

Abstract
While the constitutive, 26S proteasome plays an important role in regulating many important cellular processes, a variant form known as the immunoproteasome is thought to primarily function in adaptive immune responses. However, recent studies indicate an association of immunoproteasomes with many physiological disorders such as cancer, neurodegenerative, and inflammatory diseases. Despite this, the detailed functions of the immunoproteasome remain poorly understood. Immunoproteasome-specific probes are essential to gain insight into immunoproteasome function. Here, we describe for the first time the development of the cell-permeable, activity-based fluorescent probes, UK101-Fluor and UK101-B660, which selectively target the catalytically active LMP2/β1i subunit of the immunoproteasome. These probes will facilitate rapid detection of the cellular localization of catalytically active immunoproteasomes in living cells, providing a valuable tool to analyze immunoproteasome functions. Additionally, as LMP2/β1i may serve as a potential tumor biomarker, an LMP2/β1i-targeting fluorescent imaging probe may be applicable to a rapid readout assay to determine tumor LMP2/β1i levels.
Introduction
The important role of the ubiquitin-proteasome system (UPS) in many essential cellular processes is now well-documented.1, 2 The proteasome, a key component of the UPS, is a multi-protease complex responsible for the degradation of poly-ubiquitinated proteins involved in many important biological processes such as the cell cycle, development, and inflammatory responses. The immunoproteasome is a variant form of the constitutive proteasome. Normally, the immunoproteasome is constitutively expressed in cells of hematopoietic origin but can also be induced in normal cells by cytokines such as interferon-γ (IFN-γ) or tumor necrosis factor-α (TNF-α).3 Upon exposure to these stimuli, the immunoproteasome catalytic subunits LMP2/β1i, MECL1/β2i, and LMP7/β5i, are synthesized and incorporated, replacing their constitutive proteasome counterparts Y/β1, Z/β2 and X/β5, respectively (Fig. 1). Similar to those of the constitutive proteasome, catalytic subunits of the immunoproteasome are synthesized as inactive forms containing N-terminal propeptides: pre-LMP2, pre-LMP7 and pre-MECL1. Upon completion of proteasome assembly, the N-terminal propeptides of these inactive catalytic subunits are removed to expose the catalytic threonine residues.4, 5 In comparison with the constitutive proteasome, the capacity of the immunoproteasome to generate peptides bearing C-terminal hydrophobic amino acids is enhanced, while its capacity to produce peptides bearing C-terminal acidic residues is reduced. 6 This results in the increased production of peptides which can associate with MHC class I molecules for antigen presentation.
Figure 1.
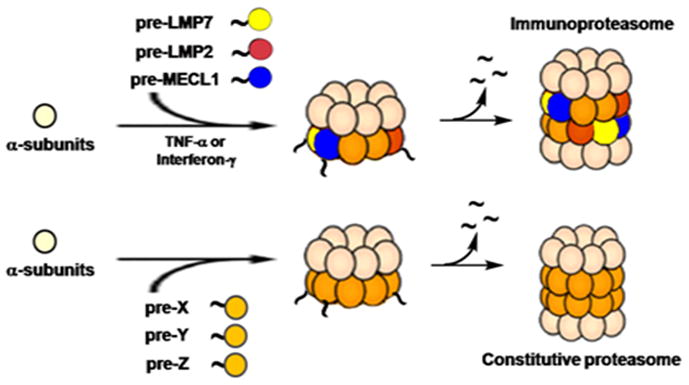
Schematic representation of immunoproteasome and constitutive proteasome assembly.
Although the immunoproteasome is thought to primarily function in adaptive immune responses, several recent studies have shown that its cellular roles are not limited to the generation of antigenic peptides but are far more multifaceted. 7–9 For example, the immunoproteasome has been implicated in a number of pathological disorders such as cancer, neurodegenerative and autoimmune diseases.10–15 More recently, the immunoproteasome has been shown to clear protein aggregates that accumulate under oxidative stress.8 Despite these advances, our understanding of immunoproteasome function still remains limited.
While recent advances in proteomics allow global proteome profiling, mechanistic dissection of the correlation between the proteome information and protein dynamics in the microcellular environment or disease states is still a major challenge in biology. One of the primary goals of chemical biology is to investigate such a biological conundrum using small molecules that perturb signaling proteins or pathways. Use of these small molecules has proven to be an effective strategy to decipher protein functions within cells.16 Pioneered by Cravatt and Bogyo, the use of small molecules has further evolved to exploit their function as active site-directed activity-based probes (ABPs), allowing the monitoring, in real time, of the activities of proteases in disease states. 17–24 Typically, an ABP targets an enzyme active site to monitor the functional enzyme in living cells. General proteasome inhibitors have also been exploited as ABP imaging agents that report proteasome activities in cells. Fluorescently labeled proteasome inhibitors 25, 26 can be used for real-time monitoring of proteasome activity for correlation with cellular processes. One example of a synthetic proteasome-targeting fluorescent probe is BODIPY-epoxomicin, which binds selectively to the β5/β1 proteasome subunits. 27 Another example is MV151,26 which was prepared by a medicinal chemistry approach and binds to all catalytic subunits of the proteasome in living cells. This kind of fluorescent probe can be used for the clinical profiling of proteasome activity, biochemical analysis of the subunit specificity of inhibitors, or cell biological analysis of proteasome functions and dynamics. Furthermore, Berkers et al. have shown that these fluorescently labeled ABPs can be used to study functionally active proteasomes in tissues.28 Despite the development of several fluorescently labeled ABPs targeting proteasomes,28, 29 there are currently no available immunoproteasome- or constitutive proteasome-specific ABPs that can be used to dissect the distinctive functions of proteasome subtypes.
In this article, we report the synthesis and characterization of fluorescent ABPs that selectively target the immunoproteasome in living cells. These probes enable fast labeling of catalytically active immunoproteasomes and can therefore be used to visualize their cellular localization. This information may provide an important clue in understanding immunoproteasome functions in living cells. Furthermore, given that LMP2/β1i is highly expressed in several types of cancer, such an LMP2-targeting ABP may potentially serve as a tool for cancer screening as well as for monitoring cancer progression.
Results and Discussion
Synthesis of fluorescein-labeled UK-101
Given that UK-101 (Fig 3), a small molecule inhibitor previously developed in our laboratory,10 selectively targets LMP2 in cells, we wanted to use UK-101 as a lead compound in developing LMP2-targeting fluorescent probes. Our molecular modeling studies 30 indicated that the alanine side chain at the P2 site of UK-101 is exposed towards the surface of LMP2 through the open channel extending from the active site (Fig. 3A). This suggests that the alanine side chain of UK-101 can be replaced by other linear groups without disrupting the specific UK-101:LMP2 interaction (Fig. 3B). Therefore, we set out to introduce a linear hydrocarbon linker at the P2 position having a free amine functional group, which can be used as a handle to attach a fluorophore to UK-101 (Fig. 3B). Specifically, the key intermediate, UK101-Lys-Cbz (7), was first prepared following a procedure similar to that reported for the synthesis of UK-101,10 replacing alanine with Cbz-protected lysine (Scheme 1). Before coupling a fluorescent residue to UK-101 via the lysine linker, we wanted to ensure that this intermediate (7) maintained binding selectivity for LMP2. This was first confirmed in EL4 mouse lymphoma cells, which highly express both the immunoproteasome and the constitutive proteasome. The cells were treated with UK101-Lys-Cbz (7) or DMSO (vehicle control), lysed, and subjected to western blotting. As shown by an increase in molecular weight resulting from covalent modification, UK101Lys-Cbz binds to LMP2 (Fig. 4A). To identify the minimum concentration of UK101-Lys-Cbz required for complete covalent modification of LMP2, EL4 cells were treated with a range of low micromolar concentrations of UK101-Lys-Cbz, DMSO, or the general proteasome inhibitor epoxomicin. The cells were then lysed and subjected to western blotting as before. The results revealed that 1 μM of UK101-Lys-Cbz is not sufficient to modify LMP2, while a 5 μM concentration of this compound labels most of the LMP2 in these cells, as visualized by a partially incomplete upward shift of the LMP2 band (Fig. 4C). A 10 μM dose of UK101-Lys-Cbz resulted in complete covalent modification of LMP2, resulting in a complete upward shift of the LMP2 band (Fig. 4C). In contrast, this intermediate (7) does not bind LMP7, Y, or X at 10 μM (Fig. 4C). Similar results were obtained using the PC-3 human prostate cancer cell line (Fig. 4B and D). It should be noted that synthesis of proteasome subunits can be induced in response to treatment with proteasome inhibitors, explaining the variation in band intensity.31 Collectively, these results suggest that derivatization of UK-101 via a linear hydrocarbon chain at the P2 position does not influence the LMP2-binding specificity of UK-101. Encouraged by this, UK101-Lys-Cbz (7) was hydrogenated and coupled to fluorescein using fluorescein-NHS (fluorescein N-succinimidyl ester) to yield UK101-Fluor (9) (Scheme 1).
Figure 3.
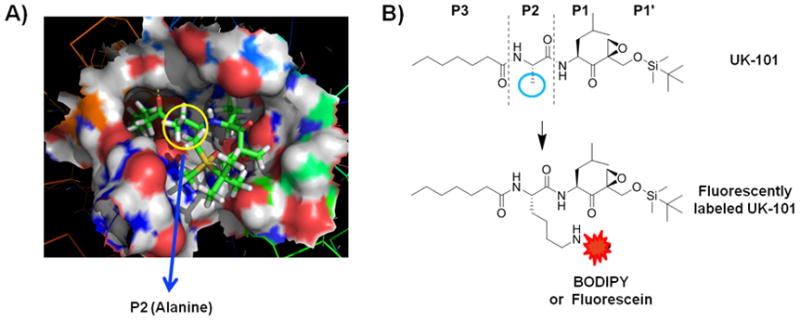
Proposed LMP2 binding mode and derivatization of UK-101. A) Molecular model depicting the binding of UK-101 (stick model) within the catalytic cleft of LMP2 (surface model). B) Structure of UK-101. The alanine side chain at the P2 position is indicated by the blue circle. This side chain was derivatized for the attachment of the fluorescein or BODIPY fluorophore to give UK101-Fluor or UK101-B660.
Scheme 1.
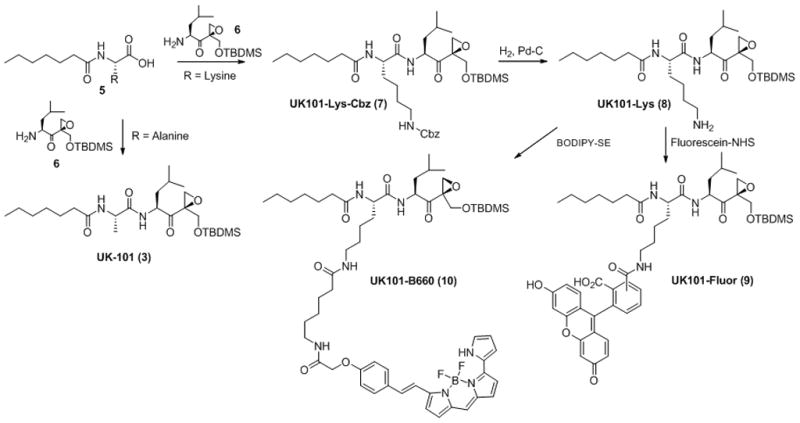
Synthesis of UK101-Fluor (9) and UK101-B660 (10). Fluorescein or BODIPY 650/665 (Invitrogen) was attached at the P2 position of UK101-Lys (8). The first coupling between compounds 5 and 6 was performed using standard peptide coupling agents: HBTU (1 eq.), HOBt (1 eq.), DIPEA (5 eq.) and CH2Cl2 (reaction solvent).
Figure 4.
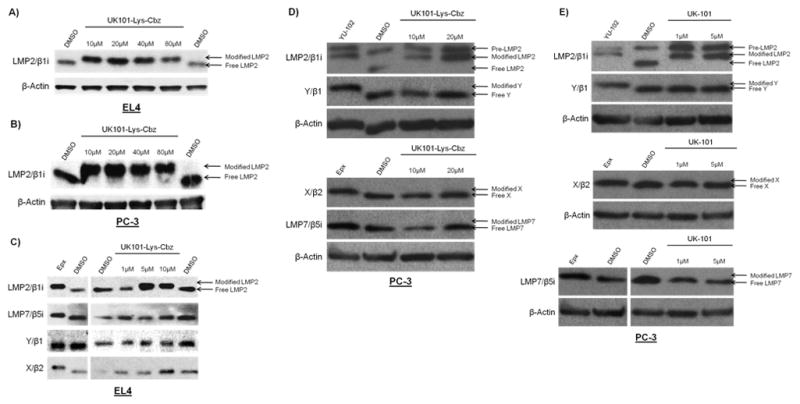
Binding specificity of the UK101-Lys-Cbz (8) and UK-101 (3). EL4 (A and C) or PC-3 (B, D, and E) cells were treated with DMSO (vehicle control) or increasing concentrations of UK101-Lys-Cbz or UK-101, lysed, and subjected to western blotting. Subunit binding specificity was determined using antibodies recognizing proteasome and immunoproteasome subunits. An upward shift of a band with respect to the DMSO control indicates an increase in molecular weight due to covalent modification. 1 μM epoxomicin-treated cells served as a positive control for LMP2, LMP7, and X band shifts in (C), and for LMP7 and X band shifts in (D and E). However, epoxomicin does not bind Y under these conditions, and therefore no shift is observed for this subunit in (C). For (D and E), 10 μM YU-102-treated cells served as a positive control for LMP2 and LMP7 band shifts.
Visualization of catalytically active LMP2 using UK101-Fluor
Next, we investigated whether UK101-Fluor (9) facilitates visualization of LMP2 in living cells. First, PC-3 cells were treated with UK101-Fluor (9), washed extensively to remove unbound fluorescent probe, and visualized via fluorescence microscopy. Binding of UK101-Fluor was observed to be dose-dependent (Fig. 5A). Based on this result, we selected a concentration of 15 μM for further studies, as this concentration gave the optimum signal to background ratio. We then wanted to ensure that the fluorescent signal observed resulted from UK101-Fluor binding to LMP2. To do this, we performed a competition assay in living cells. Specifically, PC-3 cells were preincubated with UK-101 (3) under conditions that result in complete covalent modification of LMP2, but not other proteasome subunits, as detected by a mobility shift assay (Fig 4E). Following this preincubation, the cells were treated with UK101-Fluor, washed to remove unbound probe, and visualized via fluorescence microscopy. Pretreatment with UK-101 effectively competed away the signal observed upon UK101-Fluor treatment alone, suggesting that UK101-Fluor selectively targets LMP2 (Fig. 5B). To confirm this result, PC-3 cells stably transfected with the empty pLKO vector or with the pLKO vector containing a short hairpin RNA against LMP2 were treated with UK101-Fluor as in Fig. 5A. The signal observed from UK101- Fluor in cells transfected with the empty vector was markedly decreased in the LMP2 knockdown cells, further indicating the selectivity of UK101-Fluor for LMP2 (Fig. 5C).
Figure 5.
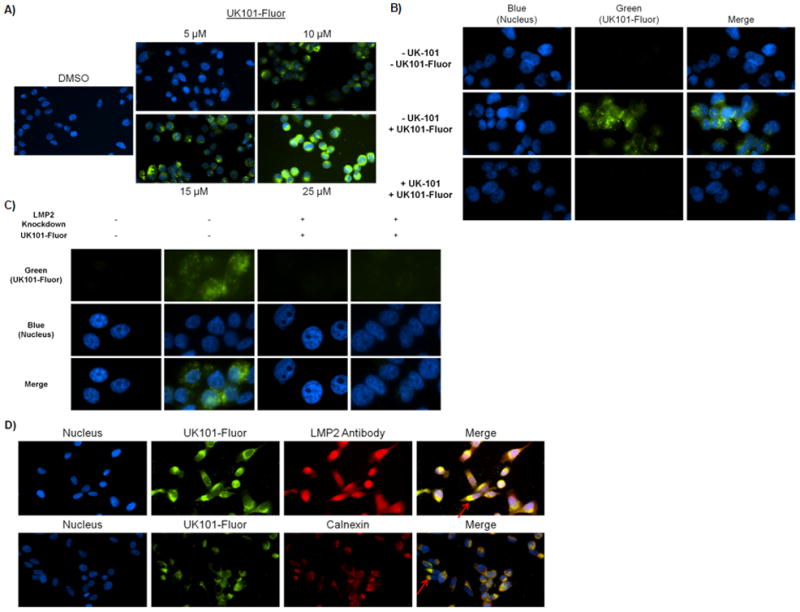
UK101-Fluor (9) selectively binds to LMP2 in PC-3 cells. A) UK101-Fluor binding with LMP2 is dose-dependent. After extensive washing to remove unbound probe, binding was examined via fluorescence microscopy. Fluorescence intensity of all images was normalized to DMSO control. Of note, LMP2 binding was also observed in cells treated with 5 μM UK101-Fluor with a longer exposure time. B) Pre-incubation with UK-101 competes away the signal observed following treatment with UK101-Fluor alone. PC-3 cells pretreated with UK-101 or DMSO were subseqently treated with UK101-Fluor, fixed, and examined via fluorescence microscopy. Fluorescence intensity in all images was normalized to control (-UK-101/-UK101-Fluor). C) LMP2 knockdown results in decreased binding of UK101-Fluor. PC-3 cells transfected with either the empty pLKO vector (LMP2 Knockdown −) or the pLKO vector containing a short hairpin RNA against LMP2 (LMP2 knockdown +) were treated with DMSO or UK101-Fluor, fixed, and examined via fluorescence microscopy. Fluorescence intensity in all images was normalized to controls (UK101-Fluor −). D) UK101-Fluor cellular localization. Cells were incubated with UK101-Fluor and subsequently stained with antibodies recognizing LMP2 or calnexin, an endoplasmic reticulum (ER) marker. The cells were then fixed and examined via fluorescence microscopy. The arrows in the merged images point to areas of overlap between green and red staining, which gives a yellow color.
To examine the cellular localization pattern of catalytically active LMP2, cells were treated with UK101-Fluor (9) as before, but were then fixed and stained with an AlexaFluor546-conjugated antibody detecting LMP2. It should be noted that UK101-Fluor (9) only detects catalytically active LMP2, which only exists within fully assembled proteasomes, in contrast to the LMP2 antibody, which detects both catalytically active as well as inactive or pre-LMP2. As shown in Fig. 5D (top panel), catalytically active LMP2 was easily detected by UK101-Fluor and does not display the diffuse distribution observed of the total cellular population of LMP2 (Fig. 5D). In contrast, the total population of LMP2 appears to be diffusely distributed throughout the cell (Fig. 5D). To investigate whether catalytically active LMP2 is localized near the endoplasmic reticulum (ER), we stained fixed UK101-Fluor-treated cells with an antibody recognizing calnexin, an ER marker. As shown in Fig. 5D (bottom panel), the majority of catalytically active LMP2 appears to be localized to the ER, as reported 32, 33. Taken together, these results demonstrate that the LMP2-specific inhibitor UK-101 can be derivatized for the development of immunoproteasome imaging probes, providing a powerful immunoproteasome imaging tools.
Synthesis of a near-infrared fluorescent probe
LMP2 is highly expressed in various types of cancer,10, 14, 15, 34, 35 and therefore a non-invasive imaging probe that selectively detects LMP2 may provide a novel diagnostic tool. Given the successful development of UK101-Fluor (Fig. 5), we next sought to develop a near-infrared fluorescent (NIRF) probe that selectively targets LMP2 and can be applied to animal model studies. Specifically, UK101-Lys-Cbz (7) was hydrogenated and coupled to a near-infrared fluorescent group (BODIPY 650/665) using BODIPY 650/665-SE to yield UK101-B660 (10) (Scheme 1).
Visualization of catalytically active LMP2 using UK101-B660
To verify that UK101-B660 (10) selectively targets LMP2, we first performed an in-gel fluorescence assay using a Typhoon imaging system. Extracts from cells treated with UK101-B660 (10) were subjected to gel electrophoresis, and the gel was imaged to determine the molecular weight of proteins covalently modified by this compound. As shown in Fig. 6A, a protein band with a size of ~23 kDa appeared, suggesting that the UK101-B660-labeled protein is likely an immunoproteasome subunit. To further confirm that this labeled protein is LMP2, a competition assay was performed with UK-101 (3) and UK101-Lys-Cbz (7). Specifically, cells were pre-treated with UK-101 (3) or UK101-Lys-Cbz (7), followed by treatment with UK101-B660 (10). Cell lysates were then prepared and analyzed as in Fig. 6A. As shown in Fig. 6B, preincubation with UK-101 or UK101-Lys-Cbz significantly attenuated the fluorescent signal resulting from UK101-B660 treatment, indicating that it really is binding LMP2 rather than another subunit. To further verify this, we performed a similar competition assay using a fluorescence microscopy readout. As shown in Fig. 6C, pretreatment of cells with UK-101 or UK101-Lys-Cbz effectively depleted fluorescent signals derived from UK101-B660, consistent with the results obtained from the in-gel fluorescence assay. Additionally, the cellular localization of UK101-B660 appeared to take on a similar pattern as that of UK101-Fluor (compare Fig. 5B and D with Fig. 6C). To ensure that LMP2 was still present and maintained a normal cellular distribution upon UK-101 treatment, cells pretreated with UK-101 followed by treatment with UK101-B660 were also stained with an antibody recognizing LMP2. As shown in Fig. 6C (bottom panel), UK-101 treatment did not impact the levels or cellular distribution of LMP2 (compare with Fig. 5D, top panel). Together, these results suggest that UK101-B660 can be used as an imaging probe to visualize catalytically active LMP2 in living cells.
Figure 6.
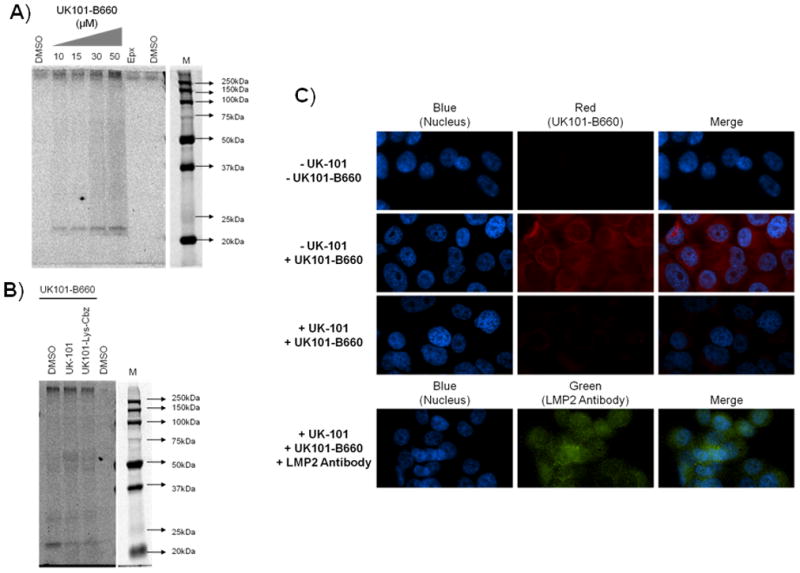
UK101-B660 (10) selectively binds to LMP2 in PC-3 cells. A and B) UK101-B660 targets the immunoproteasome catalytic subunit LMP2. A) UK101-B660 binding is dose-dependent. PC-3 cells were treated with DMSO (vehicle control), the general proteasome inhibitor epoxomicin (Epx), or increasing concentrations of UK101-B660. The cells were then lysed, and proteins were resolved by SDS-PAGE. Gels were imaged using a Typhoon imager. M=Marker. B) UK-101 or UK101-Lys preincubation competes away the fluorescent signal of UK101-B660. PC-3 cells were preincubated with DMSO (vehicle control), UK-101, or UK101-Lys, followed by incubation with DMSO or UK101-B660. The cells were then lysed, and the proteins were resolved and imaged as in (A). M=Marker. C) Cells treated with UK-101 display a red fluorescent signal that is competed away by UK-101 pretreatment. PC-3 cells were pretreated with either DMSO (vehicle control) or UK-101 and subsequently treated with DMSO or UK101-B660. UK101-B660 binding was then visualized via fluorescence microscopy.
Conclusions
In summary, for the first time, we have successfully developed fluorescent imaging agents that specifically target the catalytically active immunoproteasome subunit LMP2. Given that the immunoproteasome is gaining increasing attention as a therapeutic target for cancer, these fluorescent probes will provide a powerful tool in investigating the therapeutic potentials of targeting the immunoproteasome as well as the ever-increasing functions of the immunoproteasome. Additionally, with LMP2 serving as a potential tumor biomarker, our NIRF LMP2-targeting probe may be directly applicable to in vivo screening in animal models.
Experimental
Cell Culture
EL4 and PC-3 cell lines were obtained from the American Type Culture Collection. These cells were cultured according to the manufacturer’s instructions at 37°C in 5% CO2. PC-3 cells stably transfected with the empty pLKO vector or the pLKO vector containing a short hairpin RNA against LMP2 were maintained in F-12K medium containing 0.5 μg/mL puromycin at 37°C in 5% CO2.
Compounds
UK-101 and epoxomicin were synthesized as previously described.10, 36 Fluorescent probes (9 & 10) were synthesized following a procedure similar to the previously reported.10
Compound 9, 1H NMR (500 MHz, CDCl3): δ 8.05 (s, 1H), 7.94 (s, 1H), 6.94-6.88 (m, 1H,), 6.63-6.56 (m, 3H), 4.83 (m, 8H), 4.49-4.46 (m, 1H), 4.33-4.27 (m, 1H), 3.51-3.27 (m, 3H), 2.96 (s, 4H), 2.82 (s, 4H), 2.61 (s, 3H), 2.20-2.14 (m, 2H), 1.79-1.42 (m, 9H), 1.40-1.11 (m, 11H), 0.90-0.84 (m, 18H), 0.06-0.01 (m, 6H); MS-spec: calcd. 900.45 (M+H) +, found 900.45 (M+H) +.
Compound 10, H1 NMR (500MHz, CDCl3): δ= 7.592 (d, 2H, J = 13.5 Hz), 7.230 (d, 2H, J =13.5 Hz),7.058 (s,1H), 6.974 (m, 2H), 6.927 (m,1H), 6.889 (m,2H), 6.823 (m,1H), 6.643(s,1H), 6.399(s,1H), 5.387 (s, 1H), 5.351(m, 1H), 4.556 (m, 3H), 4.429 (dd, 2H, J = 16.0, 16.0 Hz), 3.659 (m, 4H), 3.519 (m, 1H), 2.999 (m,1H), 2.103–2.230 (m, 4H), 2.011–2.056 (m, 4H), 1.459–1.645 (m, 7H), 1.080–1.436 (m,12H), 0.851–1.029 (m, 18H), 0.046 (s, 6H). MS-spec: calcd. 1069.61 (M+H) +, found 1069.7 (M+H) +
Immunoblotting
To prepare whole cell lysates, cells were incubated for 1 hr. on ice in non-denaturing lysis buffer (50nM Tris-Cl, 150mM NaCl, 1% NP-40, 1% Triton X-100, and 1% protease inhibitor cocktail (Sigma-Aldrich). The lysates were then centrifuged at 14,000 rpm at 4°C for 10 min. (Sorvall Biofuge Primo R, Kendro Laboratory Products) and the supernatants were collected. Protein concentrations of the supernatants were determined by the Bradford method using Protein Assay Dye Reagent Concentrate (Bio-Rad) and a GENESYS 10 spectrophotometer, Thermo Spectronic (VWR). 2X Laemmli Sample Buffer (Sigma-Aldrich) was added to the supernatants, which were then heated on a heat block at 100°C for 10 min. Equal protein concentrations of the resulting samples were then loaded onto 12% polyacrylamide gels (Bio-Rad), resolved by SDS-PAGE, and transferred onto PVDF membranes (Bio-Rad). Membranes were blocked on a rotating platform for 1 hr. at room temperature in 5% nonfat dry milk (Bio-Rad). Primary and secondary antibodies were diluted in 3% BSA and incubated with membranes for 1 hr. at room temperature on a rotating platform or overnight at 4°C. Proteins were visualized on Kodak BioMax XAR Films (Sigma-Aldrich) using ECL Western Blotting Substrate (Pierce).
In-Gel Fluore scence
Denatured whole cell lysates were prepared and proteins were resolved as described for immunoblotting. Gels were then imaged using a Typhoon FLA 9000 imaging system (λex = 635 nm, (λem = 665).
Fluorescence Microscopy
Cells were plated on coverslips or chamber slides and incubated for 24–48 hrs. prior to treatment. Following treatment, the cells were fixed with 4% paraformaldehyde in PBS for 7 min. at 37°C and permeablized with 0.2% Triton-X in PBS for 30 min. at 37°C. Coverslips or slides were then blocked in 10% goat serum and 1% BSA in PBST. Where indicated, anti-LMP2 or anti-calnexin primary antibodies were diluted in DakoCytomation Antibody Diluent with Background Reducing Components and incubated with cells for 1 hr. at 37°C. Secondary antibodies were similarly diluted and incubated with cells for 30 min. at 37°C. Following this incubation, coverslips or slides were extensively washed with PBST to remove unbound ABP or antibody. Coverslips were mounted onto microscope slides using Prolong Gold antifade reagent with DAPI (Invitrogen) or VECTASHIELD Mounting Medium with DAPI (Vector Laboratories). Cells were then visualized using an inverted Nikon-Ti-U fluorescence microscope and NIS Element Research image analysis software.
UK101-Lys-Cbz Treatment
EL4 or PC-3 cells were treated with DMSO, 1 μM epoxomicin, or UK101-Lys-Cbz at the indicated concentrations for 1.5 hrs. The cells were then washed with Hank’s Balanced Salt Solution (Gibco) and harvested for immunoblotting.
UK101-Fluor and UK101-B660 treatment
PC-3 cells were treated with DMSO, UK101-Fluor or UK101-B660 at the indicated concentrations for 4 hrs. The cells were then washed with Hank’s Balanced Salt Solution (Gibco) and harvested for an in-gel fluorescence assay or prepared for fluorescence microscopy.
Competition Assays
For the in-gel fluorescence competition assay, PC-3 cells were pretreated with DMSO, 1 μM UK-101 or 20 μM UK101-Lys for 1.5 hrs. 30 μM of UK101-B660 was then added to the cell culture medium, and the cells were incubated for an additional 4 hrs. For fluorescence microscopy-based competition assays, PC-3 cells were pretreated with DMSO or 5 μM UK-101for 1.5 hrs. 15 μM of UK101-Fluor or UK101-B660 was then added to the cell culture medium, and the cells were incubated for an additional 4 hrs. The cells were then harvested for an in-gel fluorescence assay or prepared for fluorescence microscopy.
Figure 2.
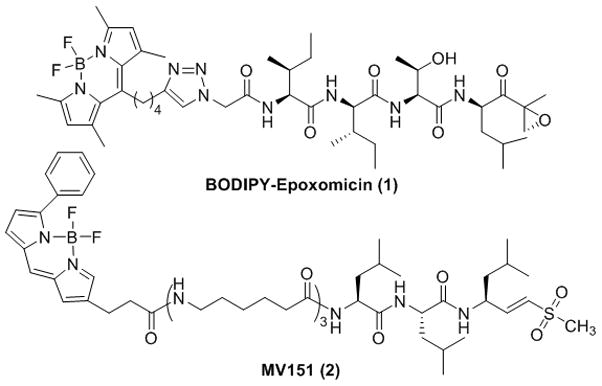
Structures of BODIPY-labeled epoxomicin and MV151. BODIPY-labeled epoxomicin selectively targets constitutive proteasome subunits β1, β5 and, to a lesser extent, LMP7, while MV151 covalently modifies all catalytically active proteasome subunits.
Acknowledgments
This work has been supported by the Markey Cancer Foundation and NIH (CA131059). We thank members of the Kim lab for their helpful comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Petroski MD. BMC Biochem. 2008;9(Suppl 1):S7. doi: 10.1186/1471-2091-9-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marques AJ, Palanimurugan R, Matias AC, Ramos PC, Dohmen RJ. Chem Rev. 2009;109:1509. doi: 10.1021/cr8004857. [DOI] [PubMed] [Google Scholar]
- 3.Kloetzel PM. Nat Rev Mol Cell Biol. 2001;2:179. doi: 10.1038/35056572. [DOI] [PubMed] [Google Scholar]
- 4.Griffin TA, Nandi D, Cruz M, Fehling HJ, Kaer LV, Monaco JJ, Colbert RA. J Exp Med. 1998;187:97. doi: 10.1084/jem.187.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De M, Jayarapu K, Elenich L, Monaco JJ, Colbert RA, Griffin TA. J Biol Chem. 2003;278:6153. doi: 10.1074/jbc.M209292200. [DOI] [PubMed] [Google Scholar]
- 6.Rivett AJ, Hearn AR. Curr Protein Pept Sci. 2004;5:153. doi: 10.2174/1389203043379774. [DOI] [PubMed] [Google Scholar]
- 7.Groettrup M, Kirk CJ, Basler M. Nat Rev Immunol. 2010;10:73. doi: 10.1038/nri2687. [DOI] [PubMed] [Google Scholar]
- 8.Seifert U, Bialy LP, Ebstein F, Bech-Otschir D, Voigt A, Schroter F, Prozorovski T, Lange N, Steffen J, Rieger M, Kuckelkorn U, Aktas O, Kloetzel PM, Kruger E. Cell. 2010;142:613. doi: 10.1016/j.cell.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 9.Yewdell JW. Proc Natl Acad Sci U S A. 2005;102:9089. doi: 10.1073/pnas.0504018102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ho YK, Bargagna-Mohan P, Mohan R, Kim KB. Chem Biol. 2007;14:419. doi: 10.1016/j.chembiol.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuhn DJ, Hunsucker SA, Chen Q, Voorhees PM, Orlowski M, Orlowski RZ. Blood. 2009;113:4667. doi: 10.1182/blood-2008-07-171637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitzpatrick LR, Khare V, Small JS, Koltun WA. Dig Dis Sci. 2006;51:1269. doi: 10.1007/s10620-006-8047-2. [DOI] [PubMed] [Google Scholar]
- 13.Fitzpatrick LR, Small JS, Poritz LS, McKenna KJ, Koltun WA. Dis Colon Rectum. 2007;50:337. doi: 10.1007/s10350-006-0796-7. [DOI] [PubMed] [Google Scholar]
- 14.Altun M, Galardy PJ, Shringarpure R, Hideshima T, LeBlanc R, Anderson KC, Ploegh HL, Kessler BM. Cancer Res. 2005;65:7896. doi: 10.1158/0008-5472.CAN-05-0506. [DOI] [PubMed] [Google Scholar]
- 15.Mishto M, Bellavista E, Santoro A, Stolzing A, Ligorio C, Nacmias B, Spazzafumo L, Chiappelli M, Licastro F, Sorbi S, Pession A, Ohm T, Grune T, Franceschi C. Neurobiol Aging. 2006;27:54. doi: 10.1016/j.neurobiolaging.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 16.Ovaa H, van Leeuwen F. Chembiochem. 2008;9:2913. doi: 10.1002/cbic.200800454. [DOI] [PubMed] [Google Scholar]
- 17.Nomura DK, Dix MM, Cravatt BF. Nat Rev Cancer. 2010;10:630. doi: 10.1038/nrc2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fonovic M, Verhelst SH, Sorum MT, Bogyo M. Mol Cell Proteomics. 2007 doi: 10.1074/mcp.M700124-MCP200. [DOI] [PubMed] [Google Scholar]
- 19.Sadaghiani AM, Verhelst SH, Bogyo M. Curr Opin Chem Biol. 2007;11:20. doi: 10.1016/j.cbpa.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 20.Blum G, Mullins SR, Keren K, Fonovic M, Jedeszko C, Rice MJ, Sloane BF, Bogyo M. Nat Chem Biol. 2005;1:203. doi: 10.1038/nchembio728. [DOI] [PubMed] [Google Scholar]
- 21.Blum G, von Degenfeld G, Merchant MJ, Blau HM, Bogyo M. Nat Chem Biol. 2007;3:668. doi: 10.1038/nchembio.2007.26. [DOI] [PubMed] [Google Scholar]
- 22.Berger AB, Vitorino PM, Bogyo M. Am J Pharmacogenomics. 2004;4:371. doi: 10.2165/00129785-200404060-00004. [DOI] [PubMed] [Google Scholar]
- 23.Cravatt BF, Wright AT, Kozarich JW. Annu Rev Biochem. 2008;77:383. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 24.Kikuchi K. Chem Soc Rev. 2010;39:2048. doi: 10.1039/b819316a. [DOI] [PubMed] [Google Scholar]
- 25.Verdoes M, Berkers CR, Florea BI, van Swieten PF, Overkleeft HS, Ovaa H. Methods Mol Biol. 2006;328:51. doi: 10.1385/1-59745-026-X:51. [DOI] [PubMed] [Google Scholar]
- 26.Verdoes M, Florea BI, Menendez-Benito V, Maynard CJ, Witte MD, van der Linden WA, van den Nieuwendijk AM, Hofmann T, Berkers CR, van Leeuwen FW, Groothuis TA, Leeuwenburgh MA, Ovaa H, Neefjes JJ, Filippov DV, van der Marel GA, Dantuma NP, Overkleeft HS. Chem Biol. 2006;13:1217. doi: 10.1016/j.chembiol.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 27.Verdoes M, Hillaert U, Florea BI, Sae-Heng M, Risseeuw MD, Filippov DV, van der Marel GA, Overkleeft HS. Bioorg Med Chem Lett. 2007;17:6169. doi: 10.1016/j.bmcl.2007.09.025. [DOI] [PubMed] [Google Scholar]
- 28.Berkers CR, van Leeuwen FW, Groothuis TA, Peperzak V, van Tilburg EW, Borst J, Neefjes JJ, Ovaa H. Mol Pharm. 2007 doi: 10.1021/mp0700256. [DOI] [PubMed] [Google Scholar]
- 29.Berkers CR, Verdoes M, Lichtman E, Fiebiger E, Kessler BM, Anderson KC, Ploegh HL, Ovaa H, Galardy PJ. Nat Methods. 2005;2:357. doi: 10.1038/nmeth759. [DOI] [PubMed] [Google Scholar]
- 30.Lei B, Abdul Hameed MD, Hamza A, Wehenkel M, Muzyka JL, Yao XJ, Kim KB, Zhan CG. J Phys Chem B. 2010 doi: 10.1021/jp1058098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meiners S, Heyken D, Weller A, Ludwig A, Stangl K, Kloetzel PM, Kruger E. J Biol Chem. 2003;278:21517. doi: 10.1074/jbc.M301032200. [DOI] [PubMed] [Google Scholar]
- 32.Rivett AJ. Curr Opin Immunol. 1998;10:110. doi: 10.1016/s0952-7915(98)80040-x. [DOI] [PubMed] [Google Scholar]
- 33.Brooks P, Murray RZ, Mason GG, Hendil KB, Rivett AJ. Biochem J. 2000;352(Pt 3):611. [PMC free article] [PubMed] [Google Scholar]
- 34.Huang YC, Chuang LY, Hung WC. Mol Pharmacol. 2002;62:1515. doi: 10.1124/mol.62.6.1515. [DOI] [PubMed] [Google Scholar]
- 35.Mishto M, Santoro A, Bellavista E, Bonafe M, Monti D, Franceschi C. Ageing Res Rev. 2003;2:419. doi: 10.1016/s1568-1637(03)00030-8. [DOI] [PubMed] [Google Scholar]
- 36.Ho A, Cyrus K, Kim KB. Eur J Org Chem. 2005:4829. [Google Scholar]