

Abstract
While interleukin (IL)-1β plays an important role in combating the invading pathogen as part of the innate immune response, its dysregulation is responsible for a number of autoinflammatory disorders. Large IL-1β activating platforms, known as inflammasomes, can assemble in response to the detection of endogenous host and pathogen-associated danger molecules. Formation of these protein complexes results in the autocatalysis and activation of caspase-1, which processes precursor IL-1β into its secreted biologically active form. Inflammasome and IL-1β activity is required to efficiently control viral, bacterial and fungal pathogen infections. Conversely, excess IL-1β activity contributes to human disease, and its inhibition has proved therapeutically beneficial in the treatment of a spectrum of serious, yet relatively rare, heritable inflammasomopathies. Recently, inflammasome function has been implicated in more common human conditions, such as gout, type II diabetes and cancer. This raises the possibility that anti-IL-1 therapeutics may have broader applications than anticipated previously, and may be utilized across diverse disease states that are linked insidiously through unwanted or heightened inflammasome activity.
Keywords: infections, inflammasome, innate immunity, interleukin-1, NLRP3
Introduction
Several families of germline encoded pattern recognition receptors (PRRs) have been identified that mediate critical aspects of the innate and protective immune responses required for control of microbial and viral infection. These include the Toll-like receptor (TLR) family, the retinoid-inducible gene 1 (RIG-I) like receptors (RLRs), the C-type lectin receptors and the nucleotide binding domain (NOD)-like receptor (NLR) family. Activation of PRRs results from the recognition of a number of conserved microbial and viral components, such as cell wall proteoglycans, pore-forming toxins and pathogen RNA and DNA [pathogen-associated molecular patterns (PAMPs)]. In addition, several host PRR activators [damage-associated molecular patterns (DAMPs)] have been identified, such as extracellular adenosine 5′-triphosphate (ATP), host DNA, hyaluronan and monosodium urate crystals. DAMPs can accumulate as a result of metabolic disorders or may be released upon cellular damage caused by trauma (i.e. myocardial infarction) and infection. They can therefore contribute to sterile-inflammation and wound responses, as well as pathogen-associated immune responses.
While TLRs detect extracellular and intracellular vacuolar stimuli, NLRs appear to respond to cytosolic disturbances. The NLR family contains several proteins (see below) that act as scaffolds and oligomerize into large protein complexes (∼700 kDa) to induce inflammasome formation via activation of the inflammatory caspase, caspase-1, which proteolyses and thereby activates precursor interleukin (IL)-1β and IL-18, and can also lead to the release of IL-1α[1,2]. Activated IL-1β is a potent endogenous pyrogen and induces flu-like symptoms such as chills, rigors, fever, nausea, vomiting, headache and fatigue when injected into humans at 1–10 nanograms/kg of body weight [3,4]. Both IL-1β and IL-1α bind to the IL-1 receptor (IL1-R) and induce the formation of a high-affinity ternary complex with the IL-1R accessory protein. The resulting downstream signalling cascade leads to transcription factor induction of proinflammatory cytokines and chemokines, and includes genes required for angiogenesis and the recruitment of immune effector cells into the extravascular space. It can also result in the activation of lymphocytes and epithelial cells. While these responses to IL-1β are critical for host protection from many types of viruses and microbes and may aid in cellular and tissue repair responses as discussed below, the dysregulation of IL-1β activity is now implicated in a variety of seemingly divergent diseases such as type II diabetes and gout. Clinical blockade of IL-1 has not only proved to be beneficial in the treatment of these conditions, but is surprisingly well tolerated, which will hopefully expedite the clinical evaluation of IL-1 activity in other pathologies in the near future.
In this review we highlight the benefits of nucleotide-binding domain (NOD)-like receptor protein 3 (NLRP3) inflammasome function in the host defence against invading pathogens (the good), the relatively rare heritable inflammasomopathies that cause excessive IL-1β activation and a number of related diseases (the bad) and the experimental and clinical evidence for unwarranted inflammasome activity contributing to common pathologies affecting millions of people, such as cancer and diabetes (the ugly).
Inflammasome structure
Twenty-two human NLRs exist that have been delineated based on their domain structure and phylogenetic relationships [5,6] (Fig. 1). In general, NLRs contain a C-terminal region made up of a variable number of leucine-rich repeats (LRRs) that are likely to autoinhibit NLR protein function in the resting state, and undergo a conformational change following the detection of an activating stimuli [7]. A central nucleotide binding and oligomerization (NACHT) domain, common to all NLRs, oligomerizes upon inflammasome activation in the presence of nucleotides such as ATP and is essential for function [8,9]. An N-terminal effector domain consisting of either a caspase activation and recruitment domain (CARD), Pyrin or baculoviral inhibitor of apoptosis protein repeat (BIR) domain precedes the NACHT domain. These N-terminal domains initiate specific downstream signalling cascades through homotypic protein interactions. In the case of inflammasome formation, the Pyrin domain containing inflammasomes (i.e. NLRP3) can bind the Pyrin domain of apoptosis speck protein (ASC), which subsequently recruits pro-caspase-1 through CARD–CARD homotypic interactions (Fig. 1). Other inflammasomes that lack a Pyrin domain (i.e. NLRC4) appear able to directly recruit caspase-1 through a C- or N-terminal CARD domain, although they may depend on ASC for optimal function [8]. Pro-caspase-1 recruitment results in proximity-induced caspase-1 oligomerization and autocatalysis, resulting in the release of the active catalytic p20 and p10 caspase-1 fragments and subsequent processing of precursor IL-1β into its biologically active 17 kDa fragment. Notably, basal levels of IL-1β are low and a nuclear factor kappa B (NF-κB)-dependent priming signal [often provided by TLR family members or tumour necrosis factor (TNF)] is required to induce precursor IL-1β expression before it can be cleaved by caspase-1, and can also enhance the expression of other inflammasome components such as NLRP3 [10]. While inflammasomes are not involved directly in the secretion of cytokines such as TNF or IL-6 (although IL-1R signalling can modulate their transcription), caspase-1 is required for non-conventional protein secretion of a variety of leaderless proteins involved in inflammation, cytoprotection and tissue repair [11]; however, any extracellular function for most of these proteins remains to be determined.
Fig. 1.
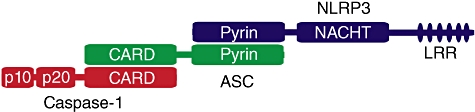
Structure of the nucleotide-binding domain (NOD)-like receptor protein 3 (NLRP3) inflammasome. NLRP3 activation leads to homotypic interactions between the NLRP3 and apoptosis speck protein (ASC) pyrin domains and complex oligomerization. The caspase activation and recruitment domain (CARD) domains of ASC and caspase-1 interact and caspase-1 dimerization results in proximity-induced autoactivation and generation of the active tetrameric caspase-1 p20 and p10 fragments.
Cleavage of IL-1β by caspase-1 is highly specific and other caspases, such as apoptotic caspases, are unlikely to activate IL-1β directly [12]. It has been suggested that a number of serine proteases may cleave IL-1β near the caspase-1 cleavage site, thus generating active IL-1β (reviewed in [13]). Similarly, activation of the TNF superfamily death receptor Fas can also induce IL-1β maturation in caspase-1 knock-out neutrophils [14]. While the relevance of IL-1β cleavage and activation by caspase-1-independent mechanisms in vivo has yet to be fully addressed, caspase-1-independent IL-1β activation can occur in murine models of arthritis, Mycobacterium tuberculosis infection and following tissue damage, suggesting that alternate protease processing of IL-1β is physiologically relevant [15–19].
The good: inflammasome protection against pathogen infection
To date, potentially five inflammasome scaffold proteins that respond to different pathogenic stimuli to activate caspase-1 and IL-1βin vivo have been characterized. The inflammasomes belonging to the NLR family include NLRP1, NLRC4 and NLRP3. The NLRP1 (NALP1/CARD7) inflammasome is activated following the detection of anthrax lethal toxin, the agent responsible for shock-death following systemic infection by Bacillus anthracis (reviewed in [6]). NLRC4 (IPAF/CARD12) detects flagellin and components of the type 3 secretion system from pathogenic bacteria such as Shigella flexneri, Salmonella typhimurium, Listeria monocytogenes and Legionella pneumophila (reviewed in [20]). Outside the NLR family, absent in melanoma 2 (AIM2) is capable of forming a caspase-1 inflammasome through its recognition of cytosolic dsDNA from viruses and bacteria, as well as the host itself [21–23], while the RIG-I inflammasome can detect cytosolic viral RNA species such as 5′-triphosphate RNA and dsRNA [24]. While we focus upon the NLRP3 inflammasome in this review, Table 1 lists the different inflammasomes involved in PAMP recognition, and whether gene knock-out murine models have established a role for inflammasome function in pathogen immunity in vivo.
Table 1.
The good: inflammasome control of pathogen infection
| Organism | PAMPS identified | Inflammasome activated | Inflammasome importance established in vivo? | Refs |
|---|---|---|---|---|
| Helminths | ||||
| Schistosoma mansoni | n.d. | NLRP3 | Yes | [138] |
| Bacteria | ||||
| Mycobacterium tuberculosis | ESX-1 secretion system | NLRP3-dependent and -independent mechanism | Yes. In vivo IL-1 production is independent of caspase-1 | [17,139–142] |
| Streptococcus pneumoniae | Pneumolysin | NLRP3 | Yes | [143,144] |
| Streptococcus pyogenes | Streptolysin O | NLRP3 | Yes. Not important in vivo | [145] |
| Streptomyces hygroscopicus | Nigericin | NLRP3 | n.a. | [146] |
| Klebsiella pneumoniae | n.d. | NLRP3 | Yes | [147] |
| Chlamycia pneumoniae | n.d. | NLRP3 | Yes | [148] |
| Salmonella typhimurium | Flagellin and type III secretion system | NLRP3 and NLRC4 | Yes | [149,150] |
| Francisella tularensis | DNA | AIM2 | Yes | [146,151,152] |
| Legionella pneumophila | Flagellin | NLRC4 | Yes | [153] |
| Listeria monocytogenes | Flagellin, Listeriolysin O, DNA | AIM2, NLRP3, NLRC4 | Yes* | [146,154,155] |
| Pseudomonas aeruginosa | Flagellin, Type III secretion system | NLRC4 | Yes | [156–158] |
| Shigella flexneri | Type III secretion system | NLRC4 | Yes | [157,159] |
| Neisseria gonorrhoeae | Lipo-oligosaccharide | NLRP3 | No | [160] |
| Staphylococcus aureus | Peptidoglycan Haemolysin | NLRP3 | Yes | [146,161–165] |
| Virbrio vulnificus and Vibrio cholerae | Haemolysins | NLRP3 | No | [166] |
| Bacillus anthracis | Anthrax lethal toxin | NLRP1 | Yes | [167–170] |
| Escherichia coli | Type III secretion system, flagellin | NLRC4 | No | [157] |
| Chlamydia trachomatis | Type III secretion system | NLRP3 | No | [171] |
| Protozoa | ||||
| Toxoplasma gondii | n.d. | NLRP1 | No | [172] |
| Plasmodium species; falciparum, berghei, chabaudi | Haemozoin, MSU | NLRP3 | Yes† | [30,136,173] |
| Fungal | ||||
| Candida albicans | Hyphae, β-glucan | NLRP3 | Yes | [174] |
| Aspergillus fumigatus | n.d., β-glucan | NLRP3 | No | [175,176] |
| Saccharomyces cerevisiae | n.d., β-glucan | NLRP3 | n.a. | [174] |
| Viruses | ||||
| Sendai virus | RNA | NLRP3 | No | [177] |
| Influenza virus | RNA, M2 ion channel | NLRP3 | Yes | [177–181] |
| Adenovirus | DNA | NLRP3 | Yes | [182] |
| Vaccinia virus | DNA, RNA | AIM2 | No | [23,155] |
| Mouse cytomegalovirus | DNA | AIM2 | Yes | [155] |
| Vesicular stomatis virus | 5′-triphosphate ssRNA | RIG-Ic, NLRP3 | Yes. Not important in vivo | [24,183] |
| Encephalomyocarditis virus | RNA | NLRP3 | Yes. Not important in vivo | [24,183] |
Flagellin importance only determined.
In a Plasmodium berghei mouse model, NLRP3 has been suggested to protect from cerebral malaria in a NLRP3-dependent, but caspase-1, apoptosis speck protein (ASC) and interleukin (IL)-1β-independent manner [184].
The requirement of retinoid-inducible gene 1 (RIG-I) for vesicular stomatis virus (VSV)-induced IL-1 is unclear as, unlike the initial finding, it was reported recently to not be required [183].
PAMP: pathogen-associated molecular pattern; ESX: early secreted antigenic target, 6 kDa secretion system; MSU: monosodium urate; n.d.: not determined; n.a.: not applicable.
The best-characterized NLR capable of forming an inflammasome complex is NLRP3 (Cryopyrin/Nalp3/Cias1/Pypaf1). The extraordinary number of NLRP3 activators suggests that it may be a general detector of cellular stresses resulting from sterile trauma, intrinsic metabolic disturbances or pathogen infection (Fig. 2). Some of the host-derived DAMPs that activate NLRP3 include hyaluronan, cholesterol crystals, extracellular ATP, β-amyloid, DNA and gout-associated monosodium urate crystals, while environmental DAMPs include asbestos, silica, nanoparticles, skin irritants and alum adjuvant. PAMPs that stimulate NLRP3 can include pathogen-associated RNA, DNA, pore-forming toxins and peptidoglycans. While potassium efflux from the cell is a general requirement for the activation of the NLRP3 inflammasome, this is not required for other inflammasomes such as the one assembled around NLRC4 [25].
Fig. 2.
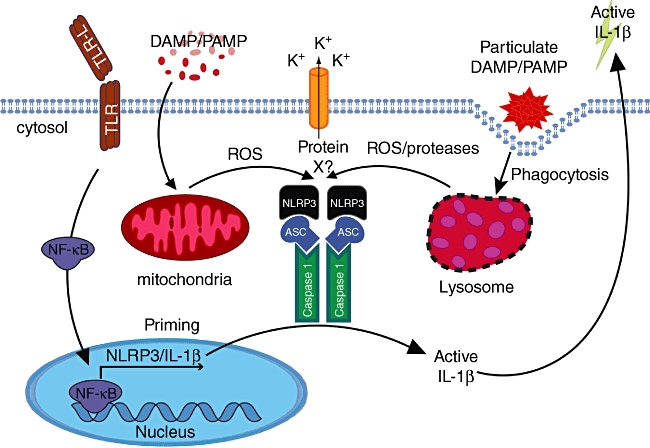
Models for nucleotide-binding domain (NOD)-like receptor protein 3 (NLRP3) inflammasome activation. Cells, typically macrophages or dendritic cells, must first be ‘primed’ by an appropriate nuclear factor kappa B (NF-κB) transcription factor activating stimuli to induce precursor interleukin (IL)-1β expression. This often occurs by activation of a Toll-like receptor (TLR) family member (depicted), tumour necrosis factor (TNF) receptor activation, or even IL-1R activation (not depicted). NLRP3 stimulants all induce K+ efflux. Although this is essential for NLRP3 activity, why and how this occurs remains a mystery. Pore-forming toxins and cell permeable NLRP3 activators can also result in mitochondrial reactive oxygen species (ROS) production that may amplify or induce NLRP3 activity, possibly via an undetermined intermediate (protein X). Particulate or crystalline matter is phagocytosed and leads to endosomal/lysosomal damage, resulting in the activation/release of lysosomal proteases (i.e. cathepsins) that can cause NLRP3 induced caspase-1 activity, which may also depend on mitochondrial ROS induction and/or processing of a NLRP3 inhibitory or activating ligand.
Stress-induced reactive oxygen species (ROS) production has been hypothesized as a common denominator that may determine NLRP3 activation status either directly via oxidation, or through an as-yet undetermined intermediate (Fig. 2) (reviewed in [26]). Other data suggest that lysosomal rupture and release of intracellular proteases, such as cathepsins, may activate NLRP3 (reviewed in [27]). This ‘frustrated phagocytosis’ scenario has been implicated in NLRP3 activation from particulate or crystalline stimulants, where their uptake leads to lysosomal damage and release of potential NLRP3 activating proteases [28,29]. While cathepsin B has been reported as taking part in NRLP3 activation based on chemical inhibitor studies, mice lacking this gene show normal NLRP3 activity in response to several NLRP3 stimuli [30]. Therefore, how lysosomal protease release/activation could induce NLRP3 activity remains unclear, although in theory it could result from the cleavage of a NLRP3 ligand or inhibitory protein [27].
While most inflammasome activators induce cellular ROS and, conversely, ROS scavenging compounds inhibit NLRP3 inflammasome function, the specificity and targets of these compounds is often questionable. Further, many ROS inducing cellular processes do not result in inflammasome activation, and the targeted deletion of ROS-producing or inhibitory genes, arguably the gold standard in analysing cellular function, has yet to result in a block in NRLP3 inflammasome function. In fact, it was demonstrated that deletion of the ROS scavenger superoxide dismutase (SOD) resulted in enhanced IL-1β activation, and it was suggested that ROS oxidizes specific caspase-1 cysteine residues resulting in caspase-1 inhibition [31]. Similarly, monocytes from chronic granulomatous disease patients who lack NADPH oxidase activity, and its associated ROS production, display increased IL-1β maturation in response to NLRP3 stimulants [32,33].
Recently it has been proposed that the mitochondria, and not NADPH oxidase activity, may be the source of ROS required for NLRP3 activation (Fig. 2) [34]. In support of this, it was reported that monocytes from patients who harbour missense mutations in TNFR1 and suffer from an autoinflammatory condition termed TNF-receptor-associated periodic syndrome (TRAPS) display increased mitochondrial respiratory capacity and ROS generation compared to normal monocytes, leading to enhanced cytokine, including IL-1β, production [35]. It will be interesting to determine if monocytes from patients harbouring activating NLRP3 mutations also display enhanced mitochondrial respiration and ROS and if this effects IL-1β maturation [35].
Understandably, a variety of both host and pathogen mechanisms have evolved to inhibit inflammasome activity. Several viral POPs (Pyrin domain-only proteins) and serpins (protease inhibitors) have been identified that can bind and inhibit caspase-1, while bacteria have been shown to interfere with caspase-1 function via type III secretion effector molecules or pore-forming virulence factors (reviewed in [36]). Host inhibitors include a secreted IL-1 receptor antagonist (Il-1Ra) that competes for receptor binding with IL-1, and an IL-1 decoy receptor (IL-1RII). Deletion of IL-1Ra in mice increases IL-1β serum levels and leads to severe inflammatory distress and disease phenotypes resembling rheumatoid arthritis [37–39]. Mutations in Pyrin cause familial Mediterranean fever and increased IL-1β activation is also observed in Pyrin knock-out mice [40]. The exact mechanism for Pyrin inhibition of inflammasome function remains unclear, although it is likely to compete with caspase-1 for binding to ASC [40]. Sequestration or inhibition of ASC and caspase-1 can also be achieved through the Pyrin or CARD-only protein families in humans (reviewed in [41]). TNF superfamily ligand stimulation, autophagy and type I IFN treatment can also result in diminished inflammasome function, although how they do so is still being unravelled [42–45].
The bad: cryopyrin-associated periodic syndromes and autoinflammatory disorders
The NLRP3 inflammasome has attracted considerable attention ever since its initial characterization due to its implication in the pathogenesis of several human inflammatory diseases [6]. In particular, mutated NLRP3 has been identified as the cause of a group of inflammatory diseases known as the cryopyrin-associated periodic syndromes (CAPS).
Cryopyrin-associated periodic syndromes
Gain of function mutations in the NACHT domain of NLRP3 were identified as being responsible for three chronic aseptic inflammatory diseases: familial cold autoinflammatory syndrome (FCAS), Muckle–Wells syndrome (MWS) and neonatal onset multi-systemic inflammatory disease/chronic infantile neurological cutaneous articular syndrome (NOMID/CINCA) [46]. These closely related conditions form the family of cryopyrin-associated periodic syndromes (CAPS), a name derived from the original designation of the NLRP3-encoding gene, then known as cryopyrin. Together, they constitute a spectrum of diseases with increasing severity, with FCAS, MWS and NOMID/CINCA representing the mildest, intermediate and most severe forms, respectively [47]. Clinically, they are characterized by recurrent fevers, urticarial-like skin rashes, joint and ocular symptoms, amyloidosis and, in the case of NOMID/CINCA, severe neurological complications [47,48] (Table 2).
Table 2.
The bad I: cryopyrin-associated periodic syndromes
| FCAS | MWS | NOMID/CINCA | |
|---|---|---|---|
| Inheritance | Autosomal dominant | Usually autosomal dominant, rare sporadic cases | Mostly sporadic, rarely autosomal dominant |
| Onset of disease | Neonatal or infancy (< 6 months) | Variable, usually infancy, up to adolescence | Neonatal or early infancy |
| Flares | Cold-triggered, usually 1–2 h after exposure, episode < 24 h | No clearly identified trigger (possibly cold exposure, stress). Spectrum of continuous symptoms to recurrent flares lasting 1–3 days | Usually continuous symptoms, with aggravation during flares |
| Relative severity | Low | Medium | High |
| Cardinal symptoms | Cold-induced fever, urticarial-like skin rash, arthralgia, conjunctivitis, sweating, fatigue, extreme thirst, nausea | Recurrent low-grade fever, urticarial-like skin rash, sensorineural hearing loss, arthritis, conjunctivitis, AA amyloidosis | Inconstant mild fever, urticarial-like skin rash, chronic aseptic meningitis, sensorineural hearing loss |
| Skin | Maculopapular, migratory, urticarial-like, usually non-pruritic skin rash | ||
| Joints | Arthralgia | Arthralgia, arthritis | Arthralgia, arthritis, bony overgrowth arthropathy of the knees (25–50%) |
| Eyes | Conjunctivitis | Conjunctivitis, uveitis | Conjunctivitis, uveitis (75%), chronic papilledema possibly progressing to blindness |
| Sensorineural hearing loss | Rare | Frequent (75%), onset during adolescence | Frequent (50%), onset during childhood |
| Central nervous system | None | None | Severe headaches, high intracranial pressure, chronic aseptic meningitis, possibly leading to mental retardation |
| Amyloidosis | Rare (< 2%) | Frequent (25–50%), adult-onset | Frequent (25%), adult-onset |
FCAS: familial cold autoinflammatory syndrome; MWS: Muckle–Wells syndrome; NOMD/CINCA: neonatal onset multi-systemic inflammatory disease/chronic infantile neurological cutaneous articular syndrome; AA amyloidosis: amyloid AA amyloidosis.
In FCAS, MWS and NOMID/CINCA, several point mutations target the NACHT domain of the NLRP3 protein, resulting in gain-of-function mutations that constitutively activate the NLRP3 inflammasome [49,50]. In accordance, monocytes and macrophages isolated from MWS patients display a basal, spontaneous secretion of mature IL-1β in the absence of any external stimulus [49]. Collectively, these observations provided a convincing rationale for a novel therapeutic approach to these patients, namely through the inhibition of inflammasome activity. Strikingly, treating these patients with anakinra, a non-glycosylated recombinant form of the naturally occurring IL-1 receptor antagonist (IL-1Ra) which blocks inflammasome-dependent IL-1β signalling, resulted in a complete cessation of clinical symptoms and biochemical changes within hours of administration [51,52]. The efficacy of anti-IL-1β therapy was demonstrated across the CAPS spectrum, even in children with the more severe phenotype of NOMID/CINCA [53,54].
The clinical availability of IL-1 inhibitors has thus been critical to the identification of the NLRP3 inflammasome as a critical regulator of inflammation in vivo. The prototypic anti-IL-1β therapy has been based so far on the daily subcutaneous injection of anakinra [55] (Table 3). Anakinra was first developed as a promising therapy for sepsis and septic shock [56], but subsequent large-scale studies have been disappointing in that regard [57–59]. However, in 2001 it was approved by the Food and Drug Administration (FDA) as a second-line treatment for rheumatoid arthritis patients suffering from a moderate to severe form of the disease that is unresponsive to at least one disease modifying anti-rheumatic drug (DMARD) therapy.
Table 3.
Current approved anti-interleukin (IL)-1 therapies
| Anakinra (Kineret) | Rilonacept (Arcalyst) | Canakinumab (Ilaris) | |
|---|---|---|---|
| Nature | Recombinant IL-1Ra | Cytokine trap (fusion protein including IL-1R and IL-1Ra) | Humanized monoclonal antibody against IL-1β |
| Target | IL-1R | IL-1β (and IL-1α, IL-1RAcP) | IL-1β |
| Half-life | 4–6 h | 8·6 days | 26 days |
| Administration | 100 mg, daily, subcutaneous | 320 mg loading dose then 160 mg maintenance, weekly, subcutaneous | 150 mg, every 8 weeks, subutaneous (or intravenous) |
| Common side-effects | Major injection site reactions, URI, infections | Minor injection site reactions, URI, infections | URI, vertigo (MWS patients), negligible injection site reactions, |
| Indications | Second-line therapy for RA (FDA 2001, EMEA 2002). CAPS (off-label use) | FCAS and MWS patients aged more than 12 years (FDA 2008, EMEA 2009) | FCAS and MWS patients aged more than 4 years (FDA and EMEA 2009) |
IL-1Ra: IL-1 receptor antagonist; IL-1R: IL-1 receptor; IL-1RAcP: IL-1 receptor accessory protein; URI, upper respiratory tract infections; CAPS; cryopyrin-associated periodic syndromes; RA: rheumatoid arthritis; FCAS: familial cold autoinflammatory syndrome; MWS: Muckle–Wells syndrome; FDA: Food and Drug Administration; EMEA: European Medicines Agency. Regarding NOMID/CINCA: the only currently approved therapy is Canakinumab (approved by the EMEA but not the FDA).
In preliminary clinical studies, anakinra has been widely successful in treating CAPS patients. Indeed, all cases reported in the literature to date responded to the therapy, albeit with various dosage requirements [47]. Overall, anakinra has a perhaps surprisingly robust record of safety [60,61], considering the prominent role of IL-1β in inflammatory and immunological responses. Conversely, patient tolerance for anakinra is often poor, due to substantial morbidity associated with injection-site reactions and upper respiratory tract infections.
Recently, two additional IL-1 antagonists have been approved for clinical use in CAPS patients (Table 3). Rilonacept is a dimeric fusion glycoprotein that acts as a soluble decoy receptor with high affinity for both IL-1α and IL-1β[62]. It has proved effective in treating FCAS and MWS patients in two clinical studies [62,63]. Rilonacept is administered subcutaneously once-weekly due to its longer half-life, and presents a safety profile comparable to that of anakinra.
Canakinumab is a fully humanized monoclonal antibody against IL-1β with a half-life of about 4 weeks that has proved safe and effective in treating CAPS patients [64]. Both the above compounds are attractive additional anti-IL-1β therapies because of their weekly (rilonacept) or bimonthly (canakinumab) schedule of administration, in contrast to the requirement for daily injections of anakinra.
Autoinflammatory syndromes
Cryopyrin-associated periodic syndromes are part of the greater family of hereditary autoinflammatory syndromes, a concept first proposed in 1999 to describe a group of inherited disorders characterized by recurrent attacks of fever and multi-systemic inflammation [65]. In contrast to autoimmune diseases, autoinflammatory disorders lack high-titre autoantibodies or antigen-specific T cells [66]. The NLRP3 inflammasome has been implicated in the pathogenesis of several additional autoinflammatory syndromes aside from CAPS, on the basis of their favourable response to anti-IL-1β therapy [6,50] (Table 4).
Table 4.
Additional autoinflammatory syndromes possibly linked to dysregulated inflammasome activity
| Disease | Cause | Cardinal symptoms | Current treatment | Successful anti-IL-1 therapy |
|---|---|---|---|---|
| Hyper IgD with periodic fever syndrome | Mutations in mevalonate kinase | Recurrent fever, lymphadenopathy, abdominal pain, diarrhoea, headaches, hepatosplenomegaly, arthralgia, skin rash (< 1 week) | None, supportive care | [185–187] |
| TNF receptor-associated periodic syndrome | Mutations in TNR receptor I | Recurrent fever, abdominal pain, severe myalgia, painful skin rash (> 1 week) | NSAIDs, corticosteroids, etanercept | [188] |
| Systemic juvenile idiopathic arthritis | Unknown | Daily recurring fever, anaemia, hepatosplenomegaly, macular salmon-coloured skin rash of trunk and extremities, myalgia, arthritis (late symptom) | NSAIDs, corticosteroids, DMARDs | [189] |
| Adult-onset Still's disease | Unknown | Daily recurring fever, hepatosplenomegaly, arthritis, salmon-colored skin rash of trunk and extremities, myalgia | NSAIDs, corticosteroids, DMARDs | [189] |
| Relapsing polychondritis | Unknown | Intermittent fever, skin rash, auricular / nasal / respiratory tract chondritis, ocular inflammation, arthritis, audiovestibular damage | Systemic corticosteroids, methotrexate | [190,191] |
| Schnitzler's syndrome | Unknown | Chronic urticarial skin rash, recurrent fever, arthralgia, myalgia | Anakinra | [192] |
| Sweet syndrome | Unknown, neutrophil-dependent | Fever, skin lesions (violet papules, plaques or nodules), pulmonary symptoms (dyspnoea, cough) | Corticosteroids | [193] |
| Behçet's disease | Unknown | Painful oral aphtous ulcers, painful genital ulcers, uveitis | Corticosteroids | [194,195] |
| Anti-synthetase syndrome | Unknown | Myositis, interstitial lung disease, arthritis, fever, Raynaud's phenomenon | Corticosteroids | [196] |
NSAIDs: non-steroidal anti-inflammatory drug; DMARD: disease-modifying anti-rheumatic drug; IL: interleukin; IgD: immunoglobulin D; TNF: tumour necrosis factor; TNR: tenascin R.
Familial Mediterranean fever (FMF) and pyogenic arthritis, pyoderma gangrenosum and acne (PAPA) syndrome are caused by mutations in the genes encoding for Pyrin and PSTPIP1, respectively [50]. While FMF is an autosomal recessive disease characterized by episodes of fever lasting 1–3 days associated with severe abdominal pain, pleuritic chest pain, arthritis and a skin rash [67], PAPA syndrome is inherited in a dominant manner, and characterized by recurrent pyogenic but sterile arthritis coupled to skin manifestations such as pyoderma gangrenosum and acne [68,69]. Interestingly, Pyrin has been shown to interact with NLRP3 inflammasome components, and the mutated form responsible for FMF was found to result in NLRP3 inflammasome activation [70]. Moreover, PSTPIP1 was shown to associate with Pyrin, and mutated forms responsible for PAPA syndrome had a higher affinity for Pyrin [71]. Taken together, these findings suggest that both FMF and PAPA syndrome might be inflammasome-dependent pathologies, and the term of ‘extrinsic inflammasomopathies’ has thus been proposed [50]. Accordingly, IL-1 antagonism has proved to be an effective treatment for both conditions [72–74]. Other autoinflammatory disorders have been linked to dysregulated inflammasome activity on the basis of their favourable response to anti-IL-1 therapy; however, in most of these cases the molecular basis for the putative link is largely unclear (Table 4).
The ugly: common human diseases linked to NLRP3 inflammasome activity
In addition to its central role in the pathogenesis of autoinflammatory disorders, the NLRP3 inflammasome has emerged recently as an unexpected sensor for metabolic danger and stress [75,76]. Indeed, it has been implicated in the development of major diseases such as gout, type 2 diabetes and obesity-induced insulin resistance. Moreover, the NLRP3 inflammasome is increasingly suspected of playing a major role in other human pathologies such as cancer, asbestosis and Alzheimer's disease.
Gout
Gout is a sterile inflammatory disease caused by monosodium urate (MSU) crystal deposition in various tissues. The prototypical clinical manifestation is acute monoarthritis, where MSU crystals precipitate in the joint, triggering an acute local inflammatory response [77]. MSU crystals were demonstrated to specifically activate the NLRP3 inflammasome, both in vitro and in vivo[78]. Uric acid is normally produced as the end result of the metabolic pathway governing the degradation of purines, and hyperuricaemia is thus a main risk factor for the development of gout [77]. Taken together, this suggests that the NLRP3 inflammasome has evolved as a sensor of metabolic endogenous danger, in addition to its pathogen-detecting functions.
Excitingly, preliminary clinical trials involving in vivo IL-1β blockade by anakinra or rilonacept in gout patients demonstrated high efficacy and the absence of adverse effects [79,80]. These findings require confirmation in large-scale controlled studies, and it will be interesting to see whether long-acting therapies such as canakinumab are able to tame chronic gout flares over time. Of special interest, anti-IL-1 therapy might be attractive to patients for which mainstream gout therapies are inefficient or contraindicated [81].
Type II diabetes
Another key metabolic danger signal resides in chronically elevated blood sugar levels and associated insulin resistance, which are hallmarks of type 2 diabetes. During recent years there has been a growing interest in the inflammatory component of the disease [75], and in particular in the role of IL-1β[82]. Indeed, IL-1β has been proposed to play a critical role in the loss of β cell mass in the course of type 2 diabetes [83], and a current hypothesis suggests that the relative balance between IL-1β and endogenous IL-1Ra regulates pancreatic islet inflammation associated with the disease [84]. Remarkably, a recent clinical trial supports the notion that IL-1β is indeed a key player in type 2 diabetes, as patients receiving IL-1β antagonists featured improved glycaemic control and β cell mass [85]. Notably, diabetic markers such as increased levels of saturated fatty acids and islet-derived amyloid polypeptide have been reported as capable of activating the NLRP3 inflammasome [86,87], and NLRP3- and ASC-deficient mice fed a high-fat diet display improved insulin sensitivity when compared to control mice [86].
Obesity-induced insulin resistance
Further experimental data suggest that the NLRP3 inflammasome is an important regulator of adipocyte differentiation and insulin sensitivity [88]. Adipocytes are rendered more metabolically active and insulin-sensitive upon NLRP3 inflammasome inhibition in murine models of obesity [88]. Strikingly, calorie restriction and exercise-mediated weight loss in obese type 2 diabetes patients is associated with a decreased NLRP3 expression in adipose tissue, coupled to decreased inflammation and improved insulin sensitivity [89]. Collectively, these findings suggest that the NLRP3 inflammasome is able to sense obesity-associated danger signals and contribute to the development of inflammation and insulin resistance [89].
Cancer
The tumour microenvironment has been likened to a non-resolving wound response, with an inflammatory milieu capable of stimulating tumour survival, growth, angiogenesis, invasion and metastasis, immune suppression and genetic mutation [90,91]. Studies suggest that IL-1, like the other key proinflammatory cytokine TNF, is often associated with tumour promotion. An evaluation of several clinical trials using recombinant IL-1β or IL-1α showed that neither had any significant therapeutic benefit when used alone against ovarian cancer, renal cell carcinoma or melanoma, and the toxicity associated with IL-1 administration is likely to outweigh any potential benefits [4].
However, immune-mediated anti-tumour responses resulting from the production of IL-1β or IL-1α have been documented. Early reports showed that IL-1 treatment (alone or in combination with chemotherapeutic treatment) of cancer cell lines or murine syngenic tumours resulted in decreased tumour cell growth and often promoted tumour regression [92–94]. Similarly, when IL-1α transgenic mice expressing 17 kDa IL-1α under the keratin 14 promoter were treated with DMBA/TPA (7,12-dimethylbenzanthracene/12-O-tetradecanoylphorbol-13-acetate), or crossed to mutant Ha-Ras expressing mice, the IL-1α expressing mice were completely resistant to papilloma and carcinoma formation due to enhanced acute inflammatory responses [95]. More recently it was also demonstrated that the NRLP3 inflammasome and subsequent IL-1β priming of T cells is critical for immune-mediated eradication of tumours following chemotherapy [96].
Several groups have also examined the role of NLRP3 and caspase-1 in inflammatory bowel diseases using the dextran sulphate sodium (DSS) mouse model of colitis. Ulcerative colitis and Crohn's disease predispose to colorectal cancer, where an inappropriate inflammatory response to commensal bacteria is believed to play a major role in the neoplastic transformation of the intestinal epithelium. Two studies have suggested that caspase-1 or NLRP3 deficiency leads to reduced colitis severity in DSS-treated mice when compared to wild-type mice [97,98]. However, opposing results reported by several groups showed that NLRP3, ASC and caspase-1 knock-out mice are all more susceptible to DSS-induced colitis and death [99–101]. In inflammasome-deficient mice, it was reported that a lack of IL-18 activation prevented the repair of the mucosal barrier following DSS-induced damage, resulting in systemic commensal bacterial spread [101]. It was also demonstrated that colitis-associated cancer, induced by DSS and azoxymethane, is enhanced significantly upon genetic deletion of either NLRP3, caspase-1 or ASC, while the role of NLRC4 remains controversial [102–104]. It has been observed previously that other TLR/IL-1R family signalling members, such as myeloid differentiation factor 88 (MydD88), also protect from DSS-associated colitis and intestinal tumorigenesis [105–107]. Therefore, the accumulative evidence suggests that appropriate innate immune signalling responses to commensal bacteria, mediated at least in part by the NLRP3 inflammasome, are a general requirement for intestinal homeostasis. Consistent with this notion, single nucleotide polymorphisms (SNPs) within the NLRP3 region that result in decreased NLRP3 expression have been identified as contributing to Crohn's disease susceptibility, suggesting that the NRLP3 inflammasome may also play a protective role in inflammatory bowel disease in humans [108].
Ulcerative colitis results from hyper-responsive inflammation, and in this context it has been demonstrated that excessive IL-1β and IL-18 production can also contribute to DSS-induced colitis and possibly cancer, as was observed when the autophagy gene ATG16L1, or the caspase-1 negative regulator, caspase-12, were deleted [43,101]. Therefore, the NLRP3 inflammasome may play an important role in cellular repair and regeneration following acute tissue damage, but if tissues are exposed to chronic or excessive inflammasome activity, NLRP3 stimulation is likely to enhance neoplastic processes.
Despite its ability to promote an immune cell-mediated anti-tumour response, high levels of IL-1 in the tumour microenvironment often correlate with a poor prognosis (reviewed in [109]). Tumour-associated macrophages and dendritic cells are likely to contribute to IL-1β levels within the tumour infiltrate, while some cancer cell lines, such as those derived from myeloma, melanoma and acute myeoblastic leukaemia, can produce active IL-1β constitutively which can contribute towards tumour cell growth and invasiveness [110–112]. It is notable that some common oncogenes, such as Ras, can induce IL-1β expression [113] and IL-1β is a known target for the transcription factor NF-κB, which is activated in many neoplastic malignancies.
IL-1 receptor signalling can induce either directly or indirectly the production genes that stimulate tumour growth, angiogenesis and metastasis [i.e. IL-6, IL-8, TNF, matrix metalloproteinases (MMPs), basic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF), vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), monocyte chemotactic protein-1 (MCP-1), CXCL-2]. Melanoma cells expressing high levels of IL-1β show reduced tumour growth and metastases when treated with IL-1Ra in murine xenograft experiments [114]. Similarly, Lewis lung cell carcinoma cells engineered to produce IL-1β showed increased tumour growth and increased expression of angiogenic factors when implanted into mice [115]. In other mouse models, murine B16 melanoma growth, invasiveness, lung metastasis and stimulation of angiogenesis is severely attenuated in IL-1β, and to a lesser extent IL-1α, knock-out mice [116], an affect which anakinra treatment appears to recapitulate somewhat in B16 melanoma injected wild-type mice [117]. Similarly, chemically induced skin carcinogenesis is severely compromised in IL-1β knock-out mice and, conversely, tumour growth accelerated upon genetic deletion of IL-1Ra [118]. The mechanisms and potential contribution of different inflammasome(s) in IL-1β activation in murine cancer models has yet to be examined in detail, although caspase-1 function does contribute to B16 melanoma hepatic metastasis [119].
Cells from acute myeloid leukaemia (AML) patients can produce and secrete IL-1β and show substantially reduced proliferation and decreased growth factor levels [i.e. granulocyte–macrophage colony-stimulating factor (GM-CSF)] when treated with IL-1Ra, although in a subpopulation of patients AML cells may also proliferate when exposed to IL-1Ra [112,120–123]. A Phase I safety trial reported no responses in patients with refractory or relapsed AML when treated with soluble decoy human IL-1R [124]. However, it was noted that the decoy IL-1R serum levels were below those that completely blocked AML cell growth in vitro and were likely to be even lower within marrow. It may therefore be worth revisiting the effects of IL-1 blockade on AML in the clinic using more efficacious IL-1 inhibitors.
Evidence for the tumorigenic role of IL-1β also comes from its association with gastric cancer, the second deadliest form of cancer worldwide after lung cancer [125]. IL-1β is induced by Helicobacter pylori within the gastric mucosa and is a potent inhibitor of gastric acid secretion, which may lead to gastric atrophy, a precursor of gastric cancer. In 2000, Rabkin et al. described IL-1 gene cluster polymorphisms that correlated with a predisposition to hypochlorhydria, gastric atrophy and gastric cancer in humans infected with H. pylori[126]. Several studies in different human populations have since confirmed these observations (reviewed in [127]), and mice engineered to express IL-1β in the stomach develop gastric inflammation and cancer [128]. However, it is still unclear how the human polymorphisms affect IL-1β production and which, if any, inflammasomes are involved.
The expression of IL-1β by either myeloma cells or innate immune cells has been associated for some time with the induction of IL-6, a key growth factor that promotes myeloma cell survival and proliferation. Recent clinical trials using IL-1Ra (combined with low-dose dexamethasone) demonstrated that IL-1 inhibition induced a chronic disease state in smouldering or indolent multiple myeloma patients, and substantially improved progression-free survival by preventing the transition to active multiple myeloma [129]. This represents the first demonstration of the therapeutic benefit of IL-1 inhibition in a human cancer.
Given the general safety of inhibiting IL-1 in vivo, and its probable role in cancer metastasis, future clinical trials examining IL-1 inhibition in cancer are deemed warranted [130]. It will also be important to determine the mechanisms and inflammasomes by which cancer cells directly or indirectly modulate IL-1β activity.
Other diseases
Numerous NLRP3 inflammasome activators have been identified and characterized in vitro[6]. In some cases, they have pointed to the unexpected implication of the NLRP3 inflammasome in the pathogenesis of various inflammatory diseases. For example, asbestosis and silicosis have been shown to activate the NLRP3 inflammasome in murine models of chronic pulmonary fibrotic disorders [131,132], raising the intriguing possibility that IL-1β may contribute to inflammation-induced lung cancer, and that anti-IL-1β therapy might be beneficial for patients suffering from these diseases. The fibrillar peptide amyloid-β, which plays a key function in the development of Alzheimer's disease, was also shown to activate the NLRP3 inflammasome [28]. Moreover, the NLRP3 inflammasome was suggested to be instrumental in the inflammatory component of the disease and its associated brain tissue damage [28]. In the skin, NLRP3 inflammasome activation has been linked to UVB-induced damage [133,134] and contact hypersensitivity [133,135]. Recent studies have shown that haemozoin, a crystal produced by plasmodium species in the course of malarial infection, activates the NLRP3 inflammasome [30,136]. More surprising still, non-coding NLRP3 mutations were linked to essential hypertension susceptibility, possibly due to increased expression of the protein [137].
In most of theses cases, including cancer, the evidence pointing to an involvement of the NLRP3 inflammasome in disease development in vivo remains preliminary and awaits further confirmation. Collectively, however, they stand as a testimony to the impressive versatility of the NLRP3 inflammasome as a danger-detection system with potentially far-reaching implications for human health.
Acknowledgments
James E. Vince is funded by an Australian NHMRC C. J. Martin Fellowship. This work was supported by the Swiss National Science Foundation through an MD–PhD grant to Philippe Menu. Thanks to Holly Anderton and James Rickard for their help with the manuscript. This review is dedicated to the memory of Professor Jürg Tschopp.
Disclosure
The authors have no conflicts of interest to declare.
References
- 1.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 2.Thornberry NA, Bull HG, Calaycay JR, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–74. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 3.Dinarello CA. IL-1: discoveries, controversies and future directions. Eur J Immunol. 2010;40:599–606. doi: 10.1002/eji.201040319. [DOI] [PubMed] [Google Scholar]
- 4.Veltri S, Smith JW., II. Interleukin 1 trials in cancer patients: a review of the toxicity, antitumor and hematopoietic effects. Stem Cells. 1996;14:164–76. doi: 10.1002/stem.140164. [DOI] [PubMed] [Google Scholar]
- 5.Ting JP, Lovering RC, Alnemri ES, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28:285–7. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 7.Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T, Alnemri ES. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J Biol Chem. 2001;276:28309–13. doi: 10.1074/jbc.C100250200. [DOI] [PubMed] [Google Scholar]
- 8.Faustin B, Lartigue L, Bruey J-M, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25:713–24. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 9.Duncan JA, Bergstralh DT, Wang Y, et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci USA. 2007;104:8041–6. doi: 10.1073/pnas.0611496104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guarda G, Zenger M, Yazdi AS, et al. Differential expression of NLRP3 among hematopoietic cells. J Immunol. 2011;186:2529–34. doi: 10.4049/jimmunol.1002720. [DOI] [PubMed] [Google Scholar]
- 11.Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–31. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 12.Luthi AU, Cullen SP, McNeela EA, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31:84–98. doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 13.Stehlik C. Multiple interleukin-1beta-converting enzymes contribute to inflammatory arthritis. Arthritis Rheum. 2009;60:3524–30. doi: 10.1002/art.24961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miwa K, Asano M, Horai R, Iwakura Y, Nagata S, Suda T. Caspase 1-independent IL-1beta release and inflammation induced by the apoptosis inducer Fas ligand. Nat Med. 1998;4:1287–92. doi: 10.1038/3276. [DOI] [PubMed] [Google Scholar]
- 15.Joosten LA, Netea MG, Fantuzzi G, et al. Inflammatory arthritis in caspase 1 gene-deficient mice: contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum. 2009;60:3651–62. doi: 10.1002/art.25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M, Corr M. Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum. 2009;60:3642–50. doi: 10.1002/art.24959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mayer-Barber KD, Barber DL, Shenderov K, et al. Caspase-1 independent IL-1beta production is critical for host resistance to mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol. 2010;184:3326–30. doi: 10.4049/jimmunol.0904189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narayan S, Pazar B, Ea HK, et al. Octacalcium phosphate (OCP) crystals induce inflammation in vivo through IL-1 but independent of the NLRP3 inflammasome. Arthritis Rheum. 2011;63:422–33. doi: 10.1002/art.30147. [DOI] [PubMed] [Google Scholar]
- 19.Fantuzzi G, Ku G, Harding MW, et al. Response to local inflammation of IL-1 beta-converting enzyme-deficient mice. J Immunol. 1997;158:1818–24. [PubMed] [Google Scholar]
- 20.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–7. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts TL, Idris A, Dunn JA, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–60. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 22.Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–13. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hornung V, Ablasser A, Charrel-Dennis M, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–18. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poeck H, Bscheider M, Gross O, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2009;11:63–9. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- 25.Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–9. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 26.Martinon F. Signaling by ROS drives inflammasome activation. Eur J Immunol. 2010;40:616–19. doi: 10.1002/eji.200940168. [DOI] [PubMed] [Google Scholar]
- 27.Hornung V, Latz E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur J Immunol. 2010;40:620–3. doi: 10.1002/eji.200940185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–65. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–56. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dostert C, Guarda G, Romero JF, et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One. 2009;4:e6510. doi: 10.1371/journal.pone.0006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meissner F, Molawi K, Zychlinsky A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat Immunol. 2008;9:866–72. doi: 10.1038/ni.1633. [DOI] [PubMed] [Google Scholar]
- 32.van de Veerdonk FL, Smeekens SP, Joosten LA, et al. Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc Natl Acad Sci USA. 2010;107:3030–3. doi: 10.1073/pnas.0914795107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood. 2010;116:1570–3. doi: 10.1182/blood-2010-01-264218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–5. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 35.Bulua AC, Simon A, Maddipati R, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS) J Exp Med. 2011;208:519–33. doi: 10.1084/jem.20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taxman DJ, Huang MT, Ting JP. Inflammasome inhibition as a pathogenic stealth mechanism. Cell Host Microbe. 2010;8:7–11. doi: 10.1016/j.chom.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma Y, Thornton S, Boivin GP, Hirsh D, Hirsch R, Hirsch E. Altered susceptibility to collagen-induced arthritis in transgenic mice with aberrant expression of interleukin-1 receptor antagonist. Arthritis Rheum. 1998;41:1798–805. doi: 10.1002/1529-0131(199810)41:10<1798::AID-ART11>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 38.Horai R, Saijo S, Tanioka H, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000;191:313–20. doi: 10.1084/jem.191.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nicklin MJ, Hughes DE, Barton JL, Ure JM, Duff GW. Arterial inflammation in mice lacking the interleukin 1 receptor antagonist gene. J Exp Med. 2000;191:303–12. doi: 10.1084/jem.191.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chae JJ, Komarow HD, Cheng J, et al. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol Cell. 2003;11:591–604. doi: 10.1016/s1097-2765(03)00056-x. [DOI] [PubMed] [Google Scholar]
- 41.Stehlik C, Dorfleutner A. COPs and POPs: modulators of inflammasome activity. J Immunol. 2007;179:7993–8. doi: 10.4049/jimmunol.179.12.7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masters SL, Mielke LA, Cornish AL, et al. Regulation of interleukin-1beta by interferon-gamma is species specific, limited by suppressor of cytokine signalling 1 and influences interleukin-17 production. EMBO Rep. 2010;11:640–6. doi: 10.1038/embor.2010.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–8. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 44.Guarda G, Dostert C, Staehli F, et al. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature. 2009;460:269–73. doi: 10.1038/nature08100. [DOI] [PubMed] [Google Scholar]
- 45.Guarda G, Braun M, Staehli F, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–23. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 46.Aganna E, Martinon F, Hawkins PN, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum. 2002;46:2445–52. doi: 10.1002/art.10509. [DOI] [PubMed] [Google Scholar]
- 47.Hoffman HM. Therapy of autoinflammatory syndromes. J Allergy Clin Immunol. 2009;124:1129–38. doi: 10.1016/j.jaci.2009.11.001. quiz 39–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neven B, Prieur A-M, Quartier dit Maire P. Medscape, cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol. 2008;4:481–9. doi: 10.1038/ncprheum0874. [DOI] [PubMed] [Google Scholar]
- 49.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity. 2004;20:319–25. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 50.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*) Annu Rev Immunol. 2009;27:621–68. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hawkins PN, Lachmann HJ, Aganna E, Mcdermott MF. Spectrum of clinical features in Muckle–Wells syndrome and response to anakinra. Arthritis Rheum. 2004;50:607–12. doi: 10.1002/art.20033. [DOI] [PubMed] [Google Scholar]
- 52.Hawkins PN, Lachmann HJ, McDermott MF. Interleukin-1-receptor antagonist in the Muckle–Wells syndrome. N Engl J Med. 2003;348:2583–4. doi: 10.1056/NEJM200306193482523. [DOI] [PubMed] [Google Scholar]
- 53.Cook GP, Savic S, Wittmann M, Mcdermott MF. The NLRP3 inflammasome, a target for therapy in diverse disease states. Eur J Immunol. 2010;40:631–4. doi: 10.1002/eji.200940162. [DOI] [PubMed] [Google Scholar]
- 54.Goldbach-Mansky R, Dailey NJ, Canna SW, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355:581–92. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 56.Fisher CJ, Jr, Opal SM, Lowry SF, et al. Role of interleukin-1 and the therapeutic potential of interleukin-1 receptor antagonist in sepsis. Circ Shock. 1994;44:1–8. [PubMed] [Google Scholar]
- 57.Fisher CJ, Jr, Dhainaut JF, Opal SM, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. 1994;271:1836–43. [PubMed] [Google Scholar]
- 58.Opal SM, Fisher CJ, Jr, Dhainaut JF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit Care Med. 1997;25:1115–24. doi: 10.1097/00003246-199707000-00010. [DOI] [PubMed] [Google Scholar]
- 59.Vincent JL, Slotman G, Van Leeuwen PA, et al. IL-1ra administration does not improve cardiac function in patients with severe sepsis. J Crit Care. 1999;14:69–72. doi: 10.1016/s0883-9441(99)90016-3. [DOI] [PubMed] [Google Scholar]
- 60.Fleischmann RM, Tesser J, Schiff MH, et al. Safety of extended treatment with anakinra in patients with rheumatoid arthritis. Ann Rheum Dis. 2006;65:1006–12. doi: 10.1136/ard.2005.048371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nuki G, Bresnihan B, Bear MB, McCabe D, Investigators EGOC. Long-term safety and maintenance of clinical improvement following treatment with anakinra (recombinant human interleukin-1 receptor antagonist) in patients with rheumatoid arthritis: extension phase of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46:2838–46. doi: 10.1002/art.10578. [DOI] [PubMed] [Google Scholar]
- 62.Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008;58:2443–52. doi: 10.1002/art.23687. [DOI] [PubMed] [Google Scholar]
- 63.Goldbach-Mansky R, Shroff SD, Wilson M, et al. A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 Trap) in patients with familial cold autoinflammatory syndrome. Arthritis Rheum. 2008;58:2432–42. doi: 10.1002/art.23620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Group CiCS Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. 2009;360:2416–25. doi: 10.1056/NEJMoa0810787. [DOI] [PubMed] [Google Scholar]
- 65.Chitkara P, Stojanov S, Kastner DL. The hereditary autoinflammatory syndromes. Pediatr Infect Dis J. 2007;26:353–4. doi: 10.1097/01.inf.0000258777.86510.da. [DOI] [PubMed] [Google Scholar]
- 66.McDermott M, Tschopp J. From inflammasomes to fevers, crystals and hypertension: how basic research explains inflammatory diseases. Trends Mol Med. 2007;13:381–8. doi: 10.1016/j.molmed.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 67.Gattorno M, Federici S, Pelagatti MA, et al. Diagnosis and management of autoinflammatory diseases in childhood. J Clin Immunol. 2008;28(Suppl. 1):S73–83. doi: 10.1007/s10875-008-9178-3. [DOI] [PubMed] [Google Scholar]
- 68.Lindor NM, Arsenault TM, Solomon H, Seidman CE, McEvoy MT. A new autosomal dominant disorder of pyogenic sterile arthritis, pyoderma gangrenosum, and acne: PAPA syndrome. Mayo Clin Proc. 1997;72:611–15. doi: 10.1016/S0025-6196(11)63565-9. [DOI] [PubMed] [Google Scholar]
- 69.Wise CA, Bennett LB, Pascual V, Gillum JD, Bowcock AM. Localization of a gene for familial recurrent arthritis. Arthritis Rheum. 2000;43:2041–5. doi: 10.1002/1529-0131(200009)43:9<2041::AID-ANR15>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 70.Papin S, Cuenin S, Agostini L, et al. The SPRY domain of Pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1beta processing. Cell Death Differ. 2007;14:1457–66. doi: 10.1038/sj.cdd.4402142. [DOI] [PubMed] [Google Scholar]
- 71.Shoham NG, Centola M, Mansfield E, et al. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc Natl Acad Sci USA. 2003;100:13501–6. doi: 10.1073/pnas.2135380100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brenner M, Ruzicka T, Plewig G, Thomas P, Herzer P. Targeted treatment of pyoderma gangrenosum in PAPA (pyogenic arthritis, pyoderma gangrenosum and acne) syndrome with the recombinant human interleukin-1 receptor antagonist anakinra. Br J Dermatol. 2009;161:1199–201. doi: 10.1111/j.1365-2133.2009.09404.x. [DOI] [PubMed] [Google Scholar]
- 73.Belkhir R, Moulonguet-Doleris L, Hachulla E, Prinseau J, Baglin A, Hanslik T. Treatment of familial Mediterranean fever with anakinra. Ann Intern Med. 2007;146:825–6. doi: 10.7326/0003-4819-146-11-200706050-00023. [DOI] [PubMed] [Google Scholar]
- 74.Dierselhuis MP, Frenkel J, Wulffraat NM, Boelens JJ. Anakinra for flares of pyogenic arthritis in PAPA syndrome. Rheumatology. 2005;44:406–8. doi: 10.1093/rheumatology/keh479. [DOI] [PubMed] [Google Scholar]
- 75.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 76.Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327:296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- 77.So A. Developments in the scientific and clinical understanding of gout. Arthritis Res Ther. 2008;10:221. doi: 10.1186/ar2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 79.So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007;9:R28. doi: 10.1186/ar2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Terkeltaub R, Sundy JS, Schumacher HR, et al. The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann Rheum Dis. 2009;68:1613–17. doi: 10.1136/ard.2009.108936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sundy JS. Progress in the pharmacotherapy of gout. Curr Opin Rheumatol. 2010;22:188–93. doi: 10.1097/BOR.0b013e3283369014. [DOI] [PubMed] [Google Scholar]
- 82.Dinarello CA, Donath MY, Mandrup-Poulsen T. Role of IL-1beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes. 2010;17:314–21. doi: 10.1097/MED.0b013e32833bf6dc. [DOI] [PubMed] [Google Scholar]
- 83.Maedler K, Sergeev P, Ehses JA, et al. Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proc Natl Acad Sci USA. 2004;101:8138–43. doi: 10.1073/pnas.0305683101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maedler K, Sergeev P, Ris F, et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002;110:851–60. doi: 10.1172/JCI15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–26. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 86.Wen H, Gris D, Lei Y, et al. Fatty acid-induced NLRP3–ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–15. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stienstra R, Joosten LAB, Koenen T, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010;12:593–605. doi: 10.1016/j.cmet.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vandanmagsar B, Youm Y-H, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–88. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–9. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 91.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 92.Nakamura S, Kashimoto S, Kajikawa F, Nakata K. Combination effect of recombinant human interleukin 1 alpha with antitumor drugs on syngeneic tumors in mice. Cancer Res. 1991;51:215–21. [PubMed] [Google Scholar]
- 93.Onozaki K, Matsushima K, Aggarwal BB, Oppenheim JJ. Human interleukin 1 is a cytocidal factor for several tumor cell lines. J Immunol. 1985;135:3962–8. [PubMed] [Google Scholar]
- 94.Nakamura S, Nakata K, Kashimoto S, Yoshida H, Yamada M. Antitumor effect of recombinant human interleukin 1 alpha against murine syngeneic tumors. Jpn J Cancer Res. 1986;77:767–73. [PubMed] [Google Scholar]
- 95.Murphy JE, Morales RE, Scott J, Kupper TS. IL-1 alpha, innate immunity, and skin carcinogenesis: the effect of constitutive expression of IL-1 alpha in epidermis on chemical carcinogenesis. J Immunol. 2003;170:5697–703. doi: 10.4049/jimmunol.170.11.5697. [DOI] [PubMed] [Google Scholar]
- 96.Ghiringhelli F, Apetoh L, Tesniere A, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–8. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 97.Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 beta-converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci USA. 2001;98:13249–54. doi: 10.1073/pnas.231473998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bauer C, Duewell P, Mayer C, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut. 2010;59:1192–9. doi: 10.1136/gut.2009.197822. [DOI] [PubMed] [Google Scholar]
- 99.Hirota SA, Ng J, Lueng A, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis. 2010;17:1359–72. doi: 10.1002/ibd.21478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–91. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dupaul-Chicoine J, Yeretssian G, Doiron K, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity. 2010;32:367–78. doi: 10.1016/j.immuni.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 102.Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti TD. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol. 2010;185:4912–20. doi: 10.4049/jimmunol.1002046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Allen IC, TeKippe EM, Woodford RM, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010;207:1045–56. doi: 10.1084/jem.20100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hu B, Elinav E, Huber S, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci USA. 2010;107:21635–40. doi: 10.1073/pnas.1016814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124–7. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- 106.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–41. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 107.Araki A, Kanai T, Ishikura T, et al. MyD88-deficient mice develop severe intestinal inflammation in dextran sodium sulfate colitis. J Gastroenterol. 2005;40:16–23. doi: 10.1007/s00535-004-1492-9. [DOI] [PubMed] [Google Scholar]
- 108.Villani AC, Lemire M, Fortin G, et al. Common variants in the NLRP3 region contribute to Crohn's disease susceptibility. Nat Genet. 2009;41:71–6. doi: 10.1038/ng285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Apte RN, Voronov E. Interleukin-1 – a major pleiotropic cytokine in tumor–host interactions. Semin Cancer Biol. 2002;12:277–90. doi: 10.1016/s1044-579x(02)00014-7. [DOI] [PubMed] [Google Scholar]
- 110.Okamoto M, Liu W, Luo Y, et al. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J Biol Chem. 2010;285:6477–88. doi: 10.1074/jbc.M109.064907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Portier M, Zhang XG, Ursule E, et al. Cytokine gene expression in human multiple myeloma. Br J Haematol. 1993;85:514–20. doi: 10.1111/j.1365-2141.1993.tb03341.x. [DOI] [PubMed] [Google Scholar]
- 112.Rambaldi A, Torcia M, Bettoni S, et al. Modulation of cell proliferation and cytokine production in acute myeloblastic leukemia by interleukin-1 receptor antagonist and lack of its expression by leukemic cells. Blood. 1991;78:3248–53. [PubMed] [Google Scholar]
- 113.Demetri GD, Ernst TJ, Pratt ES, 2nd, Zenzie BW, Rheinwald JG, Griffin JD. Expression of ras oncogenes in cultured human cells alters the transcriptional and posttranscriptional regulation of cytokine genes. J Clin Invest. 1990;86:1261–9. doi: 10.1172/JCI114833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lewis AM, Varghese S, Xu H, Alexander HR. Interleukin-1 and cancer progression: the emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J Transl Med. 2006;4:48. doi: 10.1186/1479-5876-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Saijo Y, Tanaka M, Miki M, et al. Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: in vivo analysis of tumor–stromal interaction. J Immunol. 2002;169:469–75. doi: 10.4049/jimmunol.169.1.469. [DOI] [PubMed] [Google Scholar]
- 116.Voronov E, Shouval DS, Krelin Y, et al. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci USA. 2003;100:2645–50. doi: 10.1073/pnas.0437939100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vidal-Vanaclocha F, Amezaga C, Asumendi A, Kaplanski G, Dinarello CA. Interleukin-1 receptor blockade reduces the number and size of murine B16 melanoma hepatic metastases. Cancer Res. 1994;54:2667–72. [PubMed] [Google Scholar]
- 118.Krelin Y, Voronov E, Dotan S, et al. Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 2007;67:1062–71. doi: 10.1158/0008-5472.CAN-06-2956. [DOI] [PubMed] [Google Scholar]
- 119.Vidal-Vanaclocha F, Fantuzzi G, Mendoza L, et al. IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc Natl Acad Sci USA. 2000;97:734–9. doi: 10.1073/pnas.97.2.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stosic-Grujicic S, Basara N, Dinarello CA. Modulatory in vitro effects of interleukin-1 receptor antagonist (IL-1Ra) or antisense oligonucleotide to interleukin-1 beta converting enzyme (ICE) on acute myeloid leukaemia (AML) cell growth. Clin Lab Haematol. 1999;21:173–85. doi: 10.1046/j.1365-2257.1999.00221.x. [DOI] [PubMed] [Google Scholar]
- 121.Delwel R, van Buitenen C, Salem M, et al. Interleukin-1 stimulates proliferation of acute myeloblastic leukemia cells by induction of granulocyte-macrophage colony-stimulating factor release. Blood. 1989;74:586–93. [PubMed] [Google Scholar]
- 122.Rodriguez-Cimadevilla JC, Beauchemin V, Villeneuve L, Letendre F, Shaw A, Hoang T. Coordinate secretion of interleukin-1 beta and granulocyte-macrophage colony-stimulating factor by the blast cells of acute myeloblastic leukemia: role of interleukin-1 as an endogenous inducer. Blood. 1990;76:1481–9. [PubMed] [Google Scholar]
- 123.Cozzolino F, Rubartelli A, Aldinucci D, et al. Interleukin 1 as an autocrine growth factor for acute myeloid leukemia cells. Proc Natl Acad Sci USA. 1989;86:2369–73. doi: 10.1073/pnas.86.7.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bernstein SH, Fay J, Frankel S, et al. A phase I study of recombinant human soluble interleukin-1 receptor (rhu IL-1R) in patients with relapsed and refractory acute myeloid leukemia. Cancer Chemother Pharmacol. 1999;43:141–4. doi: 10.1007/s002800050874. [DOI] [PubMed] [Google Scholar]
- 125.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 126.El-Omar EM, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 127.Perri F, Terracciano F, Gentile M, Merla A, Scimeca D, Zullo A. Role of interleukin polymorphisms in gastric cancer: ‘pros and cons’. World J Gastrointest Oncol. 2010;2:265–71. doi: 10.4251/wjgo.v2.i6.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tu S, Bhagat G, Cui G, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–19. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lust JA, Lacy MQ, Zeldenrust SR, et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1{beta}-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin Proc. 2009;84:114–22. doi: 10.4065/84.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dinarello CA. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 2010;29:317–29. doi: 10.1007/s10555-010-9229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cassel SL, Eisenbarth SC, Iyer SS, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci USA. 2008;105:9035–40. doi: 10.1073/pnas.0803933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–7. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Watanabe H, Gaide O, Pétrilli V, et al. Activation of the IL-1beta-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol. 2007;127:1956–63. doi: 10.1038/sj.jid.5700819. [DOI] [PubMed] [Google Scholar]
- 134.Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer H-D. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol. 2007;17:1140–5. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- 135.Sutterwala FS, Ogura Y, Szczepanik M, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–27. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 136.Shio MT, Tiemi Shio M, Eisenbarth SC, et al. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog. 2009;5:e1000559. doi: 10.1371/journal.ppat.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Omi T, Kumada M, Kamesaki T, et al. An intronic variable number of tandem repeat polymorphisms of the cold-induced autoinflammatory syndrome 1 (CIAS1) gene modifies gene expression and is associated with essential hypertension. Eur J Hum Genet. 2006;14:1295–305. doi: 10.1038/sj.ejhg.5201698. [DOI] [PubMed] [Google Scholar]
- 138.Ritter M, Gross O, Kays S, et al. Schistosoma mansoni triggers Dectin-2, which activates the Nlrp3 inflammasome and alters adaptive immune responses. Proc Natl Acad Sci USA. 2010;107:20459–64. doi: 10.1073/pnas.1010337107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Koo IC, Wang C, Raghavan S, Morisaki JH, Cox JS, Brown EJ. ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell Microbiol. 2008;10:1866–78. doi: 10.1111/j.1462-5822.2008.01177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Master SS, Rampini SK, Davis AS, et al. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe. 2008;3:224–32. doi: 10.1016/j.chom.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mishra BB, Moura-Alves P, Sonawane A, et al. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol. 2010;12:1046–63. doi: 10.1111/j.1462-5822.2010.01450.x. [DOI] [PubMed] [Google Scholar]
- 142.Walter K, Holscher C, Tschopp J, Ehlers S. NALP3 is not necessary for early protection against experimental tuberculosis. Immunobiology. 2010;215:804–11. doi: 10.1016/j.imbio.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 143.Kafka D, Ling E, Feldman G, et al. Contribution of IL-1 to resistance to Streptococcus pneumoniae infection. Int Immunol. 2008;20:1139–46. doi: 10.1093/intimm/dxn071. [DOI] [PubMed] [Google Scholar]
- 144.McNeela EA, Burke A, Neill DR, et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 2010;6:e1001191. doi: 10.1371/journal.ppat.1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Harder J, Franchi L, Munoz-Planillo R, Park JH, Reimer T, Nunez G. Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-kappa B activation but proceeds independently of TLR signaling and P2X7 receptor. J Immunol. 2009;183:5823–9. doi: 10.4049/jimmunol.0900444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 147.Willingham SB, Allen IC, Bergstralh DT, et al. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol. 2009;183:2008–15. doi: 10.4049/jimmunol.0900138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.He X, Mekasha S, Mavrogiorgos N, Fitzgerald KA, Lien E, Ingalls RR. Inflammation and fibrosis during Chlamydia pneumoniae infection is regulated by IL-1 and the NLRP3/ASC inflammasome. J Immunol. 2010;184:5743–54. doi: 10.4049/jimmunol.0903937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med. 2010;207:1745–55. doi: 10.1084/jem.20100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Franchi L, Amer A, Body-Malapel M, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006;7:576–82. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 151.Fernandes-Alnemri T, Yu J-W, Juliana C, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol. 2010;11:385–93. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jones JW, Kayagaki N, Broz P, et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci USA. 2010;107:9771–6. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Amer A, Franchi L, Kanneganti T-D, et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J Biol Chem. 2006;281:35217–23. doi: 10.1074/jbc.M604933200. [DOI] [PubMed] [Google Scholar]
- 154.Warren SE, Armstrong A, Hamilton MK, et al. Cutting edge: cytosolic bacterial DNA activates the inflammasome via Aim2. J Immunol. 2010;185:818–21. doi: 10.4049/jimmunol.1000724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rathinam VAK, Jiang Z, Waggoner SN, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. 2007;204:3235–45. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Miao EA, Mao DP, Yudkovsky N, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA. 2010;107:3076–80. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Franchi L, Stoolman J, Kanneganti T-D, Verma A, Ramphal R, Núñez G. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur J Immunol. 2007;37:3030–9. doi: 10.1002/eji.200737532. [DOI] [PubMed] [Google Scholar]
- 159.Suzuki T, Franchi L, Toma C, et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007;3:e111. doi: 10.1371/journal.ppat.0030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Duncan JA, Gao X, Huang MT, et al. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol. 2009;182:6460–9. doi: 10.4049/jimmunol.0802696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shimada T, Park BG, Wolf AJ, et al. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe. 2010;7:38–49. doi: 10.1016/j.chom.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Miller LS, Pietras EM, Uricchio LH, et al. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J Immunol. 2007;179:6933–42. doi: 10.4049/jimmunol.179.10.6933. [DOI] [PubMed] [Google Scholar]
- 163.Miller LS, O'Connell RM, Gutierrez MA, et al. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity. 2006;24:79–91. doi: 10.1016/j.immuni.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 164.Hultgren OH, Svensson L, Tarkowski A. Critical role of signaling through IL-1 receptor for development of arthritis and sepsis during Staphylococcus aureus infection. J Immunol. 2002;168:5207–12. doi: 10.4049/jimmunol.168.10.5207. [DOI] [PubMed] [Google Scholar]
- 165.Craven RR, Gao X, Allen IC, et al. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS One. 2009;4:e7446. doi: 10.1371/journal.pone.0007446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Toma C, Higa N, Koizumi Y, et al. Pathogenic Vibrio activate NLRP3 inflammasome via cytotoxins and TLR/nucleotide-binding oligomerization domain-mediated NF-kappa B signaling. J Immunol. 2010;184:5287–97. doi: 10.4049/jimmunol.0903536. [DOI] [PubMed] [Google Scholar]
- 167.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38:240–4. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 168.Hsu L-C, Ali SR, McGillivray S, et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci USA. 2008;105:7803–8. doi: 10.1073/pnas.0802726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Moayeri M, Crown D, Newman ZL, et al. Inflammasome sensor Nlrp1b-dependent resistance to anthrax is mediated by caspase-1, IL-1 signaling and neutrophil recruitment. PLoS Pathog. 2010;6:e1001222. doi: 10.1371/journal.ppat.1001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Terra JK, Cote CK, France B, et al. Cutting edge: resistance to Bacillus anthracis infection mediated by a lethal toxin sensitive allele of Nalp1b/Nlrp1b. J Immunol. 2010;184:17–20. doi: 10.4049/jimmunol.0903114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Abdul-Sater AA, Koo E, Hacker G, Ojcius DM. Inflammasome-dependent caspase-1 activation in cervical epithelial cells stimulates growth of the intracellular pathogen Chlamydia trachomatis. J Biol Chem. 2009;284:26789–96. doi: 10.1074/jbc.M109.026823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Witola WH, Mui E, Hargrave A, et al. NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect Immun. 2011;79:756–66. doi: 10.1128/IAI.00898-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Griffith JW, Sun T, McIntosh MT, Bucala R. Pure Hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J Immunol. 2009;183:5208–20. doi: 10.4049/jimmunol.0713552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gross O, Poeck H, Bscheider M, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459:433–6. doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- 175.Said-Sadier N, Padilla E, Langsley G, Ojcius DM. Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS One. 2010;5:e10008. doi: 10.1371/journal.pone.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kumar H, Kumagai Y, Tsuchida T, et al. Involvement of the NLRP3 inflammasome in innate and humoral adaptive immune responses to fungal beta-glucan. J Immunol. 2009;183:8061–7. doi: 10.4049/jimmunol.0902477. [DOI] [PubMed] [Google Scholar]
- 177.Kanneganti TD, Body-Malapel M, Amer A, et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006;281:36560–8. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- 178.Allen IC, Scull MA, Moore CB, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–65. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Thomas PG, Dash P, Aldridge JR, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30:566–75. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol. 2010;11:404–10. doi: 10.1038/ni.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Muruve DA, Pétrilli V, Zaiss AK, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452:103–7. doi: 10.1038/nature06664. [DOI] [PubMed] [Google Scholar]
- 183.Rajan JV, Rodriguez D, Miao EA, Aderem A. The NLRP3 inflammasome detects EMCV and VSV infection. J Virol. 2011;85:4167–72. doi: 10.1128/JVI.01687-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Reimer T, Shaw MH, Franchi L, et al. Experimental cerebral malaria progresses independently of the Nlrp3 inflammasome. Eur J Immunol. 2010;40:764–9. doi: 10.1002/eji.200939996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Rigante D, Ansuini V, Bertoni B, et al. Treatment with anakinra in the hyperimmunoglobulinemia D/periodic fever syndrome. Rheumatol Int. 2006;27:97–100. doi: 10.1007/s00296-006-0164-x. [DOI] [PubMed] [Google Scholar]
- 186.Lequerré T, Vittecoq O, Pouplin S, et al. Mevalonate kinase deficiency syndrome with structural damage responsive to anakinra. Rheumatology. 2007;46:1860–2. doi: 10.1093/rheumatology/kem258. [DOI] [PubMed] [Google Scholar]
- 187.Cailliez M, Garaix F, Rousset-Rouvière C, et al. Anakinra is safe and effective in controlling hyperimmunoglobulinaemia D syndrome-associated febrile crisis. J Inherit Metab Dis. 2006;29:763. doi: 10.1007/s10545-006-0408-7. [DOI] [PubMed] [Google Scholar]
- 188.Simon A, Bodar EJ, van der Hilst JCH, et al. Beneficial response to interleukin 1 receptor antagonist in traps. Am J Med. 2004;117:208–10. doi: 10.1016/j.amjmed.2004.02.039. [DOI] [PubMed] [Google Scholar]
- 189.Kalliolias GD, Liossis S-NC. The future of the IL-1 receptor antagonist anakinra: from rheumatoid arthritis to adult-onset Still's disease and systemic-onset juvenile idiopathic arthritis. Exp Opin Invest Drugs. 2008;17:349–59. doi: 10.1517/13543784.17.3.349. [DOI] [PubMed] [Google Scholar]
- 190.Wendling D, Govindaraju S, Prati C, Toussirot E, Bertolini E. Efficacy of anakinra in a patient with refractory relapsing polychondritis. Joint Bone Spine. 2008;75:622–4. doi: 10.1016/j.jbspin.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 191.Vounotrypidis P, Sakellariou GT, Zisopoulos D, Berberidis C. Refractory relapsing polychondritis: rapid and sustained response in the treatment with an IL-1 receptor antagonist (anakinra) Rheumatology. 2006;45:491–2. doi: 10.1093/rheumatology/kel041. [DOI] [PubMed] [Google Scholar]
- 192.Pizzirani C, Falzoni S, Govoni M, et al. Dysfunctional inflammasome in Schnitzler's syndrome. Rheumatology. 2009;48:1304–8. doi: 10.1093/rheumatology/kep222. [DOI] [PubMed] [Google Scholar]
- 193.Delluc A, Limal N, Puéchal X, Francès C, Piette JC, Cacoub P. Efficacy of anakinra, an IL1 receptor antagonist, in refractory Sweet syndrome. Ann Rheum Dis. 2008;67:278–9. doi: 10.1136/ard.2006.068254. [DOI] [PubMed] [Google Scholar]
- 194.Bilginer Y, Ayaz NA, Ozen S. Anti-IL-1 treatment for secondary amyloidosis in an adolescent with FMF and Behçet's disease. Clin Rheumatol. 2010;29:209–10. doi: 10.1007/s10067-009-1279-8. [DOI] [PubMed] [Google Scholar]
- 195.Botsios C, Sfriso P, Furlan A, Punzi L, Dinarello CA. Resistant Behçet disease responsive to anakinra. Ann Intern Med. 2008;149:284–6. doi: 10.7326/0003-4819-149-4-200808190-00018. [DOI] [PubMed] [Google Scholar]
- 196.Furlan A, Botsios C, Ruffatti A, Todesco S, Punzi L. Antisynthetase syndrome with refractory polyarthritis and fever successfully treated with the IL-1 receptor antagonist, anakinra: a case report. Joint Bone Spine. 2008;75:366–7. doi: 10.1016/j.jbspin.2007.07.010. [DOI] [PubMed] [Google Scholar]