

Abstract
Phenethyl isothiocyanate (PEITC) is a highly promising cancer chemopreventive constituent of cruciferous vegetables (e.g., watercress) with in vivo efficacy in experimental rodent cancer models. Research thus far implicates apoptosis induction in cancer chemopreventive response to PEITC, but the mechanism of proapoptotic effect is not fully understood. The present study demonstrates that p53 upregulated modulator of apoptosis (PUMA)-independent apoptosis by PEITC is mediated by B-cell lymphoma 2 interacting mediator of cell death (Bim). Exposure of a cell line (BRI-JM04) derived from spontaneously developing mammary tumor of a MMTV-neu transgenic mouse to pharmacological concentrations of PEITC resulted in decreased cell viability coupled with apoptosis induction, characterized by release of histone-associated DNA fragments into the cytosol and cleavage of poly-(ADP-ribose)-polymerase and procaspase-3. The PEITC-induced apoptosis in BRI-JM04 cells was associated with up-regulation of Bak, PUMA, and Bim (long and short forms of Bim), increased S65 phosphorylation of BimEL (extra-long form), and down-regulation of Bcl-xL and Bcl-2. On the other hand, a non-tumorigenic human mammary epithelial cell line (MCF-10A) was significantly more resistant to PEITC-induced apoptosis compared with BRI-JM04 despite induction of Bax and PUMA due to concomitant overexpression of anti-apoptotic proteins, including Bcl-xL, Bcl-2, and Mcl-1. Wild-type HCT-116 cells and its isogenic PUMA knockout variant exhibited comparable sensitivity to PEITC-induced apoptosis. On the other hand, siRNA knockdown of Bim protein imparted partial but statistically significant protection against PEITC-induced apoptosis in BRI-JM04, MCF-7, and MDA-MB-231 cells. In conclusion, the present study provides novel insight into the mechanism of PEITC-induced apoptosis involving Bim.
Keywords: Phenethyl Isothiocyanate, Bim, Apoptosis, Chemoprevention
INTRODUCTION
Phenethyl isothiocyanate (PEITC), which occurs naturally as a thioglucoside conjugate (gluconasturtiin) in a variety of edible cruciferous vegetables including watercress, has emerged as a promising cancer chemopreventive agent [1, 2]. The gluconasturtiin is converted to PEITC upon processing (cutting or chewing) of cruciferous vegetables through catalytic mediation of myrosinases [1, 3]. Cancer chemopreventive potential of PEITC was initially recognized by demonstration of its ability to inhibit chemically-induced cancer in experimental rodents. For example, it was shown by Wattenberg [4] that PEITC administration 4 hour before carcinogen challenge inhibited 7,12-dimethylbenz[a]anthracene-induced mammary cancer development in Sprague-Dawley rats. Dietary administration of PEITC also inhibited forestomach and pulmonary adenomas in female ICR/Ha mice [4]. Administration of 3 μmol PEITC/g in the diet in rats before and during treatment with the tobacco-derived carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone resulted in significant inhibition of lung tumorigenesis compared with rats on control diet [5]. A subsequent structure-activity study involving alkyl and arylalkyl isothiocyanates concluded that both lipophilicity and lower reactivity of the isothiocyanates were important determinants of their cancer preventive efficacy against cancer in A/J mice induced by the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone [6]. Stoner et al [7] showed that N-nitrosobenzylmethylamine-induced esophageal carcinogenesis in rats was inhibited significantly by PEITC. More recent studies, including those from our laboratory have revealed that PEITC can inhibit spontaneous cancer development in transgenic mouse models. For example, ApcMin/+ mice fed a diet containing 0.05% of PEITC for 3 weeks developed significantly less (~32% decrease) and smaller polyps in comparison to mice fed control AIN-76A diet [8]. Administration of 0.05% PEITC in the diet significantly decreased incidence of prostate tumor formation in Transgenic Adenocarcinoma of Mouse Prostate transgenic mice, which was associated with decreased proliferation and increased apoptotic index [9]. We have also shown recently that dietary feeding of PEITC (3 μmol PEITC/g diet) significantly decreased incidence as well as burden (affected area) of poorly differentiated cancer in the dorsolateral prostate of Transgenic Adenocarcinoma of Mouse Prostate transgenic mice in association with induction of autophagy, overexpression of E-cadherin, and suppression of plasma clusterin level [10].
Apoptosis induction is one of the mechanisms implicated in anticancer response to PEITC both in cultured cancer cells and in vivo rodent models of cancer [8, 9, 11–15]. Proapoptotic potential of PEITC was initially recognized in the late nineties [11, 12]. While Chen et al [11] documented involvement of c-Jun N-terminal kinase (JNK) in PEITC-induced apoptosis, a critical role for p53 in apoptotic cell death was shown by Dong and colleagues [12]. Elucidation of the mechanism by which PEITC causes apoptosis has been the subject of intense research over the past decade [16–18]. A mechanistic model emerging from these studies involves generation of reactive oxygen species due to inhibition of complex III of the mitochondrial respiratory chain leading to activation of Bax and mitochondria-mediated apoptosis [16–18, 19, 20]. Despite these advances, however, the mechanism of PEITC-induced apoptosis downstream of ROS generation and upstream of caspase activation is not fully understood.
Mitochondria-mediated apoptosis upstream of caspase activation is regulated by Bcl-2 family proteins, which serve to either inhibit (e.g., Bcl-2, Bcl-xL, and Mcl-1) or facilitate (e.g., Bak, Bax, and Bim) apoptosis [21–25]. Previous studies from our laboratory have shown that SV40 immortalized mouse embryonic fibroblasts derived from Bax and Bak knockout mice are significantly more resistant to PEITC-induced apoptosis compared with those derived from the wild-type mice [26]. The PEITC was shown to trigger apoptosis in Jurkat cells made resistant by overexpression of Bcl-2 [27]. The present study extends these findings and investigates the role of B-cell lymphoma 2 interacting mediator of cell death (Bim) and p53 upregulated modulator of apoptosis (PUMA) in regulation of PEITC-mediated apoptotic cell death. This was a worthy research objective considering both these molecules have emerged as critical regulators of apoptosis by different stimuli [23–25].
MATERIALS AND METHODS
Cell Culture and Reagents
The BRI-JM04 cells, derived from spontaneously developing mammary tumor of a MMTV-neu mouse were generously provided by Dr. Anne Lenferink (Biotechnology Research Institute, Montreal, Canada) and cultured in Dulbecco’s modified essential medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and penicillin-streptomycin antibiotic mixture. Spontaneously immortalized non-tumorigenic human mammary epithelial cell line MCF-10A was obtained from American Type Culture Collection (Manassas, VA) and maintained in serum-free Mammary Epithelial Growth Medium (Clonetics, San Diego, CA) supplemented with 100 ng/mL cholera toxin (Calbiochem, La Jolla, CA). The MDA-MB-231 and MCF-7 human breast cancer cell lines were purchased from the American Type Culture Collection. Monolayer cultures of MDA-MB-231 cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum and penicillin-streptomycin antibiotic mixture. Monolayer cultures of MCF-7 cells were maintained in minimum essential medium supplemented with non-essential amino acids, 1 mM sodium pyruvate, 10 μg/mL insulin, 10% fetal bovine serum, and penicillin-streptomycin antibiotic mixture. The HCT-116 human colon cancer cell line and its isogenic PUMA-knockout variant were generously provided by Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD) and cultured in McCoy’s 5A modified medium supplemented with 10% fetal bovine serum and penicillin-streptomycin antibiotic mixture. Each cell line was cultured at 37°C in an atmosphere of 95% air and 5% CO2. Cell culture media, antibiotic mixture, and fetal bovine serum were purchased from Invitrogen-Life Technologies (Carlsbad, CA). The PEITC (purity ≥ 98%) was purchased from LKT laboratories (St. Paul, MN). An antibody specific for detection of full and cleaved poly-(ADP-ribose)-polymerase (PARP) as well as anti-PUMA, anti-Bak, anti-Bax, and anti-Bcl-xL antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); an antibody specific for detection of S65 phosphorylated BimEL was purchased from Millipore (Billerica, MA); anti-total Bim and anti-caspase-3 antibodies were purchased from Cell Signaling (Danvers, MA). Bim-targeted siRNA was purchased from Santa Cruz Biotechnology, whereas a nonspecific control siRNA was from Qiagen (Valencia, CA). The 4′,6-diamidino-2-phenylindole (DAPI) and anti-actin antibody were purchased from Sigma-Aldrich (St. Louis, MO).
Trypan Blue Dye Exclusion Assay
Cell viability was determined by trypan blue dye exclusion assay. Briefly, the cells were seeded at a density of 1×105 cells per well in 12-well plates, allowed to attach by overnight incubation, and then treated with 0.008% dimethyl sulfoxide (DMSO; vehicle for PEITC) or the indicated concentrations of PEITC for 24 or 36 hours. After treatment, the cells were trypsinized and stained with trypan blue solution, and viable cells were counted under an inverted microscope using a hemocytometer.
Determination of Apoptosis
Apoptosis induction was assessed by DAPI assay or determination of histone-associated DNA fragment release into the cytosol. Release of histone-associated DNA fragments into the cytosol in control (DMSO-treated) and PEITC-treated cells was determined using a kit from Roche Applied Sciences (Indianapolis, IN) and by following the supplier’s instructions. For DAPI assay, the cells were plated in triplicate onto coverslips in 12-well plates. After treatment with DMSO or PEITC, cells were fixed with 2% paraformaldehyde (Sigma-Aldrich) for 1 hour at room temperature and permeabilized using 0.5% Triton X-100 (Sigma-Aldrich) for 15 minutes at room temperature. After washing with phosphate-buffered saline (PBS), the cells were incubated with 50 ng/mL DAPI in PBS for 10 minutes at room temperature. Subsequently, the cells were mounted and air-dried. Apoptotic cells with condensed and fragmented DNA were scored under a Leica fluorescence microscope (100× objective magnification). At least 100 cells were scored from each replicate of control and PEITC-treated samples.
Immunoblotting
Briefly, cells were treated with DMSO (control) or PEITC for specified time and lysed on ice with a solution consisting of 50 mM Tris, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 150 mM NaCl, protease inhibitors, and phosphatase inhibitors. The cell lysate was cleared by centrifugation at 14,000 rpm for 30 minutes. Protein concentration was measured using Bradford reagent (Bio-Rad, Hercules, CA). Lysate proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then wet-transferred onto polyvinylidene fluoride membrane (Perkin Elmer, Boston, MA). After blocking with 5% nonfat dry milk in Tris-buffered saline containing 0.05% Tween-20 (Bio-Rad, Hercules, CA), the membrane was incubated with primary antibody overnight at 4°C or 2 hours at room temperature. Subsequently, the membrane was incubated with an appropriate secondary antibody, and the immunoreactive protein bands were visualized by the use of the Enhanced Chemiluminescence reagent (NEN Life Science Products, Boston, MA). Each membrane was stripped and re-probed with anti-actin antibody to correct for differences in protein loading. The western blotting experiments were repeated at least twice using independently prepared cell lysates.
RNA Interference of Bim
Desired cells (50% confluency) were transiently transfected with a control (non-specific) small interfering RNA (siRNA) or a Bim-targeted siRNA for 24 hours with the use of OligoFECTAMINE. The cells were then treated with DMSO (control) or the indicated concentrations of PEITC for 48 hours and used for determination of apoptosis by DAPI staining. Knockdown of Bim protein in transfected cells was confirmed by western blotting.
Statistical Analysis
Each experiment was repeated at least twice to ensure reproducibility of the results. One-way ANOVA followed by Bonferroni’s multiple comparison test or Dunnett’s adjustment was used to determine statistical significance of difference in measured variables between groups. Difference was considered significant at P<0.05.
RESULTS
PEITC Treatment Caused Apoptosis in BRI-JM04 Cells
We used BRI-JM04 cell line derived from mammary tumor of a MMTV-neu transgenic mouse to determine proapoptotic response to PEITC. This cell line was selected for two reasons: (a) we reasoned that effectiveness of PEITC against BRI-JM04 cells would provide impetus to test its efficacy for prevention of mammary cancer in MMTV-neu mice, and (b) the BRI-JM04 cells and MMTV-neu mice collectively provide a model to directly test in vivo relevance of the cellular mechanistic findings. Viability of BRI-JM04 cells was reduced significantly and in a dose-dependent manner upon a 36-hour exposure to PEITC (Figure 1A). The PEITC-mediated inhibition of BRI-JM04 cell viability was accompanied by apoptosis induction as judged by a significant increase in release of histone-associated DNA fragments into the cytosol, which is a well-accepted measure of apoptotic cell death (Figure 1B). Consistent with these results, the PEITC treatment (36-hours) caused cleavage of PARP in BRI-JM04 cells, which was negligible in DMSO-treated control cells (Figure 1C).
Figure 1.
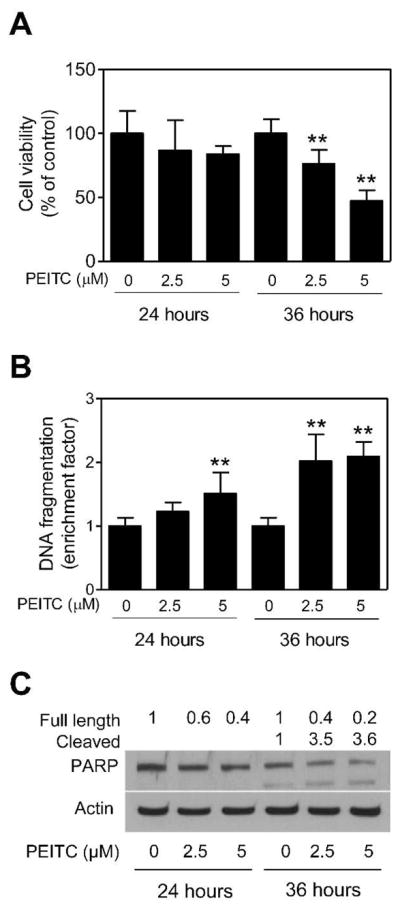
The PEITC treatment decreased viability of BRI-JM04 cells. (A) Viability of BRI-JM04 cells after 24- or 36-hour treatment with DMSO (control) or the indicated concentrations of PEITC as determined by trypan blue dye exclusion assay. Percent cell viability relative to DMSO-treated control is shown for each treatment. (B) Cytoplasmic histone-associated DNA fragment release into the cytosol after 24- or 36-hour treatment of BRI-JM04 cells with DMSO (control) or the indicated concentrations of PEITC. Results are expressed as enrichment factor relative to DMSO-treated control. (C) Western blotting for PARP cleavage using lysates from BRI-JM04 cells after 24- or 36-hour treatment with DMSO (control) or the indicated concentrations of PEITC. Numbers above bands represent changes in levels of full length and cleaved PARP relative to DMSO-treated control at each time point. Quantitative experiments (panels A and B) were done twice in triplicate and combined data from both experiments are presented as mean ± SD (n = 6). **Significantly different (P<0.01) compared with corresponding control by one-way ANOVA with Dunnett’s adjustment.
Proapoptotic response to PEITC was confirmed by DAPI assay and determination of procaspase-3 cleavage. Figure 2A depicts microscopic images of control (DMSO-treated) and PEITC-treated BRI-JM04 cells after staining with DAPI. Cells with DNA condensation and fragmentation were clearly visible in the PEITC-treated BRI-JM04 cultures, but rarely seen in DMSO-treated controls. Quantitatively, fraction of apoptotic cells was increased significantly over DMSO-treated control in BRI-JM04 cells after treatment with 2.5 and 5 μM PEITC for 24- and 36-hours (Figure 2B). Moreover, PEITC treatment resulted in a marked increase in levels of cleaved caspase-3 in comparison with DMSO-treated control BRI-JM04 cells (Figure 2C). Collectively, these results indicated that the PEITC-treated BRI-JM04 cells were driven to apoptotic cell death.
Figure 2.

The PEITC treatment caused apoptosis in BRI-JM04 cells. (A) Representative microscopic images depicting apoptotic cells with condensed and fragmented DNA (DAPI assay) in BRI-JM04 cultures after 48-hour treatment with DMSO (control) or the indicated concentrations of PEITC. (B) Quantitation of apoptotic cells (DAPI assay) in BRI-JM04 cultures after 24- or 36-hour treatment with DMSO (control) or the indicated concentrations of PEITC. Results are expressed as enrichment factor relative to DMSO-treated control. Experiment was repeated twice in triplicate and combined data from both experiments are shown as mean ± SD (n = 6). **Significantly different (P<0.01) compared with corresponding control by one-way ANOVA with Dunnett’s adjustment. (C) Western blotting for cleaved caspase-3 using lysates from BRI-JM04 cells after 24- or 36-hour treatment with DMSO (control) or the indicated concentrations of PEITC. Numbers above bands represent changes in levels of cleaved caspase-3 relative to DMSO-treated control at each time point.
Effect of PEITC Treatment on Levels of Bcl-2 Family Proteins in BRI-JM04 Cells
To gain insights into the mechanism of PEITC-induced apoptosis in BRI-JM04 cells, we determined its effects on a panel of anti-apoptotic and pro-apoptotic members of Bcl-2 family proteins. As can be seen in Figure 3, the PEITC-treated BRI-JM04 cells exhibited a concentration-dependent and marked decrease in proteins levels of Bcl-2 and Bcl-xL. While protein level of Bax was unaffected, dose-dependent increase in expression of several proapoptotic Bcl-2 family member proteins was clearly discernible in PEITC-treated BRI-JM04 cells, including Bak, PUMA, and Bim (especially the long and short forms) (Figure 3). Moreover, S65 phosphorylation of BimEL, which is known to regulate its apoptotic activity [23, 24], was increased by PEITC treatment in BRI-JM04 cells. These results suggested that the proapoptotic response to PEITC was likely mediated by alterations in protein levels of Bcl-2 family proteins.
Figure 3.
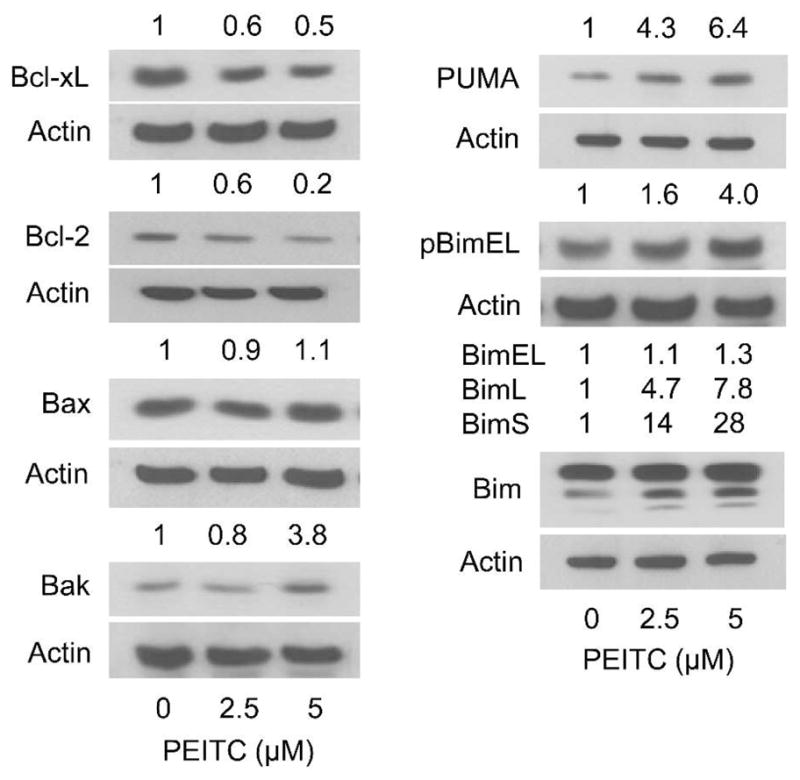
Effect of PEITC treatment on levels of Bcl-2 family proteins in BRI-JM04 cells. Western blotting for Bcl-xL, Bcl-2, Bax, Bak, PUMA, S65 phosphorylated BimEL, and total Bim using lysates from BRI-JM04 cells treated for 24 hours with DMSO (control) or the indicated concentrations of PEITC. Numbers above bands represent changes in levels of proteins relative to DMSO-treated control. Western blotting for each protein was repeated twice with independently prepared lysates and the results were comparable.
MCF-10A Cells were Resistant to PEITC-induced Apoptosis
Next, we tested whether proapoptotic response to PEITC was selective towards cancer cells using a non-tumorigenic normal human mammary epithelial cell line (MCF-10A). The MCF-10A cell line, which has intact proliferation controls [28], was isolated from fibrocystic breast disease and spontaneously immortalized. As can be seen in Figure 4A, the PEITC-mediated enrichment of apoptotic cells (DAPI assay) was much less pronounced in the MCF-10A cell line compared with BRI-JM04 cells (Figure 2B). For example, the MCF-10A cultures treated for 24 hours with 5 μM PEITC revealed about 2-fold enrichment of apoptotic cells over DMSO-treated control (Figure 4A), whereas an enrichment of >10-fold was observed in similarly treated BRI-JM04 cells (Figure 2B).
Figure 4.

A non-tumorigenic human mammary epithelial cell line (MCF-10A) was relatively more resistant to PEITC-induced apoptosis compared with BRI-JM04 cells. (A) Quantitation of apoptotic cells (DAPI assay) in MCF-10A cultures after 24-hour treatment with DMSO (control) or the indicated concentrations of PEITC. Results are expressed as enrichment factor relative to DMSO-treated control. Experiment was repeated twice in triplicate and representative data from one experiment are shown as mean ± SD (n = 3). (B) Western blotting for Bcl-xL, Bcl-2, Mcl-1, Bax, Bak, PUMA, S65 phosphorylated BimEL, and total Bim using lysates from MCF-10A cells treated for 24 hours with DMSO (control) or the indicated concentrations of PEITC. Numbers above bands represent changes in levels of proteins relative to DMSO-treated control. Western blotting for each protein was repeated twice with independently prepared lysates and the results were comparable.
The effect of PEITC treatment on levels of Bcl-2 family proteins was determined using MCF-10A cells. Unlike BRI-JM04 cells, the PEITC-treated MCF-10A cells (24-hour treatment) exhibited induction of several anti-apoptotic Bcl-2 family members including Bcl-2, Bcl-xL, and Mcl-1 (Figure 4B). Moreover, an increase in short form of Mcl-1 (32 kDa) upon treatment of MCF-10A cells with PEITC was also discernible. In addition, treatment of MCF-10A cells with PEITC resulted in induction of Bax, but not Bak, and PUMA. A modest increase in S65 phosphorylation of BimEL, but not total Bim, was evident in PEITC-treated MCF-10A cells (Figure 4B). Collectively, these results indicated that the effect of PEITC treatment on expression of Bcl-2 family proteins was markedly different in BRI-JM04 versus MCF-10A cells, respectively characterized by suppression of anti-apoptotic proteins (Bcl-2 and Bcl-xL) but induction of proapoptotic proteins (Bak, PUMA, and Bim) versus induction of both anti-apoptotic (Bcl-2, Bcl-xL, and Mcl-1) and proapoptotic proteins (Bax and PUMA).
PUMA was Dispensable for PEITC-induced Apoptosis in HCT-116 Cells
Because the effect of PEITC treatment was extremely robust on PUMA expression in BRI-JM04 cells, we designed experiments to test contribution of this protein in proapoptotic response to PEITC. An isogenic HCT-116 variant lacking PUMA expression (Figure 5A) and its wild-type counterpart were used to address this question. The HCT-116 cell-pair was a better experimental model over RNA interference because siRNA transfection often fails to achieve complete knockdown. To our surprise, PUMA knockout cells were as sensitive to PEITC-induced apoptosis as the wild-type HCT-116 cells. These results indicated that PUMA protein was dispensable for PEITC-induced apoptosis.
Figure 5.

The PEITC-induced apoptosis in BRI-JM04 cells was regulated by Bim. (A) Western blotting for PUMA using lysates from wild-type HCT-116 cells (HCT-116-PUMA-WT; lane 1) or its isogenic variant lacking PUMA (HCT-116-PUMA-KO; lane 2). Number above band represents PUMA protein level relative to HCT-116-PUMA WT cells. (B) Quantitation of apoptotic cells (DAPI assay) in HCT-116-PUMA-WT and HCT-116-PUMA-KO cultures after 24-hour treatment with DMSO (control) or the indicated concentrations of PEITC. Results are expressed as enrichment factor relative to DMSO-treated control HCT-116-PUMA-WT cells. Experiment was repeated twice in triplicate and representative data from one experiment are shown as mean ± SD (n = 3). aSignificantly different (P<0.05) compared with corresponding DMSO-treated control by one-way ANOVA followed by Bonferroni’s test. (C) Western blotting for BimEL using lysates from BRI-JM04 cells transiently transfected with a control siRNA (lane 1) or a Bim-targeted siRNA (lane 2). Number above band represents BimEL protein level relative to BRI-JM04 cells transfected with control siRNA. (D) Quantitation of apoptotic cells (DAPI assay) in BRI-JM04 cells transiently transfected with a control siRNA or a Bim-targeted siRNA and treated for 48 hours with DMSO or the indicated concentrations of PEITC. Results are expressed as enrichment factor relative to DMSO-treated control siRNA transfected BRI-JM04 cells. Experiment was repeated twice in triplicate and representative data from one experiment are shown as mean ± SD (n = 3). Significantly different (P<0.05) compared with acorresponding DMSO-treated control, and bbetween PEITC-treated groups by one-way ANOVA followed by Bonferroni’s test.
Effect of Bim Knockdown on PEITC-induced Apoptosis in Breast Cancer Cells
We have shown previously that Bax and Bak deficiency confers significant protection against PEITC-induced apoptosis (26). Likewise, the role of Bcl-2 in proapoptotic response to PEITC has been studied previously (27). In the present study, we extended these observations to determine the role of Bim in regulation of PEITC-induced apoptosis using siRNA technology. As shown in Figure 5C, the level of BimEL protein was reduced by about 80% after transfection of BRI-JM04 cells with a Bim-targeted siRNA. The PEITC-mediated enrichment of apoptotic cells (DAPI assay) over DMSO-treated control was significantly higher in BRI-JM04 cells transfected with a control siRNA in comparison with Bim knockdown cells (Figure 5D). Lack of full protection may be attributable to incomplete knockdown of the Bim protein (Figure 5D). Alternatively, the possibility that PEITC-induced apoptosis is only partially dependent on Bim can’t be excluded.
We used a pair of well-characterized human breast cancer cell lines (MCF-7 and MDA-MB-231 cells) to confirm contribution of Bim in PEITC-induced apoptosis. The level of BimEL protein was reduced by 80–90% in MCF-7 and MDA-MB-231 cells upon transfection with a Bim-targeted siRNA in comparison with corresponding control siRNA transfected cells (Figure 6A). Similar to BRI-JM04 cells, siRNA-mediated knockdown of Bim protein conferred partial but statistically significant protection against PEITC induced apoptosis in both MCF-7 and MDA-MB-231 cells (Figure 6B). Collectively, these results indicated that Bim protein contributed to PEITC-induced apoptosis, which was not a cell line-specific response.
Figure 6.

RNA interference of Bim partially protected against PEITC-induced apoptosis in human breast cancer cells. (A) Western blotting for BimEL using lysates from MCF-7 and MDA-MB-231 cells transiently transfected with a control siRNA (lane 1) or a Bim-targeted siRNA (lane 2). (B) Quantitation of apoptotic cells (DAPI assay) in MCF-7 and MDA-MB-231 cells transiently transfected with a control siRNA or a Bim-targeted siRNA and treated for 48 hours with DMSO or the indicated concentrations of PEITC. Results are expressed as enrichment factor relative to corresponding DMSO-treated control siRNA transfected cells (mean ± SD; n = 3). Significantly different (P<0.05) compared with acorresponding DMSO-treated control, and bbetween PEITC-treated groups by one-way ANOVA followed by Bonferroni’s test.
DISCUSSION
Major objective of the present study was two-fold: (a) to test whether PEITC treatment reduced viability of BRI-JM04 cells to provide justification for a future in vivo efficacy study using MMTV-neu transgenic mice, and (b) to identify mechanistic correlates that could serve as in vivo biomarkers of PEITC response in future animal and human studies. We found that PEITC treatment decreases viability of BRI-JM04 cells in association with apoptosis induction, characterized by DNA fragmentation and cleavage of PARP and procaspase-3. The proapoptotic effect of PEITC against BRI-JM04 cells is observed at pharmacologically relevant concentrations [29, 30]. For example, the Cmax of PEITC in rat plasma is 2.0 and 4.7 μM after oral treatment with 0.5 mg/kg and 5 mg/kg PEITC, respectively [30]. It is fascinating to note that pharmacokinetic behavior of PEITC is altered after repeated oral administrations with higher plasma Cmax concentrations and decreased plasma clearance leading to enhanced bioavailability [30].
The present study shows that a non-tumorigenic human mammary epithelial cell line MCF-10A) is significantly more resistant to PEITC-induced apoptosis compared with BRI-JM04 cells as judged by DAPI assay. Previous studies have shown that the MCF-10A cell line is also resistant to reduction in cell viability by PEITC or structurally-related isothiocyanates compared with breast cancer cells [31, 32]. However, the mechanism underlying selectivity of PEITC for apoptosis induction in cancer cells was not clear. The present study offers some explanations to fill this gap in our knowledge. The PEITC treatment decreases protein levels of anti-apoptotic Bcl-2 and Bcl-xL in BRI-JM04 cells (Figure 3). Suppression of Bcl-2 protein level after 24-hour treatment with 3, 10 and 30 μM PEITC has also been observed in the MCF-7 cells [32]. Interestingly, the levels of Bcl-2 and Bcl-xL are markedly increased in MCF-10A cells after 24-hour treatment with 2.5 and 5 μM PEITC (Figure 4B). Thus, the ratio of anti-apoptotic/proapoptotic Bcl-2 family protein levels by PEITC treatment is markedly different between BRI-JM04 versus MCF-10A cells. For example, the Bcl-xL/Bak ratio in BRI-JM04 cells treated with 2.5 and 5 μM PEITC is about 0.8 and 0.1, respectively. To the contrary, treatment of MCF-10A cells with 2.5 and 5 μM PEITC results in Bcl-xL/Bak ratio of about 1.3 and 1.9, respectively. Similarly, the Bcl-2/Bak ratio in BRI-JM04 cells treated with 2.5 and 5 μM PEITC is about 0.8 and 0.05, respectively. Treatment of MCF-10A cells with 2.5 and 5 μM PEITC results in Bcl-2/Bak ratio of about 1.5 and 1.4, respectively. Another noticeable difference between BRI-JM04 cells and MCF-10A cells is alteration of Bim expression. The PEITC treatment causes robust induction of Bim protein expression, especially the long and short forms of this protein, in BRI-JM04 cells, which is not observed in the MCF-10A cell line. However, further studies are needed to determine if difference in sensitivity of normal versus cancerous breast cells to PEITC-induced apoptosis is due to differential alterations in Bcl-2 family proteins.
The present study reveals PEITC-mediated induction of Bak, PUMA, and Bim in BRI-JM04 cells. Role of Bax, Bak, and Bcl-2 in regulation of PEITC-induced apoptosis has been studied previously in other cellular systems [26, 27], but no such information was available for PUMA or Bim. The PUMA is a Bcl-2 homology 3 (BH3)-only Bcl-2 family member and regarded as a critical mediator of both p53-dependent and p53-independent apoptosis by different stimuli including genotoxic insult, oncogenic alterations, growth factor and cytokine withdrawal, and infections [25]. Analogous to other BH3-only Bcl-2 family members, PUMA transduces death signals to the mitochondria by directly or indirectly activating multidomain proapoptotic proteins Bax and/or Bak [25]. The present study reveals that PUMA is dispensable for PEITC-induced apoptosis because (a) PUMA is induced more robustly in the MCF-10A cell line compared with BRI-JM04 cells, and (b) the wild-type HCT-116 cells and its PUMA knockout variant exhibit comparable sensitivity to PEITC-induced apoptosis.
Bim is another BH3-only member of the Bcl-2 family that is transcriptionally up-regulated in cells undergoing apoptosis [33–35]. Three alternative splice variants of Bim have been described [35]. The BimS (short form) induces apoptosis, but the apoptotic activity of the BimL (long form) and BimEL (extra long form) is suppressed by binding to the dynein motor complex [36]. The BimL and BimEL forms bind to dynein light chain through a short peptide motif (DKSTQTP) that is absent in BimS [36]. We found that PEITC treatment causes induction of all three forms of Bim in BRI-JM04 cells, whereas none of the Bim forms are induced in the MCF-10A cell line by a similar PEITC exposure. Furthermore, the PEITC-mediated apoptosis in BRI-JM04, MCF-7, and MDA-MB-231 cells is partially but statistically significantly attenuated by siRNA knockdown of Bim protein. Partial protection conferred against PEITC-induced apoptosis in each cell line is likely attributable to lack of complete knockdown. An alternate possibility relates to Bim-independent mechanisms in regulation of PEITC-induced apoptosis. Nonetheless, the present study suggests that Bim induction may be a useful biomarker of PEITC response in future in vivo studies. However, the mechanism underlying PEITC-mediated induction of Bim remains to be determined.
Proapoptotic activity of Bim is also regulated by its phosphorylation by extracellular signal-regulated kinase (ERK) and JNK [23, 24]. Both ERK and JNK have been implicated in regulation of PEITC-induced apoptosis in other cancer cell types [11, 37]. For example, we have shown previously that the PEITC-induced apoptosis in PC-3 human prostate cancer cell line is associated with rapid and sustained phosphorylation of ERK. Similar experimental evidence exists for JNK in regulation of PEITC-mediated apoptotic cell death [11]. The present study shows that PEITC treatment causes an increase in S65 phosphorylation of BimEL in the BRI-JM04 cell line, but this effect is much less pronounced in MCF-10A cells. It would be interesting to determine if S65 phosphorylation of BimEL in our model is mediated by ERK. Further work is also needed to determine if PEITC-mediated S65 phosphorylation of BimEL is unique to the BRI-JM04 cells.
The main conclusions of the present study include: (a) the PEITC treatment causes Bim-mediated apoptosis in cultured breast cancer cells at pharmacological concentrations, which provides a valid rationale to determine efficacy of PEITC for prevention of mammary cancer in MMTV-neu mice in vivo; (b) PUMA is dispensable for PEITC-induced apoptosis, at least in HCT-116 cells; (c) BRI-JM04 and MCF-10A cells respond differently to PEITC-mediated alterations in expression of Bcl-2 family proteins, which partly explains relative resistance of MCF-10A cells to PEITC-induced apoptosis compared with cancerous breast cells.
Acknowledgments
This study was supported by the United States Public Health Service grants RO1 CA101753-07 and RO1 CA129347-05, awarded by the National Cancer Institute.
Abbreviations
- PEITC
phenethyl isothiocyanate
- JNK
c-Jun N-terminal kinase
- Bim
B-cell lymphoma 2 interacting mediator of cell death
- BimEL
extra-long form of Bim
- BimL
long form of Bim
- BimS
short form of Bim
- PUMA
p53 upregulated modulator of apoptosis
- PARP
poly-(ADP-ribose)-polymerase
- DAPI
4′,6-diamidino-2-phenylindole
- DMSO
dimethyl sulfoxide
- PBS
phosphate-buffered saline
- siRNA
small interfering RNA
- BH3
Bcl-2 homology 3
- ERK
extracellular signal-regulated kinase
References
- 1.Hecht SS. Chemoprevention by isothiocyanates. J Cell Biochem. 1995;22(suppl):195–209. doi: 10.1002/jcb.240590825. [DOI] [PubMed] [Google Scholar]
- 2.Hecht SS. Inhibition of carcinogenesis by isothiocyanates. Drug Metab Rev. 2000;32:395–411. doi: 10.1081/dmr-100102342. [DOI] [PubMed] [Google Scholar]
- 3.Fahey JW, Zalcmann AT, Talalay P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry. 2001;56(1):5–51. doi: 10.1016/s0031-9422(00)00316-2. [DOI] [PubMed] [Google Scholar]
- 4.Wattenberg LW. Inhibition of carcinogenic effects of polycyclic hydrocarbons by benzyl isothiocyanate and related compounds. J Natl Cancer Inst. 1977;58:395–398. doi: 10.1093/jnci/58.2.395. [DOI] [PubMed] [Google Scholar]
- 5.Morse MA, Wang CX, Stoner GD, et al. Inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced DNA adduct formation and tumorigenicity in the lung of F344 rats by dietary phenethyl isothiocyanate. Cancer Res. 1989;49:549–553. [PubMed] [Google Scholar]
- 6.Jiao D, Eklind KI, Choi CI, Desai DH, Amin SG, Chung FL. Structure-activity relationships of isothiocyanates as mechanism-based inhibitors of 4- (methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis in A/J mice. Cancer Res. 1994;54:4327–4333. [PubMed] [Google Scholar]
- 7.Stoner GD, Morrissey DT, Heur YH, Daniel EM, Galati AJ, Wagner SA. Inhibitory effects of phenethyl isothiocyanate on N-nitrosobenzylmethylamine carcinogenesis in the rat esophagus. Cancer Res. 1991;51:2063–2068. [PubMed] [Google Scholar]
- 8.Khor TO, Cheung WK, Prawan A, Reddy BS, Kong AN. Chemoprevention of familial adenomatous polyposis in Apc (Min/+) mice by phenethyl isothiocyanate (PEITC) Mol Carcinog. 2008;47:321–325. doi: 10.1002/mc.20390. [DOI] [PubMed] [Google Scholar]
- 9.Barve A, Khor TO, Hao X, et al. Murine prostate cancer inhibition by dietary phytochemicals- curcumin and phenyethylisothiocyanate. Pharm Res. 2008;25:2181–2189. doi: 10.1007/s11095-008-9574-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Powolny AA, Bommareddy A, Hahm ER, et al. Chemopreventative potential of the cruciferous vegetable constituent phenethyl isothiocyanate in a mouse model of prostate cancer. J Natl Cancer Inst. 2011;103:571–584. doi: 10.1093/jnci/djr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen YR, Wang W, Kong AN, Tan TH. Molecular mechanisms of c-Jun N-terminal kinase-mediated apoptosis induced by anticarcinogenic isothiocyanates. J Biol Chem. 1998;273:1769–1775. doi: 10.1074/jbc.273.3.1769. [DOI] [PubMed] [Google Scholar]
- 12.Huang C, Ma WY, Li J, Hecht SS, Dong Z. Essential role of p53 in phenethyl isothiocyanate-induced apoptosis. Cancer Res. 1998;58:4102–4106. [PubMed] [Google Scholar]
- 13.Xiao D, Johnson CS, Trump DL, Singh SV. Proteasome-mediated degradation of cell division cycle 25C and cyclin-dependent kinase 1 in phenethyl isothiocyanate-induced G2/M phase cell cycle arrest in PC-3 human prostate cancer cells. Mol Cancer Ther. 2004;3:567–575. [PubMed] [Google Scholar]
- 14.Bommareddy A, Hahm ER, Xiao D, et al. Atg5 regulates phenethyl isothiocyanate- induced autophagic and apoptotic cell death in human prostate cancer cells. Cancer Res. 2009;69:3704–3712. doi: 10.1158/0008-5472.CAN-08-4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferguson LR, Philpott M. Cancer prevention by dietary bioactive components that target the immune response. Curr Cancer Drug Targets. 2007;7:459–464. doi: 10.2174/156800907781386605. [DOI] [PubMed] [Google Scholar]
- 16.Cheung KL, Kong AN. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. AAPS J. 2010;12:87–97. doi: 10.1208/s12248-009-9162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stan SD, Kar S, Stoner GD, Singh SV. Bioactive food components and cancer risk reduction. J Cell Biochem. 2008;104:339–356. doi: 10.1002/jcb.21623. [DOI] [PubMed] [Google Scholar]
- 18.Antosiewicz J, Ziolkowski W, Kar S, Powolny AA, Singh SV. Role of reactive oxygen intermediates in cellular responses to dietary cancer chemopreventive agents. Planta Med. 2008;74:1570–1579. doi: 10.1055/s-2008-1081307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trachootham D, Zhou Y, Zhang H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Xiao D, Powolny AA, Moura MB, et al. Phenethyl isothiocyanate inhibits oxidative phosphorylation to trigger reactive oxygen species-mediated death of human prostate cancer cells. J Biol Chem. 2010;285:26558–26569. doi: 10.1074/jbc.M109.063255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chao DT, Korsmeyer SJ. Bcl-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 22.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akiyama T, Dass CR, Choong PF. Bim-targeted cancer therapy: a link between drug action and underlying molecular changes. Mol Cancer Ther. 2009;8:3173–3180. doi: 10.1158/1535-7163.MCT-09-0685. [DOI] [PubMed] [Google Scholar]
- 24.Gillings AS, Balmanno K, Wiggins CM, Johnson M, Cook SJ. Apoptosis and autophagy: BIM as a mediator of tumour cell death in response to oncogene-targeted therapeutics. FEBS J. 2009;276:6050–6062. doi: 10.1111/j.1742-4658.2009.07329.x. [DOI] [PubMed] [Google Scholar]
- 25.Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene. 2008;27:S71–S83. doi: 10.1038/onc.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao D, Zeng Y, Choi S, Lew KL, Nelson JB, Singh SV. Caspase-dependent apoptosis induction by phenethyl isothiocyanate, a cruciferous-vegetable derived cancer chemopreventive agent, is mediated by Bak and Bax. Clin Cancer Res. 2005;11:2670–2679. doi: 10.1158/1078-0432.CCR-04-1545. [DOI] [PubMed] [Google Scholar]
- 27.Thomson SJ, Brown KK, Pullar JM, Hampton MB. Phenethyl isothiocyanate triggers apoptosis in Jurkat cells made resistant by the overexpression of Bc1–2. Cancer Res. 2006;66:6772–6777. doi: 10.1158/0008-5472.CAN-05-3809. [DOI] [PubMed] [Google Scholar]
- 28.Morse DL, Gray H, Payne CM, Gillies RJ. Docetaxel induces cell death through mitotic catastrophe in human breast cancer cells. Mol Cancer Ther. 2005;4:1495–1504. doi: 10.1158/1535-7163.MCT-05-0130. [DOI] [PubMed] [Google Scholar]
- 29.Ji Y, Kuo Y, Morris ME. Pharmacokinetics of dietary phenethyl isothiocyanate in rats. Pharm Res. 2005;22:1658–1666. doi: 10.1007/s11095-005-7097-z. [DOI] [PubMed] [Google Scholar]
- 30.Konsue N, Kirkpatrick J, Kuhnert N, King LJ, Ioannides C. Repeated oral administration modulates the pharmacokinetic behavior of the chemopreventive agent phenethyl isothiocyanate in rats. Mol Nutr Food Res. 2010;54:426–432. doi: 10.1002/mnfr.200900090. [DOI] [PubMed] [Google Scholar]
- 31.Xiao D, Vogel V, Singh SV. Benzyl isothiocyanate-induced apoptosis in human breast cancer cells is initiated by reactive oxygen species and regulated by Bax and Bak. Mol Cancer Ther. 2006;5:2931–2945. doi: 10.1158/1535-7163.MCT-06-0396. [DOI] [PubMed] [Google Scholar]
- 32.Lee JW, Cho MK. Phenethyl isothiocyanate induced apoptosis via down regulation of Bcl-2/XIAP and triggering of the mitochondrial pathway in MCF-7 cells. Arch Pharm Res. 2008;31:1604–1612. doi: 10.1007/s12272-001-2158-2. [DOI] [PubMed] [Google Scholar]
- 33.Putcha GV, Moulder KL, Golden JP, et al. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron. 2001;29:615–28. doi: 10.1016/s0896-6273(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 34.Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 2001;29:629–43. doi: 10.1016/s0896-6273(01)00239-2. [DOI] [PubMed] [Google Scholar]
- 35.O’Reilly LA, Cullen L, Visvader J, et al. The proapoptotic BH3-only protein bim is expressed in hematopoietic, epithelial, neuronal, and germ cells. Am J Pathol. 2000;157:449–61. doi: 10.1016/S0002-9440(10)64557-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puthalakath H, Huang DC, O’Reilly LA, King SM, Strasser A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell. 1999;3:287–96. doi: 10.1016/s1097-2765(00)80456-6. [DOI] [PubMed] [Google Scholar]
- 37.Xiao D, Singh SV. Phenethyl isothiocyanate-induced apoptosis in p53-deficient PC-3 human prostate cancer cell line is mediated by extracellular signal-regulated kinases. Cancer Res. 2002;62:3615–3619. [PubMed] [Google Scholar]