| [3] |
| [4] |
CBS, and particularly, CGL, catalyze a multitude of H2S-generating reactions using cysteine and homocysteine as substrates (Scheme 2). While the reactivity of CBS is limited to β-replacement reactions (equations 5–8), CGL catalyzes chemical transformations at both the β-(equations 7–9) and γ-carbons (equations 10–11). Of these, the β-replacement of serine with homocysteine (equation 5) and the α,γ-elimination of cystathionine (equation 12), represent the canonical reactions catalyzed by CBS and CGL in the reverse transsulfuration pathway that converts homocysteine, derived from methionine, to cysteine.
Scheme 2.

Reactions catalyzed by CBS (5–8) and CGL (7–8)
| [5] |
| [6] |
| [7] |
| [8] |
| [9] |
| [10] |
| [11] |
| [12] |
H2S biogenesis by CBS and CGL in the context of the redox biochemistry of H2S has been reviewed recently [7], and only the PLP-relevant aspects of H2S production are emphasized in this review.
B. Kinetics of H2S Generation
(i) The CAT/AAT-MST Pathway
The CAT/AAT-MST pathway is involved in cysteine degradation and CAT exhibits a low affinity for cysteine. The KMs for cysteine and 2-oxoglutarate are 22 mM and 0.06 mM, respectively [22] and the specific activity of rat liver CAT/AAT at pH 8.0 is estimated from the specific activity reported by Akagi et al [22] to be 1.2 s−1 at 37 °C. In contrast, the KM for aspartate is lower (1.6 mM) and the enzyme exhibits a 20-fold higher kcat with this substrate [22]. Since aspartate (~730 μM in mouse liver and ~4 mM in mouse brain (Vitvitsky, V. and Banerjee, R, unpublished data)) is significantly more abundant than cysteine (~130 μM in mouse liver and 90 μM in mouse brain [31]), and the KM for aspartate is ~14-fold lower than for cysteine, the enzyme is expected to function primarily as an AAT.
Meister and coworkers were the first to characterize MST and demonstrated the formation of H2S from 3-mercaptopyruvate in the presence of a reducing agent [18]. Stipanuk and coworkers attempted to estimate the relative contributions of CBS, CGL and CAT/MST to cysteine desulfhydration in rat liver and kidney extracts by using assays varying in substrate composition, pH and inhibitors or activators for these enzymes [17]. Their results pointed to the predominance of CGL to H2S production from cysteine in both tissues at pH 7.4, although the concentration of cysteine used in the assay (2 mM) was not “physiological” as noted, at least for liver. The contribution of the CAT/MST reactions to H2S production, assessed at a very high concentration of cysteine (160 mM), was only observed under alkaline conditions (pH 9.7) and in the presence of a reductant (3 mM dithiothreitol), and was negligible at a physiologically relevant pH. For rat liver MST, the KM for mercaptopyruvate is 1.2 mM and kcat is 4.0 × 103 s−1 at 25 °C and pH 9.55 in the presence of 25 mM mercaptoethanol [32, 33]. For bovine kidney MST, a similar value for the KM for mercaptopyruvate (2.8 mM) but a significantly lower kcat (713 s−1) at pH 9.55 and 30 °C has been reported [34]. The contribution of the CAT/MST pathway to H2S generation at physiologically relevant substrate concentrations and pH remains to be assessed. This pathway might be more important for H2S production in tissues where cysteine concentrations are high (e.g. kidney where cysteine and aspartate concentrations are ~520 μM and 1.7 mM respectively (Vitvitsky V and Banerjee, R, unpublished results) and/or in tissues where the transsulfuration enzymes are missing or present at very low concentrations.
(ii) The Reverse Transsulfuration Pathway
Although cysteine desulfhydration is referred to as the primary mechanism for H2S generation, as discussed above (equations 6–12 and Scheme 2) and shown in Figure 1, several alternative routes can be used by CBS and CGL due to their relatively low substrate stringencies. We have recently reported a detailed steady-state kinetic characterization of the multiple H2S-generating reactions catalyzed by human CBS and CGL and used these kinetic parameters to simulate their relative contributions at physiologically relevant substrate concentrations and under hyperhomocysteinemic conditions [35, 36]. CBS catalyzes the condensation of two amino acids, and the two substrate binding pockets are referred to as site 1 (i.e. where the PLP external aldimine is formed) and site 2, where the nucleophilic amino acid is bound, respectively. The KM values for the H2S-generating substrates for human CBS are: 3.2 mM for homocysteine, which binds only to site 2, and 6.8 and 27 mM for cysteine for sites 1 and 2, respectively [36]. Kinetic analyses reveal that under maximal velocity conditions, the preferred route for H2S generation by human CBS is via condensation of cysteine and homocysteine to generate cystathionine and H2S (kcat = 19.6 ± 1.2 s−1 at 37 °C, equation 7) followed by condensation of two moles of cysteine to form lanthionine and H2S (kcat = 0.86 ± 0.08 s−1, equation 8). The least efficient route is the β-replacement of cysteine to give serine and H2S (kcat = 0.36 ± 0.04 s−1, equation 6). In these reactions, only cysteine binds to the PLP site to form the external aldimine. Homocysteine binds to site 2 and condenses with the PLP-aminoacrylate species. Since the KM for cysteine is high (6.8 mM) relative to the intracellular concentration of this amino acid, the rate of reaction 7, i.e., the condensation of cysteine and homocysteine, is limited by the concentration of cysteine and is relatively insensitive to increasing concentrations of homocysteine [36].
The kinetics of H2S generation by CGL are considerably more complex. Cysteine and homocysteine bind in various combinations to either the PLP site or to the second site and at least five H2S-generating reactions result (equations 7–11 and Scheme 2). The KM values for the H2S-generating substrates for human CGL are: 2.7 mM and 5.9 mM for homocysteine for sites 1 and 2, respectively, and 1.7 and 33 mM for cysteine for sites 1 and 2, respectively [35]. Under maximal velocity conditions, the following order of turnover numbers for H2S production is observed: γ-condensation of 2 moles of homocysteine (kcat = 4.9 s−1, equation 11) > α,γ–elimination of homocysteine (0.92 s−1, equation 10) ≈ condensation of two moles of cysteine (0.85 s−1, equation 8) α,β–elimination of cysteine (0.47 s−1, equation 9) > β–replacement of cysteine by homocysteine (0.15 s−1, equation 7). Since homocysteine can bind either to the first (PLP) or the second amino acid-binding site, CGL-catalyzed H2S generation is sensitive to changes in the intracellular concentration of homocysteine and is predicted to increase between 20–200-fold under hyperhomocysteinemic conditions (simulated at 200 μM homocysteine) [35].
Based on the steady-state kinetic parameters, simulations at physiologically relevant substrate concentrations (cysteine: 100 μM, homocysteine: 10 μM, and serine: 560 μM) predict that CBS would account for ~25–70% of total H2S production, depending on the extent of its activation by the allosteric regulator, S-adenosylmethionine (AdoMet) [36]. However, the assumption of equimolar concentrations of CBS and CGL is an important limitation of the simulations as borne out by recent quantitative estimates of these two enzymes in select tissues [37]. For instance, in murine liver, CGL is 60-fold more abundant than CBS. Taking this difference in enzyme levels into account, we predict that liver CGL accounts for ~97% of murine liver H2S production at physiologically relevant substrate concentrations [37]. Similar estimates for other tissues await quantitative determination of the relative (or absolute) amounts of CBS and CGL in each tissue. Under conditions of moderate and severe hyperhomocysteinemia, the contribution of CGL to H2S production is predicted to increase.
Determination of maximal tissue H2S generation rates at high concentrations of substrates (20 mM cysteine and/or 20 mM homocysteine) and in the presence or absence of the CGL inhibitor, propargylglycine, allow estimation of the relative contributions of CGL versus CBS to tissue H2S production capacity. From this study, it was concluded that CBS accounts for ~50%, ~80% and ~95%, respectively of the total H2S output in liver, kidney and brain at saturating substrate concentrations. A similar conclusion about the relative importance of CBS to kidney H2S production was reported previously [38]. The approach of assessing H2S production at high substrate concentrations is useful for studying influences of effectors (e.g. nutritional, gene knockout, stress) on the metabolic capacity of the pathway.
C. Structures of H2S-generating Enzymes
PLP enzymes are loosely classified based on their sequence similarity and structural folds into five groups [39, 40] and on the basis of the carbon atom involved in chemistry, into three groups [41]. According to the former classification, CAT/AAT and CGL belong to different subfamilies within the fold type I family, while CBS belongs to the fold type II family (Figure 2). Based on the latter classification, CAT/AAT, CBS and CGL belong to the α, β and γ families respectively.
Figure 2.

Overall folds for PLP enzymes involved in H2S generation pathways. The structures of chicken AAT/CAT (A), Drosophila CBS (B) and human CGL (C) are shown. The subunits in each enzyme are shown in different colors and the PLP is shown in ball representation in navy. PDB files 1MAQ, 3PC2 and 3COG were employed to generate the structures in A, B and C, respectively.
(i) CAT/AAT
CAT/AAT is one of the best-characterized enzymes in the fold type I family with multiple structures of the cytoplasmic and mitochondrial forms in the presence and absence of substrates or analogs being available [29, 42, 43]. The reaction mechanism of CAT/AAT has been reviewed from a structural perspective [44] and only the salient structural features are discussed in this section. Each monomer in the homodimeric protein consists of a large and a small domain with the active site being located at the interface where the PLP is covalently linked to a lysine residue donated by the large domain (Figure 2A). In the absence of substrate, the subunits exist in an open conformation, while the binding of substrate induces domain closure resulting in substrate sequestration from the solvent. Since a crystal structure of AAT/CAT with cysteine is not available, the interactions that have been noted with dicarboxylate substrates are discussed here. Two arginine residues move by several angstroms and engage in salt-bridge interactions with the α and β-carboxylates of aspartate, in the closed and active conformation of the enzyme [42, 43] (Figure 3). The active site lysine that is involved in a Schiff base linkage with PLP is also believed to serve as the base for abstraction of the α-proton in the first half reaction and as the general acid that later donates a proton to the ketimine intermediate [44]. The ε-amino group of this lysine residue is located 2.9 Å from the C4′ of the external aldimine of the substrate analog, methylaspartate [42]. The O-3′ atom of PLP is hydrogen bonded to an asparagine side chain while the pyridine ring nitrogen is within 2.6 Å of the β-carboxylate oxygen of an aspartate residue. The aspartate stabilizes the carbanion generated upon α-proton abstraction, contributing ~5 kcal/mol to lowering the barrier for this rate-determining step in the first half reaction. In contrast, the interaction between the aspartate and pyridine nitrogen is less significant for the second half reaction, i.e., the transfer of the amino group from PMP to 2-ketoglutarate [45].
Figure 3.
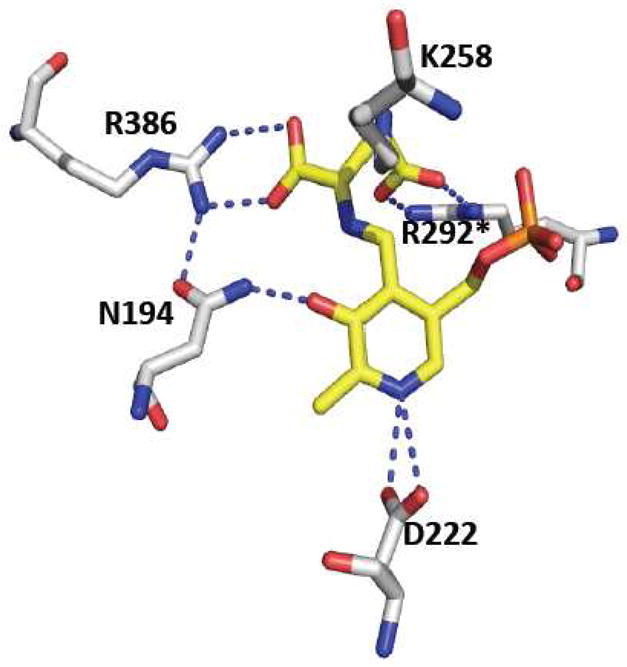
Close-up of the active site structure of chicken CAT/AAT with a glutamate ketimine intermediate. The residues that interact with the O3′ and pyridine nitrogen of PLP, the lysine residue that forms the Schiff base in the internal aldimine, and the arginine residues that interact with the proximal and distal carboxylates of the glutamate ketimine are shown. One of the two arginine residues, marked with the asterisk, is donated by the adjacent subunit. The figure was generated using the PDB file 1MAQ and the residues follow the numbering for the chicken sequence.
(ii) CBS
CBS from higher eukaryotes is unique in the family of PLP enzymes in also being a hemeprotein [46]. Its organization is consequently more complex with an N-terminal heme-binding domain, a central PLP-binding catalytic domain and a C-terminal regulatory domain, which binds the allosteric activator, AdoMet [47]. The first structures of full-length CBS from Drosophila have been reported recently in which a reactive carbanion and an aminoacrylate intermediate have been captured [48]. The mammalian enzyme tends to forms aggregates ranging from 2- to 16-mers in solution [49, 50]. It is not known if the multiple oligomeric states of CBS exist in vivo and if so, whether they have a regulatory role. In contrast to human CBS, the Drosophila enzyme is less prone to aggregation and the crystal structure of the full-length dimeric and active form of CBS has been solved (Figure 2B). Earlier structures of the catalytic core of human CBS had revealed the placement of the heme some 20 Å from PLP [51, 52]. The catalytic core comprises two subdomains that bind PLP at the interface. Substrate binding induces the closed conformation and causes an active site serine residue to move in towards the PLP cofactor. The structure of the carbanion intermediate of serine at 1.7 Å resolution captures the zwitterionic species in which the Cα proton has been abstracted by the active site lysine (Figure 4A) that is originally involved in Schiff base formation in the internal aldimine. The 2.1 Å distance between the εNH3+ group of the lysine and the Cα, reveals how the negative charge is stabilized with additional stabilization provided by PLP’s C4A, the Schiff base nitrogen, and the side chain of the serine residue (Ser116 in the Drosophila sequence) that is repositioned upon substrate binding. In the structure of the aminoacrylate intermediate, the εNH3+ group is seen to move away from Cα, and interacts instead with the phosphate group of PLP (Figure 4B). The presence of the heme in CBS from higher organisms limits direct visualization of PLP-based reaction intermediates and much of the spectroscopic characterization has been limited to either an N-terminal truncated form of human CBS lacking the heme domain [53] or yeast CBS [54–57]. Recently, the aminoacrylate intermediate has been observed by difference stopped-flow spectroscopy in the heme-containing Drosophila CBS [48]. The side chain of a serine residue (Ser318 in the Drosophila sequence) is involved in a hydrogen-bonding interaction with the pyridine nitrogen of PLP. Mutation of this serine to alanine in yeast CBS results in an 80-fold decrease in catalytic efficiency while mutation to aspartate, which leads to loss of the PLP cofactor, results in undetectable activity [57]. The modest effect of the Ser→Ala mutation is consistent with ylide-type electrostatic stabilization of the carbanion intermediate being provided primarily by the Schiff base. These results are consistent with the crystal structure of the carbanion intermediate in Drosophila CBS and also with computational studies that suggest a more prominent role for the Schiff base in carbanion stabilization rather than delocalization through the extended conjugated ring system of PLP [58, 59]. An asparagine (Asn118 in the Drosophila sequence) residue interacts with the O3′ of PLP and its mutation to alanine or histidine in yeast CBS results in an ~100- and 1000-fold decrease, respectively in catalytic efficiency.
Figure 4.
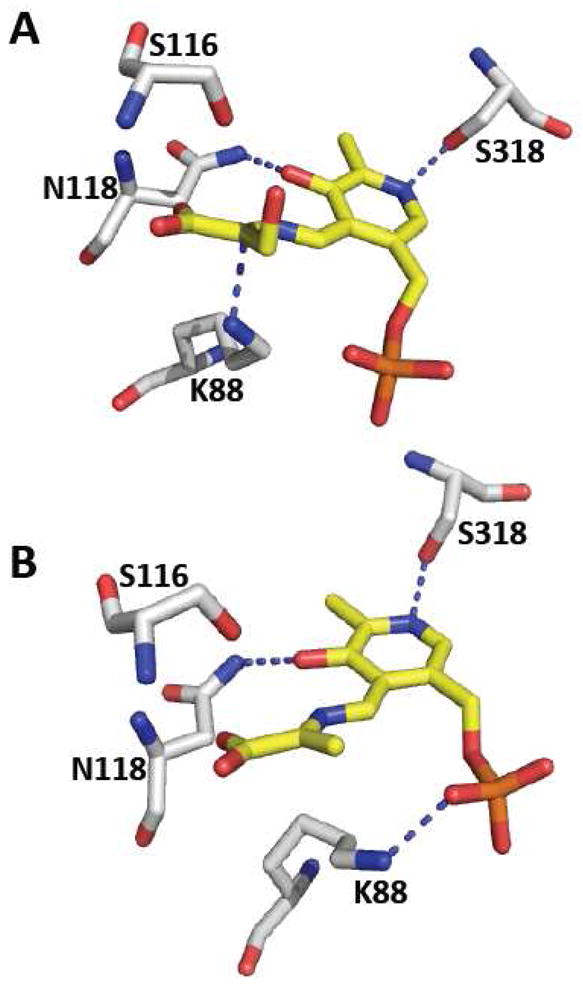
Close-up of the active site structure of Drosophila CBS. A. Structure of the carbanion intermediate showing the close distance between K88 and Cα and the hydrogen bonding interactions between O3′ and the pyridine nitrogen of PLP. B. Structure of the aminoacrylate intermediate reveals that K88 has repositioned away from Cα and is engaged in hydrogen bonding interactions with the phosphate group of PLP. The figure was generated using the PDB files 3PC4 (A) and 3PC3 (B) and the numbering for the Drosophila sequence is reported.
The N-terminal heme domain, which is part of the catalytic core, reveals the presence of the histidine and cysteine residues that serve as axial heme ligands in both the ferric and ferrous oxidation states (Figure 6). Perturbation in the heme domain is transmitted over a long range to the PLP site and modulates CBS activity [60]. The responsiveness of the PLP site to redox changes at the heme site is revealed by 31P NMR spectroscopy by changes in the phosphate chemical shifts in ferric versus ferrous CBS [61]. The structural pathway for communication between the sites is not known. The regulatory domain of CBS has a tandem repeat of two CBS domains, a secondary structure motif that is named after this protein and found in all kingdoms of life and is commonly use to bind adenine nucleotides [62]. Unlike human CBS, Drosophila CBS exhibits high basal activity, which does not increase further in the presence of AdoMet. Hence, it has been inferred that that the conformation of Drosophila CBS is locked in an activated state and corresponds to the conformation of human CBS that is achieved upon binding of AdoMet [48].
Figure 6.

The heme environment influences the tautomeric equilibrium at the PLP site in human CBS. In the ferric state, the active ketoenimine tautomer predominates in the active site. Formation of the ferrous-CO CBS form results in displacement of the endogenous C52 heme ligand by CO and is correlated with a shift to the inactive enolimine form of the internal aldimine. A possible route for communication between the two cofactors is via an alpha helix in which R266 at one end forms a salt bridge with the heme ligand, C52. At the other end, two threonine residues, T257 and T260 engage in hydrogen bonding interactions with the phosphate group of PLP.
(iii) CGL
The structures of apo- (lacking PLP) and holo-human CGL, and in complex with the suicide inhibitor, propargylglycine and of yeast CGL have been reported [63, 64]. Human CGL containing bound PLP is a homotetramer (Figure 2C) while the apo-enzyme is a mixture of the monomeric and tetrameric species with the latter being the dominant form. Each subunit consists of two subdomains as is typical of the fold I family manifold [65]. The PLP is bound to the larger of the two subdomains, which is at the N-terminus and is in Schiff base linkage to Lys212 (human sequence numbering) (Figure 5). Tyr114 swings 19.3 Å upon PLP binding to occupy a position above the pyridine ring and is involved in an aromatic π-stacking interaction. The tyrosine is postulated to play a role in activating cystathionine for the transschiffization reaction [64]. While its mutation to alanine impairs catalysis, mutation to phenylalanine activates H2S generation 3.6-fold, suggesting that the π-stacking interaction might be important for properly orienting the PLP in the active site [66]. Two residues, Tyr60 and Arg62 make hydrogen-bonding contacts with the phosphate group of PLP and are donated by an adjacent monomer. A pathogenic mutation, Arg62His has been described in patients who have elevated levels of plasma cystathionine, consistent with a role for this residue in cofactor binding and therefore, CGL function [67]. The O3′ atom of PLP is hydrogen bonded to an asparagine residue while Asp187 interacts with the pyridine nitrogen, stabilizing the positive charge on PLP and enhancing its electrophilicity. Mutation of Asp187 to either alanine or glutamate results in complete loss of H2S producing activity [66].
Figure 5.
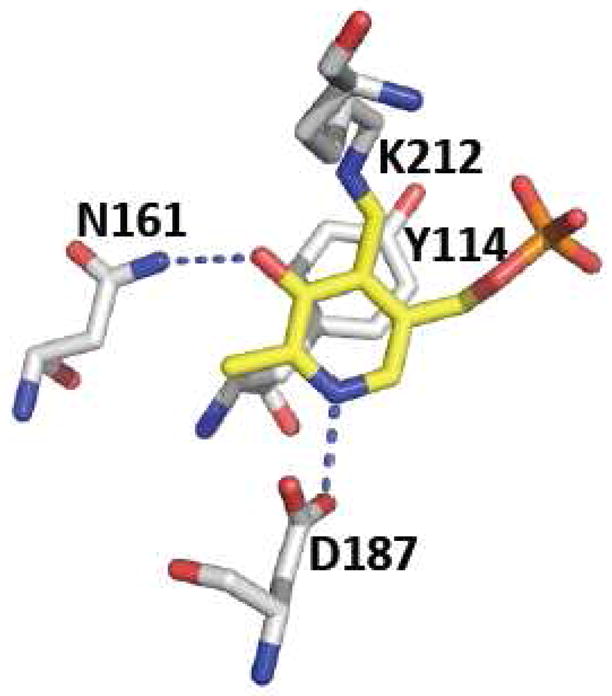
Close-up of the active site structure of human CGL. The residues that interact with the O3′ and pyridine nitrogen of PLP, the Schiff-base forming lysine residue and the tyrosine that lids the pyridine ring are shown. The figure was generated using the PDB file 3COG and the residues follow the numbering for the human sequence.
D. Regulation of H2S producing enzymes
(i) CAT/AAT-MST
The lax substrate specificity of aspartate aminotransferase makes transamination of cysteine possible [21, 22]. Little is known about the regulation of this enzyme except that the CAT activity is potently inhibited by aspartate, as might be expected from the significantly lower KM for aspartate (1.6 mM) compared to cysteine (22 mM) [22]. Interestingly, while CAT activity is highest in heart and liver, MST activity is highest in liver and kidney.
MST catalyzes the degradation of mercaptopyruvate in the cysteine catabolic pathway, detoxifies cyanide, and serves as an antioxidant enzyme. Human MST has five cysteine residues but lacks a CXXC motif. Rat MST contains three exposed cysteines: Cys247, Cys154 and Cys263 and is reported to be redox regulated [68]. Treatment of MST with a stoichiometric amount of hydrogen peroxide leads to oxidation of the active site residue Cys247 residue to cysteine sulfenate and to enzyme inhibition. The enzymatic activity is restored upon treatment with reducing agents like dithiothreitol or NADPH/thioredoxin reductase/thioredoxin. The metabolic logic of this regulation is that under oxidative stress conditions, reduced MST activity spares the cysteine pool, making it more available for the synthesis of the antioxidant, glutathione [68]. In addition to the redox-sensitive active site cysteine, the two surface-exposed cysteines, Cys154 and Cys263, are involved in an intermolecular disulfide giving rise to the monomer-dimer equilibrium [69]. The dimeric enzyme is activated with reducing agents like thioredoxin and serves as an additional redox-sensing switch for the regulation of MST activity. The cellular relevance of redox regulation of MST needs to be established.
(ii) CBS
Although human CBS and Drosophila CBSs are heme-containing PLP-dependent enzymes [46, 48], CBS isolated from lower organisms like yeast and protozoans are devoid of heme [70, 71]. The exact role of heme in CBS is still not clear [60]. The Fe3+/Fe2+ heme redox potential for human CBS is −350 ± 4 mV for the full-length protein [72] and −291 ± 5 mV for the truncated protein missing the C-terminal regulatory domain [73]. It is not known whether the ferrous form of the enzyme exists in vivo and if so, what is the identity of its redox partner. The ferric heme in CBS is inert towards most ligands except mercuric chloride [74]. In contrast, ferrous heme binds to exogenous ligands like CO, NO, HgCl2 and isonitriles [75–77]. Binding of these exogenous ligands to the heme leads to inhibition of CBS activity in both the ferric and ferrous states. Binding of CO to heme induces a tautomeric shift from the ketoenamine to the enolimine form of PLP, which is correlated with loss of CBS activity [78] (Figure 6). The communication is predicted to result from disruption of a salt bridge between the Cys52 ligand to heme and Arg266 at the bottom of a helix, which extends to the active site and makes hydrogen-bonding contacts through Thr257 and Thr260 to the phosphate group of PLP.
Earlier studies had suggested that the activity of CBS is regulated by the redox state of heme [52, 79]. However, more recent studies have shown that the activity of ferrous CBS is similar to that of the ferric form [72, 80–82] and that loss in activity observed under reducing conditions is associated with formation of a species with Soret peak at 424 nm (also known as the C-424 species). When CBS is reduced under alkaline pH conditions and at a high temperature, the C-424 species is formed quite readily and represents a heme state in which the lower axial cysteine ligand has been replaced by an unidentified and charge-neutral ligand [82]. The C-424 species represents a catalytically inactive state of the enzyme. We have demonstrated that mutation of a second coordination sphere ligand, Arg266Met, facilitates formation of the C-424 species [72]. The physiological relevance, if any, of C-424 species remains to be elucidated. If the C-424 species can revert to the ferrous (450 nm) or ferric (428 nm) state via interaction with another protein or a small molecule effector in the cell, it could lead to yet another mode of regulation of CBS.
In addition to heme, CBS has a second redox-active center, a CXXC motif (Cys272-Pro273-G274-Cys275 in human CBS) that have been captured in the dithiol and disulfide states in the crystal structures of truncated human CBS [52, 83]. Like the heme cofactor, the CXXC motif is not conserved in lower organisms like yeast and protozoans. The CXXC motif is ~ 20 Å away from both the PLP and the heme cofactors and its ability to regulate CBS remains to be explored. Under cell culture conditions, flux through the transsulfuration pathway is increased under oxidative stress conditions resulting in increased synthesis of cysteine, the limiting amino acid for the synthesis of glutathione [84, 85]. Since CBS activity limits transsulfuration flux, regulation of CBS via redox changes in the CXXC motif is a possibility that remains to be assessed. Mutation of the cysteines in the CXXC motif had no influence on the heme-dependent inhibition of CBS via formation of the C-424 species under reducing conditions [52]. However, the study was not designed to independently evaluate a redox regulatory role for the CXXC motif since the control assay contained high concentrations of the thiol substrate, homocysteine, which would reduce the disulfide form of the CXXC motif thus simulating the reduced state achieved with the mutations of cysteine to either alanine or serine. A nonthiol substrate analog would be needed to evaluate the role of the CXXC motif as a redox switch.
H2S can reversibly modify cysteine side chains to persulfide (−SSH) in some proteins, potentially increasing the reactivity of cysteines [86]. This posttranslational modification was also reported for CBS and CGL although the identity of the modified cysteines was not reported [86]. Our laboratory has been unable to detect persulfidation of CBS and CGL (unpublished results) and the in vivo existence and regulatory potential of this modification for these two enzymes, remains to be established.
CBS has a C-terminal regulatory domain, which binds the allosteric effector, AdoMet. The C-terminal regulatory region contains two hydrophobic CBS domains named after this protein [62], namely CBS1 and CBS2. AdoMet binding results in an ~2–3 fold increase in specific activity of the enzyme [87] and also in H2S production [36]. Furthermore, binding of AdoMet to CBS stabilizes the protein against proteolysis in vitro and increases its half-life from 18 to 49 h under cell culture conditions [88]. The C-terminal regulatory domain of CBS exerts intrasteric inhibition that is alleviated either by its deletion [89, 90], proteolytic cleavage [91], thermal activation [92], and selective mutations in the C-terminal domain [89, 92] or by binding of AdoMet to CBS [87]. Deletion of this regulatory domain generates a more active form of the enzyme, with an ~4-fold higher kcat than the full-length enzyme.
Treatment of human embryonic kidney 293 cells with TNF-α (tumor necrosis factor), which enhances reactive oxygen species (ROS) levels, leads to formation of the truncated form of CBS [93]. TNF-α treatment induced an ~50% increase in CBS activity, which was AdoMet unresponsive, consistent with loss of the regulatory domain during targeted proteolysis of the protein. A corresponding increase in intracellular glutathione concentration was also seen under these conditions. Injection of mice with lipopolysaccharide, a pro-inflammatory agent that is known to induce TNF-α and increase ROS levels, also induced CBS cleavage and resulted in increased glutathione levels, suggesting a possible physiological relevance of the truncated CBS form. Although the mechanism of TNF-α-induced cleavage of CBS is not known, the role of superoxide in this process was implicated by the observation that the truncation was abrogated by inhibitors of superoxide production or by overexpression of superoxide dismutase but not catalase [94].
CBS levels are down regulated in response to testosterone in human prostate cancer cells by a post-transcriptional mechanism [95]. The diminished CBS levels are paralleled by corresponding decreases in flux through the transsulfuration pathway and in glutathione levels and result in increased susceptibility of androgen responsive prostate cancer cells to oxidative stress [95]. CBS is a target of in vivo and in vitro sumoylation, a modification that decreases the activity of the protein [96, 97]. In the presence of human polycomb protein 2 (Pc2), sumoylation of CBS was enhanced, indicating that Pc2 functions as a SUMO E3 ligase for CBS. The H2S-generating substrates, cysteine and homocysteine, inhibit sumoylation of CBS, but only in the presence of Pc2 [97]. Although the role of SUMO modification of CBS is not known, the observation that CGL is also a substrate for sumoylation at least under in vitro conditions, has led to the suggestion that this modification might serve to relocalize the cysteine-generating enzymes to the nucleus under conditions where the demand for glutathione in this compartment are high, e.g. during proliferation [98].
The multiple levels of regulation in CBS emphasize its critical role in sulfur homeostasis. The activity of CBS gates the flow of sulfur from the methionine cycle via the transsulfuration pathway to cysteine, and thereon, to glutathione biosynthesis. Under oxidative stress conditions, the traffic of sulfur through the transsulfuration pathway increases several fold, depending on the cell type [84, 85, 99] and modulation of CBS activity is an important mechanism for tuning the cellular response to oxidizing conditions and for glutathione homeostasis [100].
(iii) CGL
CGL cleaves cystathionine via an α,γ-elimination into cysteine, α-ketobutyrate and ammonia. It is believed to be primarily responsible for H2S production in the vasculature and CGL−/− mice have ~80 % lower H2S levels in the heart and aorta [101]. However, while age-dependent hypertension in CGL−/− mice has been reported by one group [101] it has not been observed by another [102] and remains controversial. Although the activity of CGL was reportedly increased ~2-fold in the presence of calmodulin [101], we have not been able to observe this effect using purified and active human CGL. Unlike CBS, regulation of CGL activity is not well understood.
Human CGL has 10 cysteine residues, of which four are present in two CXXC motifs, (Cys252-Tyr253-Leu254-Cys255 and Cys307-Thr308-Gly309-Cys310). The CXXC motifs are conserved in mammals (e.g. human, rat and mouse) but not in lower organisms (e.g. nematode and yeast). The influence, if any of the oxidation state of the CXXC motifs on CGL activity is not known. Purified human CGL has been shown to be a target of sumoylation [97]. The presence of the substrate, cystathionine, did not influence sumoylation of CGL. The in vivo relevance of CGL sumoylation and its possible effect on nuclear localization remain to be assessed.
Summary
The growing interest in the biology of H2S signaling focuses attention on the structures, mechanisms and regulation of the enzymes that biosynthesize it. And in these early days, large gaps exist in our understanding of fundamental aspects of H2S biochemistry such as which enzymes are primarily responsible for H2S production in different tissues and under different conditions and how are they regulated? H2S is generated from the sulfur containing amino acids, cysteine and homocysteine, and for all three PLP enzymes, the KMs for the substrates are very high (in the millimolar range) in contrast to their intracellular concentrations (typically in the tens to hundreds of micromolar range). This raises the obvious question as to whether modulators of these enzymes exist in the cell and influence their affinity for substrate under specific conditions. Studies on the relative contributions of the H2S generating reactions tend to primarily use cysteine as substrate to the exclusion of homocysteine, which leads to an underestimation of CBS-dependent H2S production. Furthermore, the use of nonspecific inhibitors e.g. iodoacetate (a general alkylating agent), hydroxylamine (that releases the cofactor from PLP enzymes), aminooxyacetate (a general transaminase inhibitor) or even propargylglycine (a suicide inhibitor of CGL, which has some off-target activity), to decipher the contributions of the individual H2S-generating enzymes, demonstrates the limitations of the published studies and emphasizes the need for developing reagents that are enzyme specific. Multiple structures of all three PLP-dependent enzymes involved either indirectly (AAT/CAT) or directly (CBS and CGL) in H2S production are available and could, in combination with high throughput screening, be used for guiding rational inhibitor design. One of the obvious challenges for the latter is the similarity in the mode of substrate binding (i.e. via a Schiff base to PLP) and the similarity in the reactivity of the external aldimine for all three PLP enzymes.
Some other aspects of H2S biochemistry that warrant mechanistic scrutiny are the form of this molecule that mediates its physiological effects, i.e. H2S versus HS− and the identity of the cellular targets themselves. Since H2S is derived from amino acids whose concentrations are far below the KMs for the respective enzymes, strategies for stimulating or dampening production in response to cellular signaling needs, await elucidation. Alternatively, changes in H2S levels might be effected primarily by regulation of its catabolic pathway, which is strictly oxygen dependent and scavenges H2S efficiently. Under hypoxic conditions, sulfide oxidation is inhibited, but what about other conditions that might modulate flux through this pathway? Along these lines, the potential role of the “bound” sulfane-sulfur pool as an H2S reservoir needs to be assessed and if pertinent, the physiologically relevant conditions under which this pool is mobilized, need to be identified. In a field where insights into the pleiotropic physiological effects of H2S continue to emerge, rigorous biochemical studies on its production, mechanisms of action and removal are urgently needed to support realization of its pharmacological potential.
Research highlights
H2S is a gaseous signaling molecule, with neuromodulatory and cardioprotective effects, which is synthesized by enzymes involved in sulfur metabolism. T
Three of the H2S-producing enzymes are pyridoxal phosphate-dependent and utilize the suflur amino acids, cysteine and homocysteine as subtrates.
The structure, function and regulation of the H2S producing enzymes are the focus of this review article.
Footnotes
This work was supported by grants from the National Institutes of Health (HL58984).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kimura H. Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem Biophys Res Commun. 2000;267:129–133. doi: 10.1006/bbrc.1999.1915. [DOI] [PubMed] [Google Scholar]
- 3.Evans CL. The toxicity of hydrogen sulphide and other sulphides. Q J Exp Physiol Cogn Med Sci. 1967;52:231–248. doi: 10.1113/expphysiol.1967.sp001909. [DOI] [PubMed] [Google Scholar]
- 4.Truong DH, Eghbal MA, Hindmarsh W, Roth SH, O’Brien PJ. Molecular mechanisms of hydrogen sulfide toxicity. Drug Metab Rev. 2006;38:733–744. doi: 10.1080/03602530600959607. [DOI] [PubMed] [Google Scholar]
- 5.Wang R. Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 2002;16:1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- 6.Kimura H. Hydrogen sulfide: from brain to gut. Antioxid Redox Signal. 2010;12:1111–1123. doi: 10.1089/ars.2009.2919. [DOI] [PubMed] [Google Scholar]
- 7.Kabil O, Banerjee R. The redox biochemistry of hydrogen sulfide. J Biol Chem. 2010;285:21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Predmore BL, Lefer DJ. Development of hydrogen sulfide-based therapeutics for cardiovascular disease. J Cardiovasc Transl Res. 2010;3:487–498. doi: 10.1007/s12265-010-9201-y. [DOI] [PubMed] [Google Scholar]
- 9.Gadalla MM, Snyder SH. Hydrogen Sulfide as a Gasotransmitter. J Neurochem. 2010 doi: 10.1111/j.1471-4159.2010.06580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blackstone E, Morrison M, Roth MB. H2S induces a suspended animation-like state in mice. Science. 2005;308:518. doi: 10.1126/science.1108581. [DOI] [PubMed] [Google Scholar]
- 11.Miller DL, Roth MB. Hydrogen sulfide increases thermotolerance and lifespan in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2007;104:20618–20622. doi: 10.1073/pnas.0710191104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olson KR. Is hydrogen sulfide a circulating “gasotransmitter” in vertebrate blood? Biochim Biophys Acta. 2009;1787:856–863. doi: 10.1016/j.bbabio.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 13.Furne J, Saeed A, Levitt MD. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1479–1485. doi: 10.1152/ajpregu.90566.2008. [DOI] [PubMed] [Google Scholar]
- 14.Levitt MD, Abdel-Rehim MS, Furne J. Free and Acid-labile Hydrogen Sulfide Concentrations in Mouse Tissues: Anomalously High Free Hydrogen Sulfide in Aortic Tissue. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3525. [DOI] [PubMed] [Google Scholar]
- 15.Vorobets VS, Kovach SK, Kolbasov GY. Distribution of Ion Species and Formation of Ion Pairs in Concentrated Polysulfide Solutions in Photoelectrochemical Transducers. Russian J Applied Chem. 2002;75:229–234. [Google Scholar]
- 16.Bouillaud F, Blachier F. Mitochondria and sulfide: A very old story of poisoning, feeding and signaling? Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3678. [DOI] [PubMed] [Google Scholar]
- 17.Stipanuk MH, Beck PW. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem J. 1982;206:267–277. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meister A, Fraser PE, Tice SV. Enzymatic desulfuration of beta-mercaptopyruvate to pyruvate. J Biol Chem. 1954;206:561–575. [PubMed] [Google Scholar]
- 19.Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, Kimura H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal. 2009;11:703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- 20.Nagahara N, Ito T, Kitamura H, Nishino T. Tissue and subcellular distribution of mercaptopyruvate sulfurtransferase in the rat: confocal laser fluorescence and immunoelectron microscopic studies combined with biochemical analysis. Histochem Cell Biol. 1998;110:243–250. doi: 10.1007/s004180050286. [DOI] [PubMed] [Google Scholar]
- 21.Ubuka T, Umemura S, Yuasa S, Kinuta M, Watanabe K. Purification and characterization of mitochondrial cysteine aminotransferase from rat liver. Physiol Chem Phys. 1978;10:483–500. [PubMed] [Google Scholar]
- 22.Akagi R. Purification and characterization of cysteine aminotransferase from rat liver cytosol. Acta Med Okayama. 1982;36:187–197. doi: 10.18926/AMO/30697. [DOI] [PubMed] [Google Scholar]
- 23.Cronin CN, Kirsch JF. Role of arginine-292 in the substrate specificity of aspartate aminotransferase as examined by site-directed mutagenesis. Biochemistry. 1988;27:4572–4579. doi: 10.1021/bi00412a052. [DOI] [PubMed] [Google Scholar]
- 24.Goldberg JM, Swanson RV, Goodman HS, Kirsch JF. The tyrosine-225 to phenylalanine mutation of Escherichia coli aspartate aminotransferase results in an alkaline transition in the spectrophotometric and kinetic pKa values and reduced values of both kcat and Km. Biochemistry. 1991;30:305–312. doi: 10.1021/bi00215a041. [DOI] [PubMed] [Google Scholar]
- 25.Inoue K, Kuramitsu S, Okamoto A, Hirotsu K, Higuchi T, Morino Y, Kagamiyama H. Tyr225 in aspartate aminotransferase: contribution of the hydrogen bond between Tyr225 and coenzyme to the catalytic reaction. J Biochem. 1991;109:570–576. doi: 10.1093/oxfordjournals.jbchem.a123421. [DOI] [PubMed] [Google Scholar]
- 26.Malcolm BA, Kirsch JF. Site-directed mutagenesis of aspartate aminotransferase from E. coli. Biochem Biophys Res Commun. 1985;132:915–921. doi: 10.1016/0006-291x(85)91894-7. [DOI] [PubMed] [Google Scholar]
- 27.McPhalen CA, Vincent MG, Jansonius JN. X-ray structure refinement and comparison of three forms of mitochondrial aspartate aminotransferase. J Mol Biol. 1992;225:495–517. doi: 10.1016/0022-2836(92)90935-d. [DOI] [PubMed] [Google Scholar]
- 28.Toney MD, Kirsch JF. The K258R mutant of aspartate aminotransferase stabilizes the quinonoid intermediate. J Biol Chem. 1991;266:23900–23903. [PubMed] [Google Scholar]
- 29.Malashkevich VN, Strokopytov BV, Borisov VV, Dauter Z, Wilson KS, Torchinsky YM. Crystal structure of the closed form of chicken cytosolic aspartate aminotransferase at 1.9 A resolution. J Mol Biol. 1995;247:111–124. doi: 10.1006/jmbi.1994.0126. [DOI] [PubMed] [Google Scholar]
- 30.Malashkevich VN, Toney MD, Jansonius JN. Crystal structures of true enzymatic reaction intermediates: aspartate and glutamate ketimines in aspartate aminotransferase. Biochemistry. 1993;32:13451–13462. doi: 10.1021/bi00212a010. [DOI] [PubMed] [Google Scholar]
- 31.Vitvitsky V, Dayal S, Stabler S, Zhou Y, Wang H, Lentz SR, Banerjee R. Perturbations in homocysteine-linked redox homeostasis in a murine model for hyperhomocysteinemia. Am J Physiol Regul Integr Comp Physiol. 2004;287:R39–46. doi: 10.1152/ajpregu.00036.2004. [DOI] [PubMed] [Google Scholar]
- 32.Nagahara N, Okazaki T, Nishino T. Cytosolic mercaptopyruvate sulfurtransferase is evolutionarily related to mitochondrial rhodanese. Striking similarity in active site amino acid sequence and the increase in the mercaptopyruvate sulfurtransferase activity of rhodanese by site-directed mutagenesis. J Biol Chem. 1995;270:16230–16235. doi: 10.1074/jbc.270.27.16230. [DOI] [PubMed] [Google Scholar]
- 33.Nagahara N, Nishino T. Role of amino acid residues in the active site of rat liver mercaptopyruvate sulfurtransferase. CDNA cloning, overexpression, and site-directed mutagenesis. J Biol Chem. 1996;271:27395–27401. doi: 10.1074/jbc.271.44.27395. [DOI] [PubMed] [Google Scholar]
- 34.Jarabak R, Westley J. Steady-state kinetics of 3-mercaptopyruvate sulfurtransferase from bovine kidney. Arch Biochem Biophys. 1978;185:458–465. doi: 10.1016/0003-9861(78)90189-3. [DOI] [PubMed] [Google Scholar]
- 35.Chiku T, Padovani D, Zhu W, Singh S, Vitvitsky V, Banerjee R. H2S biogenesis by cystathionine gamma -lyase leads to the novel sulfur metabolites, lanthionine and homolanthionine, and is responsive to the grade of hyperhomocysteinemia. J Biol Chem. 2009;284:11601–11612. doi: 10.1074/jbc.M808026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J Biol Chem. 2009;284:22457–22466. doi: 10.1074/jbc.M109.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kabil O, Vitvitsky V, Xie P, Banerjee R. The Quantitative Significance of the Two Transsulfuration Enzymes for Tissue H2S Production. 2010 doi: 10.1089/ars.2010.3781. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Z, Prathapasinghe G, Wu N, Hwang SY, Siow YL, KO Ischemia-reperfusion reduces cystathionine-beta-synthase-mediated hydrogen sulfide generation in the kidney. Am J Physiol Renal Physiol. 2009;297:F27–35. doi: 10.1152/ajprenal.00096.2009. [DOI] [PubMed] [Google Scholar]
- 39.Grishin NV, Phillips MA, Goldsmith EJ. Modeling of the spatial structure of eukaryotic ornithine decarboxylases. Protein Sci. 1995;4:1291–1304. doi: 10.1002/pro.5560040705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jansonius JN. Structure, evolution and action of vitamin B6-dependent enzymes. Curr Opin Struct Biol. 1998;8:759–769. doi: 10.1016/s0959-440x(98)80096-1. [DOI] [PubMed] [Google Scholar]
- 41.Alexander FW, Sandmeier E, Mehta PK, Christen P. Evolutionary relationships among pyridoxal-5′-phosphate-dependent enzymes. Regio-specific alpha, beta and gamma families. Eur J Biochem. 1994;219:953–960. doi: 10.1111/j.1432-1033.1994.tb18577.x. [DOI] [PubMed] [Google Scholar]
- 42.Rhee S, Silva MM, Hyde CC, Rogers PH, Metzler CM, Metzler DE, Arnone A. Refinement and comparisons of the crystal structures of pig cytosolic aspartate aminotransferase and its complex with 2-methylaspartate. J Biol Chem. 1997;272:17293–17302. [PubMed] [Google Scholar]
- 43.McPhalen CA, Vincent MG, Picot D, Jansonius JN, Lesk AM, Chothia C. Domain closure in mitochondrial aspartate aminotransferase. J Mol Biol. 1992;227:197–213. doi: 10.1016/0022-2836(92)90691-c. [DOI] [PubMed] [Google Scholar]
- 44.Kirsch JF, Eichele G, Ford GC, Vincent MG, Jansonius JN, Gehring H, Christen P. Mechanism of action of aspartate aminotransferase proposed on the basis of its spatial structure. J Mol Biol. 1984;174:497–525. doi: 10.1016/0022-2836(84)90333-4. [DOI] [PubMed] [Google Scholar]
- 45.Yano T, Kuramitsu S, Tanase S, Morino Y, Kagamiyama H. Role of Asp222 in the catalytic mechanism of Escherichia coli aspartate aminotransferase: the amino acid residue which enhances the function of the enzyme-bound coenzyme pyridoxal 5′-phosphate. Biochemistry. 1992;31:5878–5887. doi: 10.1021/bi00140a025. [DOI] [PubMed] [Google Scholar]
- 46.Kery V, Bukovska G, Kraus JP. Transsulfuration depends on heme in addition to pyridoxal 5′-phosphate. Cystathionine b-synthase is a heme protein. J Biol Chem. 1994;269:25283–25288. [PubMed] [Google Scholar]
- 47.Banerjee R, Evande R, Kabil O, Ojha S, Taoka S. Reaction mechanism and regulation of cystathionine beta-synthase. Biochim Biophys Acta. 2003;1647:30–35. doi: 10.1016/s1570-9639(03)00044-x. [DOI] [PubMed] [Google Scholar]
- 48.Koutmos M, Kabil O, Smith JL, Banerjee R. Structural basis for substrate activation and regulation by cystathionine beta-synthase (CBS) domains in cystathionine {beta}-synthase. Proc Natl Acad Sci U S A. 2010;107:20958–20963. doi: 10.1073/pnas.1011448107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sen S, Banerjee R. A Pathogenic Linked Mutation in the Catalytic Core of Human Cystathionine beta-Synthase Disrupts Allosteric Regulation and Allows Kinetic Characterization of a Full-Length Dimer. Biochemistry. 2007;46:4110–4116. doi: 10.1021/bi602617f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frank N, Kery V, Maclean KN, Kraus JP. Solvent-accessible cysteines in human cystathionine beta-synthase:Crucial role of cysteine 431 in S-adenosyl L-methionine binding. Biochemistry. 2006;45:11021–11029. doi: 10.1021/bi060737m. [DOI] [PubMed] [Google Scholar]
- 51.Meier M, Janosik M, Kery V, Kraus JP, Burkhard P. Structure of human cystathionine beta-synthase: a unique pyridoxal 5′-phosphate-dependent heme protein. EMBO J. 2001;20:3910–3916. doi: 10.1093/emboj/20.15.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taoka S, Lepore BW, Kabil O, Ojha S, Ringe D, Banerjee R. Human cystathionine beta-synthase is a heme sensor protein. Evidence that the redox sensor is heme and not the vicinal cysteines in the CXXC motif seen in the crystal structure of the truncated enzyme. Biochemistry. 2002;41:10454–10461. doi: 10.1021/bi026052d. [DOI] [PubMed] [Google Scholar]
- 53.Evande R, Ojha S, Banerjee R. Visualization of PLP-bound intermediates in hemeless variants of human cystathionine beta-synthase: evidence that lysine 119 is a general base. Arch Biochem Biophys. 2004;427:188–196. doi: 10.1016/j.abb.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 54.Jhee KH, Niks D, McPhie P, Dunn MF, Miles EW. The reaction of yeast cystathionine beta-synthase is rate-limited by the conversion of aminoacrylate to cystathionine. Biochemistry. 2001;40:10873–10880. doi: 10.1021/bi011087j. [DOI] [PubMed] [Google Scholar]
- 55.Taoka S, Banerjee R. Stopped-flow kinetic analysis of the reaction catalyzed by the full length yeast cystathionine beta synthase. J Biol Chem. 2002;277:22421–22425. doi: 10.1074/jbc.M202513200. [DOI] [PubMed] [Google Scholar]
- 56.Aitken SM, Kirsch JF. Role of active-site residues Thr81, Ser82, Thr85, Gln157, and Tyr158 in yeast cystathionine beta-synthase catalysis and reaction specificity. Biochemistry. 2004;43:1963–1971. doi: 10.1021/bi035496m. [DOI] [PubMed] [Google Scholar]
- 57.Quazi F, Aitken SM. Characterization of the S289A,D mutants of yeast cystathionine beta-synthase. Biochim Biophys Acta. 2009;1794:892–897. doi: 10.1016/j.bbapap.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 58.Bach RD, Canepa C, Glukhovstev MN. Influence of Electrostatic Effects on Activation Barriers in Enzymatic Reactions:Pyridoxal 5′-Phosphate-Dependent Decarboxylation of α-Amino Acids. J Am Chem Soc. 1999;121:6542–6555. [Google Scholar]
- 59.Toney MD. Computational studies on nonenzymatic and enzymatic pyridoxal phosphate catalyzed decarboxylations of 2-aminoisobutyrate. Biochemistry. 2001;40:1378–1384. doi: 10.1021/bi0012383. [DOI] [PubMed] [Google Scholar]
- 60.Singh S, Madzelan P, Banerjee R. Properties of an unusual heme cofactor in PLP-dependent cystathionine beta-synthase. Nat Prod Rep. 2007;24:631–639. doi: 10.1039/b604182p. [DOI] [PubMed] [Google Scholar]
- 61.Kabil Ö, Taoka S, LoBrutto R, Shoemaker R, Banerjee R. The pyridoxal phosphate binding sites are similar in human heme-dependent and yeast heme-independent cystathionine beta synthases. Evidence from 31P NMR and pulsed EPR spectroscopy that the heme and the PLP cofactors are not proximal in the human enzyme. J Biol Chem. 2001;276:19350–19355. doi: 10.1074/jbc.M100029200. [DOI] [PubMed] [Google Scholar]
- 62.Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci. 1997;22:12–13. doi: 10.1016/s0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- 63.Sun Q, Collins R, Huang S, Holmberg-Schiavone L, Anand GS, Tan CH, van-den-Berg S, Deng LW, Moore PK, Karlberg T, Sivaraman J. Structural basis for the inhibition mechanism of human cystathionine gamma-lyase, an enzyme responsible for the production of H(2)S. J Biol Chem. 2009;284:3076–3085. doi: 10.1074/jbc.M805459200. [DOI] [PubMed] [Google Scholar]
- 64.Messerschmidt A, Worbs M, Steegborn C, Wahl MC, Huber R, Laber B, Clausen T. Determinants of enzymatic specificity in the Cys-Met-metabolism PLP-dependent enzymes family: crystal structure of cystathionine gamma-lyase from yeast and intrafamiliar structure comparison. Biol Chem. 2003;384:373–386. doi: 10.1515/BC.2003.043. [DOI] [PubMed] [Google Scholar]
- 65.Schneider G, Kack H, Lindqvist Y. The manifold of vitamin B6 dependent enzymes. Structure. 2000;8:R1–6. doi: 10.1016/s0969-2126(00)00085-x. [DOI] [PubMed] [Google Scholar]
- 66.Huang S, Chua JH, Yew WS, Sivaraman J, Moore PK, Tan CH, Deng LW. Site-directed mutagenesis on human cystathionine-gamma-lyase reveals insights into the modulation of H2S production. J Mol Biol. 2010;396:708–718. doi: 10.1016/j.jmb.2009.11.058. [DOI] [PubMed] [Google Scholar]
- 67.Kraus JP, Hasek J, Kozich V, Collard R, Venezia S, Janosikova B, Wang J, Stabler SP, Allen RH, Jakobs C, Finn CT, Chien YH, Hwu WL, Hegele RA, Mudd SH. Cystathionine gamma-lyase: Clinical, metabolic, genetic, and structural studies. Mol Genet Metab. 2009;97:250–259. doi: 10.1016/j.ymgme.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nagahara N, Katayama A. Post-translational regulation of mercaptopyruvate sulfurtransferase via a low redox potential cysteine-sulfenate in the maintenance of redox homeostasis. J Biol Chem. 2005;280:34569–34576. doi: 10.1074/jbc.M505643200. [DOI] [PubMed] [Google Scholar]
- 69.Nagahara N, Yoshii T, Abe Y, Matsumura T. Thioredoxin-dependent enzymatic activation of mercaptopyruvate sulfurtransferase. An intersubunit disulfide bond serves as a redox switch for activation. J Biol Chem. 2007;282:1561–1569. doi: 10.1074/jbc.M605931200. [DOI] [PubMed] [Google Scholar]
- 70.Jhee KH, McPhie P, Miles EW. Yeast cystathionine beta-synthase is a pyridoxal phosphate enzyme but, unlike the human enzyme, Is not a heme protein. J Biol Chem. 2000;275:11541–11544. doi: 10.1074/jbc.c000056200. [DOI] [PubMed] [Google Scholar]
- 71.Nozaki T, Shigeta Y, Saito-Nakano Y, Imada M, Kruger WD. Characterization of transsulfuration and cysteine biosynthetic pathways in the protozoan hemoflagellate, Trypanosoma cruzi. Isolation and molecular characterization of cystathionine beta-synthase and serine acetyltransferase from Trypanosoma. J Biol Chem. 2001;276:6516–6523. doi: 10.1074/jbc.M009774200. [DOI] [PubMed] [Google Scholar]
- 72.Singh S, Madzelan P, Stasser J, Weeks CL, Becker D, Spiro TG, Penner-Hahn J, Banerjee R. Modulation of the heme electronic structure and cystathionine beta-synthase activity by second coordination sphere ligands: The role of heme ligand switching in redox regulation. J Inorg Biochem. 2009;103:689–697. doi: 10.1016/j.jinorgbio.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carballal S, Madzelan P, Zinola CF, Grana M, Radi R, Banerjee R, Alvarez B. Dioxygen reactivity and heme redox potential of truncated human cystathionine beta-synthase. Biochemistry. 2008;47:3194–3201. doi: 10.1021/bi700912k. [DOI] [PubMed] [Google Scholar]
- 74.Taoka S, Green EL, Loehr TM, Banerjee R. Mercuric Chloride-Induced Spin or Ligation State Changes in Ferric or Ferrous Human Cystathionine b-synthase Inhibit Enzyme Activity. J Inorg Bioc. 2001;87:253–259. doi: 10.1016/s0162-0134(01)00336-1. [DOI] [PubMed] [Google Scholar]
- 75.Taoka S, Banerjee R. Activity of human cystathionine beta synthase is regulated by CO and NO: Possible role for the hemeprotein in CO sensing. J Inorg Bioc. 2001;87:245–251. doi: 10.1016/s0162-0134(01)00335-x. [DOI] [PubMed] [Google Scholar]
- 76.Taoka S, West M, Banerjee R. Characterization of the heme and pyridoxal phosphate cofactors of human cystathionine beta synthase reveals nonequivalent active sites. Biochemistry. 1999;38:2738–2744. doi: 10.1021/bi9826052. [DOI] [PubMed] [Google Scholar]
- 77.Vadon-Le Goff S, Delaforge M, Boucher JL, Janosik M, Kraus JP, Mansuy D. Coordination chemistry of the heme in cystathionine beta-synthase: formation of iron(II)-isonitrile complexes. Biochem Biophys Res Commun. 2001;283:487–492. doi: 10.1006/bbrc.2001.4807. [DOI] [PubMed] [Google Scholar]
- 78.Weeks CL, Singh S, Madzelan P, Banerjee R, Spiro TG. Heme regulation of human cystathionine beta-synthase activity: insights from fluorescence and Raman spectroscopy. J Am Chem Soc. 2009;131:12809–12816. doi: 10.1021/ja904468w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Taoka S, Ohja S, Shan X, Kruger WD, Banerjee R. Evidence for heme-mediated redox regulation of human cystathionine beta-synthase activity. J Biol Chem. 1998;273:25179–25184. doi: 10.1074/jbc.273.39.25179. [DOI] [PubMed] [Google Scholar]
- 80.Cherney MM, Pazicni S, Frank N, Marvin KA, Kraus JP, Burstyn JN. Ferrous human cystathionine beta-synthase loses activity during enzyme assay due to a ligand switch process. Biochemistry. 2007;46:13199–13210. doi: 10.1021/bi701159y. [DOI] [PubMed] [Google Scholar]
- 81.Pazicni S, Cherney MM, Lukat-Rodgers GS, Oliveriusova J, Rodgers KR, Kraus JP, Burstyn JN. The heme of cystathionine beta-synthase likely undergoes a thermally induced redox-mediated ligand switch. Biochemistry. 2005;44:16785–16795. doi: 10.1021/bi051305z. [DOI] [PubMed] [Google Scholar]
- 82.Pazicni S, Lukat-Rodgers GS, Oliveriusova J, Rees KA, Parks RB, Clark RW, Rodgers KR, Kraus JP, Burstyn JN. The redox behavior of the heme in cystathionine beta-synthase is sensitive to pH. Biochemistry. 2004;43:14684–14695. doi: 10.1021/bi0488496. [DOI] [PubMed] [Google Scholar]
- 83.Meier M, Oliveriusova J, Kraus JP, Burkhard P. Structural insights into mutations of cystathionine beta-synthase. Biochim Biophys Acta. 2003;1647:206–213. doi: 10.1016/s1570-9639(03)00048-7. [DOI] [PubMed] [Google Scholar]
- 84.Mosharov E, Cranford MR, Banerjee R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry. 2000;39:13005–13011. doi: 10.1021/bi001088w. [DOI] [PubMed] [Google Scholar]
- 85.Vitvitsky V, Thomas M, Ghorpade A, Gendelman HE, Banerjee R. A functional transsulfuration pathway in the brain links to glutathione homeostasis. J Biol Chem. 2006;281:35785–35793. doi: 10.1074/jbc.M602799200. [DOI] [PubMed] [Google Scholar]
- 86.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Finkelstein JD, Kyle WE, Martin JL, Pick AM. Activation of cystathionine synthase by adenosylmethionine and adenosylethionine. Biochem Biophys Res Commun. 1975;66:81–87. doi: 10.1016/s0006-291x(75)80297-x. [DOI] [PubMed] [Google Scholar]
- 88.Prudova A, Bauman Z, Braun A, Vitvitsky V, Lu SC, Banerjee R. S-adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity. Proc Natl Acad Sci U S A. 2006;103:6489–6494. doi: 10.1073/pnas.0509531103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Evande R, Boers GHJ, Blom HJ, Banerjee R. Alleviation of Intrasteric Inhibition by the Pathogenic Activation Domain Mutation, D444N, in Human Cystathionine beta-synthase. Biochemistry. 2002;41:11832–11837. doi: 10.1021/bi026248d. [DOI] [PubMed] [Google Scholar]
- 90.Shan X, Kruger WD. Correction of disease causing CBS mutations in yeast. Nature Genetics. 1998;19:91–93. doi: 10.1038/ng0598-91. [DOI] [PubMed] [Google Scholar]
- 91.Kery V, Poneleit L, Kraus JP. Trypsin cleavage of human cystathionine beta-synthase into an evolutionarily conserved active core: structural and functional consequences. Arch Biochem Biophys. 1998;355:222–232. doi: 10.1006/abbi.1998.0723. [DOI] [PubMed] [Google Scholar]
- 92.Janosik M, Kery V, Gaustadnes M, Maclean KN, Kraus JP. Regulation of human cystathionine beta-synthase by S-adenosyl-L- methionine: evidence for two catalytically active conformations involving an autoinhibitory domain in the C-terminal region. Biochemistry. 2001;40:10625–10633. doi: 10.1021/bi010711p. [DOI] [PubMed] [Google Scholar]
- 93.Zou CG, Banerjee R. Tumor necrosis factor-a-induced targeted proteolysis of cystathionine beta-synthase modulates redox homeostasis. J Biol Chem. 2003;278:16802–16808. doi: 10.1074/jbc.M212376200. [DOI] [PubMed] [Google Scholar]
- 94.Zou CG, Banerjee R. Tumor necrosis factor-alpha-induced targeted proteolysis of cystathionine beta-synthase modulates redox homeostasis. J Biol Chem. 2003;278:16802–16808. doi: 10.1074/jbc.M212376200. [DOI] [PubMed] [Google Scholar]
- 95.Prudova A, Albin M, Bauman Z, Lin A, Vitvitsky V, Banerjee R. Testosterone regulation of homocysteine metabolism modulates redox status in human prostate cancer cells. Antioxid Redox Signal. 2007;9:1875–1881. doi: 10.1089/ars.2007.1712. [DOI] [PubMed] [Google Scholar]
- 96.Kabil O, Zhou Y, Banerjee R. Human cystathionine beta-synthase is a target for sumoylation. Biochemistry. 2006;45:13528–13536. doi: 10.1021/bi0615644. [DOI] [PubMed] [Google Scholar]
- 97.Agarwal N, Banerjee R. Human Polycomb 2 Protein Is a SUMO E3 Ligase and Alleviates Substrate-Induced Inhibition of Cystathionine β-Synthase Sumoylation. PLoS ONE. 2008;3:e4032. doi: 10.1371/journal.pone.0004032. doi: 4010.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Markovic J, Borras C, Ortega A, Sastre J, Vina J, Pallardo FV. Glutathione is recruited into the nucleus in early phases of cell proliferation. J Biol Chem. 2007;282:20416–20424. doi: 10.1074/jbc.M609582200. [DOI] [PubMed] [Google Scholar]
- 99.Garg SK, Yan Z, Vitvitsky V, Banerjee R. Differential Dependence on Cysteine from Transsulfuration versus Transport during T cell Activation. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Martinov MV, Vitvitsky VM, Banerjee R, Ataullakhanov FI. The logic of the hepatic methionine metabolic cycle. Biochim Biophys Acta. 2009;1804:89–96. doi: 10.1016/j.bbapap.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ishii I, Akahoshi N, Yamada H, Nakano S, Izumi T, Suematsu M. Cystathionine gamma-Lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J Biol Chem. 2010;285:26358–26368. doi: 10.1074/jbc.M110.147439. [DOI] [PMC free article] [PubMed] [Google Scholar]