

Abstract
Immunoglobulin (Ig) therapy is constantly evolving. Advances in the basic and clinical science of immunoglobulins have provided new perspectives in using polyclonal IgG to treat patients with primary immunodeficiencies. Recent meta-analyses of patient data and outcomes, optimization of IgG administration and better understanding of the IgG receptor variability and clinical effect are new concepts which practising immunologists can use in tailoring their approach to treating patients with primary immunodeficiencies. This manuscript presents the proceedings of a satellite symposium, held in conjunction with the European Society for Immunodeficiencies (ESID) 2010 meeting, to inform attendees about new scientific concepts in IgG therapy, with the goal of empowering expert level evaluation of what optimal IgG therapy is today.
Keywords: IgG receptor, immunoglobulin, intravenous IgG, primary immunodeficiencies, subcutaneous IgG
Introduction
Primary immunodeficiencies (PI) disorders predispose patients to recurrent infections and chronic lung disease, requiring patients to undergo immunoglobulin (Ig) replacement therapy. Immunoglobulin formulations can be administered subcutaneously (SCIG) or intravenously (IVIG). Immunologists in the United States were asked if they thought their patients would be better served by SCIG compared to IVIG [1]. The most common response was that 25–50% of patients would be better served by SCIG (Fig. 1). European immunologists, however, are more likely to hold that greater percentages of patients will be better served by SCIG (Hernandez-Trujillo et al., manuscript in preparation). Nevertheless, even with having a perceived advantageous benefit, SCIG therapy, as well as IVIG therapy, have the potential to be improved with further innovation in the areas of formulation, administration, optimization of the choice of clinical end-point and advances in understanding mechanisms of IgG receptor function relative to IgG therapy.
Fig. 1.
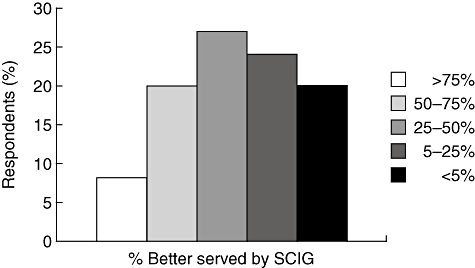
Survey of benefits of subcutaneous immunoglobulin (SCIG) for patients. Percentage of US immunologists believing that patients would ultimately be better served by SCIG compared to intravenous immunoglobulin (IVIG). Physician members of the American Academy of Allergy Asthma and Immunology (AAAAI) were asked their opinion regarding IVIG and SCIG benefit in a web-based survey conducted in 2005–06. Importantly, this effort started before the US Food and Drug Administration (FDA) approved the use of SCIG in primary immunodeficiencies (PI) in the United States. Data adapted from Yong et al. 2010.
Change in formulation to a higher IgG concentration represents a straightforward means to offer patients with PI a more convenient subcutaneous infusion option. A prospective, open-label, multi-centre, single-arm, Phase III study was conducted to evaluate the efficacy and safety of a 20% liquid SCIG stabilized with l-proline in patients with PI over 15 months, and the results underscore positive aspects of SCIG therapy [2]. A mean serum IgG of 12·5 g/l was achieved using weekly doses that added up to approximately 153% of the monthly IVIG dosage given before study entry. There was a total of 96 non-serious infections, corresponding to a rate of 2·76 infections/patient/year, and no serious bacterial infections (SBI) were observed. In addition to the overall infection rate, the rate of missed work/school days (2·06 days/patient/year) was also low over the duration of the study relative to that described in a study with 16% SCIG [3]. No serious adverse events (AEs) related to study medication were reported. The formulation allows storage at 25°C, which may improve convenience for patients. In a study of healthy volunteers, 20% SCIG and 16% SCIG (Vivaglobin®, CSL Behring GmbH, Marburg, Germany) were evaluated for comparative local tolerance. At the same IgG dose, lower scores for both mean and maximal local pain at the injection site were observed for the 20% SCIG formulation (P = 0·0205 and P = 0·0801, respectively; Fig. 2). Optimization of IgG formulation can lead potentially to practical improvements for patients in reducing the infusion volume and, consequently, shortening the infusion time.
Fig. 2.
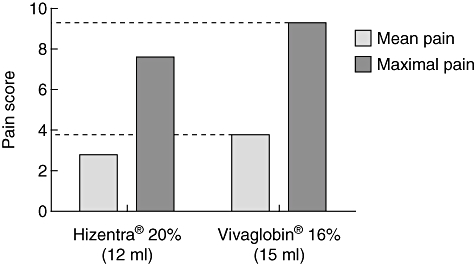
Comparative local tolerability of SCIG formulations. Mean (grey) and maximal (black) pain scores in 28 healthy volunteers after an equivalent immunoglobulin (Ig)G dose of Hizentra® (12 ml) or Vivaglobin® (15 ml) are shown. Dashed lines highlight differences between pain scores. Data were adapted from Hagan et al. 2010.
IgG therapy may be optimized by knowledge of the serum IgG levels required to minimize infection risk. A meta-analysis of 17 studies (mean of 34 patients per study) evaluating serum IgG levels and pneumonia incidence in patients with PI receiving IVIG was the first of its kind across PI studies [4]. The study revealed that average serum IgG levels increased by 1·21 g/l for every 100 mg/kg IVIG dose increase. Pneumonia incidence declined by 27% with each 1·00 g/l increment in serum IgG levels (for data up to 10 g/l IVIG) (Fig. 3) [4]. Pneumonia incidence with maintenance of 5 g/l serum IgG levels was fivefold higher than that with 10 g/l. Sufficient data were not available within the studies to allow predictions for IgG levels > 10 g/l. The analysis also identified that across studies there was a lack of standardization in diagnosing infections and reporting of end-points relevant to the therapy. The results of a recent prospective study of patients with PI followed over 22 years showed that a broad range of serum IgG levels was required to bring patients into an infection-free state [5]. For patients with common variable immunodeficiency (CVID, n = 90) the replacement immunoglobulin dose required to prevent breakthrough infections was 0·2–1·2 g/kg/month and resultant serum IgG levels were 5–16 g/l. Patients with X-linked agammaglobulinaemia (XLA; n = 15) remained infection free, with an immunoglobulin dose ranging from 0·5–0·9 g/kg/month, and resultant serum IgG levels were 8–13 g/l. Patients with XLA required a significantly higher mean dose (0·67 ± 0·12 g/kg) to prevent all infections compared with patients with CVID (0·53 ± 0·19 g/kg; P = 0·01). This observation is likely to reflect the greater severity of antibody deficiency in XLA patients; evidence suggests that high serum IgG levels probably protect against the development of enteroviral meningoencephalitis [6]. That the optimal serum IgG levels required to prevent breakthrough infection varied from patient to patient suggests that therapy efficacy should be evaluated by clinical outcomes and not simply the achievement of a particular serum IgG level, a conclusion shared by many investigators [5,7–9].
Fig. 3.
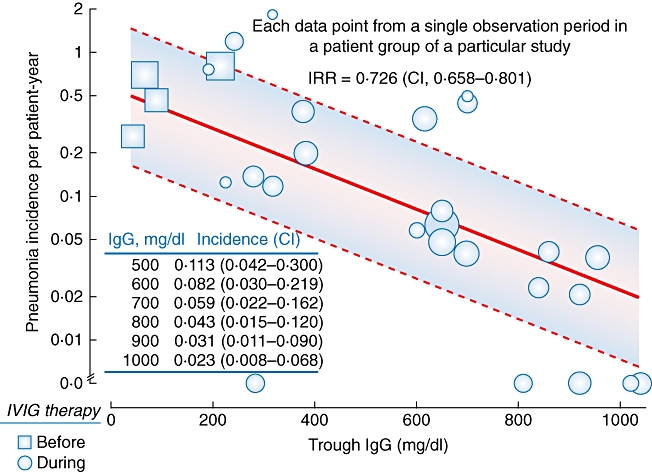
Relation of serum immunoglobulin (Ig)G level to pneumonia incidence in a meta-analysis. Pneumonia incidence data were extracted from 17 studies and are represented by individual circles or squares, the size of which corresponds to observed patient years in the study. Squares represent the historical incidence of pneumonia (where reported) in patients from a particular study, prior to being started on intravenous immunoglobulin (IVIG) therapy (thus serum IgG levels nominal). The bold line represents multi-level model predictions and dashed lines the meta-regression 95% confidence interval (CI). Interval values are provided in the inset table. Data were modified from Orange et al. 2010.
In this satellite symposium sponsored by CSL Behring, Chair Jordan Orange described current immunoglobulin therapy trends and practice based on results from various clinical studies. Bodo Grimbacher discussed results from well-organized, extensive, statistically evaluated patient data from the European Society for Immunodeficiencies (ESID) online patient registry. Siraj Misbah presented insights from clinical interventions and outcomes with immunoglobulin administered through the subcutaneous route. Finally, Taco Kuijpers showed that the variability in IgG Fc receptor genes can have an impact upon therapy with polyclonal IgG. Together, these advances in the basic and clinical science of immunoglobulins provide new perspectives in using polyclonal IgG therapy and enable physicians to provide today optimal IgG therapy for patients with PI.
Understanding outcomes in immunodeficiencies: end-points and targets
Immunoglobulin replacement therapy has improved the lives of patients with PI in measureable ways. Since the initiation of immunoglobulin therapy in the 1950s, mortality of patients with PI has decreased and life expectancy has increased substantially to the present day. Clinicians have searched for suitable end-points for evaluating the efficacy of IgG therapy. IgG therapy has improved morbidity as measured by a reduction in the number of pneumonia events from 0·82 to 0·12 per patient/year (P = 0·006) [10]. This is a substantial improvement in the treatment of primary immunodeficiencies, despite that this rate is still higher than that for the general population (five to 11 cases per 1000 individuals [11–13]). An improved health-related quality of life (HRQL) for patients with CVID receiving immunoglobulin replacement compared to those not receiving immunoglobulin therapy has been shown through fewer days in hospital (12·5 versus 19·8 days/year, respectively) and days missed off work or school (6·1 versus 23·3 days/year, respectively) [14]. The number and severity of infections, along with monitoring of serum IgG levels, are other obvious measures for evaluating therapy efficacy.
Various other end-points evaluating the efficacy of IgG therapy in patients with PI have been explored. Pulmonary function has been studied [15–20], but the lack of sensitivity of the available methods has prevented the wide use of this measure. The Chest CT in ADS Group (http://www.chest-ct-group.eu/), an international group of immunologists, pulmonologists and radiologists, has developed a methodology for improving the diagnosis of disease in patients with antibody deficiency syndrome. This group uses high-resolution chest computed tomography (CT) scanning along with a battery of lung function tests which are used to give a CT score to track the progression of lung disease.
The potential use of C-reactive protein (CRP) as an indicator of IgG therapy efficacy was discussed. CRP is an acute-phase protein produced in response to various stimuli involving tissue damage such as inflammation and infection. Serum CRP has been used extensively as a marker of bacterial infection [21]. However, due to its low specificity, its true diagnostic value in clinical practice has been questioned [22,23]. A retrospective, single-centre study was carried out to examine the association between CRP levels and clinical outcomes in patients with CVID on immunoglobulin replacement. The cohort consisted of 112 CVID patients and was divided into three groups based on median CRP values (0–5, 5–10 and > 10 mg/l). There were 10 patients in the > 10 mg/l group. There were a large number of patients in both 0–5 and 5–10 mg/l groups and 12 patients were selected randomly from each group for the analysis. Five outcome parameters were investigated: number of infections, number of serious infections, number of antibiotic courses, days off sick and days in hospital. These parameters are also part of the quality of life data set in the ESID database [14]. The working hypothesis was that these outcome parameters would correlate positively with serum CRP levels. However, when considering CRP on a continuous scale, no strong evidence of an association between CRP and any of the parameters examined was found (Table 1). Only weak evidence of an association between CRP and the number of serious infections was observed, but this was not statistically significant (P = 0·08). The Spearman's rank correlation coefficient between the two variables was positive, suggesting that the number of serious infections increased with increasing serum CRP level. When the CRP measurements were divided into three categories (0–5, 5–10 and > 10 mg/l), the Kruskal–Wallis analysis suggested that there was not enough evidence that any of the outcome parameters varied between CRP categories (Table 1). The results of this study suggest that serum CRP value is not a good surrogate marker for the evaluation of the clinical efficacy of immunoglobulin replacement in adult patients with CVID and, therefore, a routine follow-up of CRP in patients with PI is not recommended. However, its judicious use helps in the assessment of selected patients with PI associated with chronic infective or inflammatory disease.
Table 1.
Analysis of the association between serum C-reactive protein (CRP) values and outcome parameters in patients with common variable immunodeficiency (CVID)
| Correlation of serum CRP values with outcome parameters* | Analysis of outcome parameters in patients grouped according to serum CRP values† | |||||
|---|---|---|---|---|---|---|
| Outcome | Correlation coefficient | P-value | CRP (mg/l) | Median (IQR) | Mean (s.d.) | P-value |
| Number of infections | 0·20 | 0·28 | < 5 | 3·1 (2·6, 12·2) | 6·0 (5·9) | 0·47 |
| 5–10 | 9·8 (2·3, 14·4) | 11·4 (11·8) | ||||
| > 10 | 7·5 (4·0, 17·7) | 11·2 (12·5) | ||||
| Number of serious infections | 0·31 | 0·08 | < 5 | 0·0 (0·0, 0·0) | 0·1 (0·4) | 0·39 |
| 5–10 | 0·0 (0·0, 0·0) | 0·8 (2·6) | ||||
| > 10 | 0·0 (0·0, 2·6) | 2·0 (4·3) | ||||
| Antibiotic courses | 0·09 | 0·62 | < 5 | 5·0 (0·0, 10·3) | 6·4 (6·4) | 0·30 |
| 5–10 | 13·6 (3·5, 31·2) | 15·5 (13·8) | ||||
| > 10 | 8·6 (4·0, 11·8) | 9·6 (8·9) | ||||
| Days sick | −0·12 | 0·51 | < 5 | 4·7 (0·0, 26·3) | 10·9 (13·0) | 0·39 |
| 5–10 | 16·9 (0·0, 34·5) | 46·7 (102·2) | ||||
| > 10 | 1·4 (0·0, 16·2) | 13·5 (29·0) | ||||
| Days hospitalized | 0·21 | 0·24 | < 5 | 0·0 (0·0, 0·5) | 0·8 (2·0) | 0·23 |
| 5–10 | 0·0 (0·0, 0·0) | 0·0 (0·0) | ||||
| > 10 | 0·0 (0·0, 5·4) | 22·7 (55·0) | ||||
IQR: interquartile range; s.d.: standard deviation.
Spearman's rank coefficient
Kruskal–Wallis analysis.
Optimizing SCIG usage: recent insights from clinical studies
SCIG is becoming well established as a viable alternative to IVIG for patients with primary antibody deficiency. SCIG is as efficacious as IVIG in infection prophylaxis and in achieving satisfactory serum IgG levels as has been demonstrated in several recent key clinical studies of a 16% SCIG versus IVIG formulation. A total of 158 patients with PI were assessed in three different studies and no difference in mean infection scores and in duration of infections was observed for SCIG versus IVIG [3,24,25]. Of particular interest is that for European-based studies, the Vivaglobin® dose given is equivalent when switching patients from IVIG to SCIG, whereas in North American studies the United States Food and Drug Administration (US FDA) requires the initial SCIG dose at switching to be 1·37 times the previous IVIG dose, in order to achieve a similar area under the IgG concentration–time curve. Despite this, no difference between the rate of SBIs was observed in these European versus North American studies. There were, however, differences in the overall infection rate, an observation which should generate further evaluation. The European Hizentra trial showed that an increase in IgG dose upon switching from IVIG to SCIG is not necessary to maintain a low frequency of SBIs, but is beneficial in reduction of the rate of non-serious infections and the associated rates of hospitalization and antibiotic use [7]. As SCIG is given more frequently in smaller doses compared with IVIG, it allows increasing the total monthly dose more easily without risk of compromising tolerability. Additionally, SCIG has a very favourable AE profile. In contrast to IVIG, there have been no reports of associated renal impairment, aseptic meningitis or anaphylaxis. Moreover, SCIG has been used successfully in cases of IVIG-induced anaphylaxis associated with anti-IgA antibodies.
In a recent US study, 49 patients previously on IVIG were switched to IgPro20, a 20% liquid SCIG stabilized with l-proline [2]. No SBIs (defined as per US FDA criteria) were observed and the rate of non-serious infections was low (2·76 infections/patient/year). Subcutaneous administration allows infusion of up to 1·2 g/kg/month and a 20% SCIG formulation enables administration of even higher doses [2]. Furthermore, SCIG therapy results in more stable serum IgG levels over time, as smaller doses are given more frequently compared to the larger IVIG boluses given every 3–4 weeks [26]. A maintenance of serum IgG levels can be achieved with SCIG even with a reduction in total monthly dose compared to the previously IVIG administered dose [27]. Which of these aspects will be validated as advantageous to SCIG therapy will be determined in the course of future studies, and these have the potential to drive greater use of SCIG. SCIG offers many patients a viable, convenient alternative to IVIG.
A logical step forward from the successful use of SCIG in replacement therapy is the use of SCIG in the setting of immunomodulation. Multi-focal motor neuropathy (MMN) is known to be responsive to IVIG therapy. MMN is a serious autoimmune neuropathy characterized by segmental demyelination, conduction block and asymmetric weakness, with relatively preserved muscle bulk. MMN is associated with anti-GM1 antibodies in 50–80% of cases. Three recent studies of SCIG in patients with MMN who were switched from IVIG show that SCIG was as efficacious as IVIG, as measured by combined dynamometric [28] and the Medical Research Council (MRC) muscle strength scores [29]. In a more recent study, patients were switched gradually over 3 weeks from IVIG to SCIG [30]. The majority of patients maintained MRC muscle strength score over the 6-month study. In all three studies, the majority of patients elected to continue SCIG administration at the end of the study (Table 2). One patient who experienced muscle strength deterioration also continued to use this form of administration [29]. Thus, SCIG showed good efficacy, was preferred by patients with MMN and its use in immunomodulation should be investigated further. SCIG may also be effective in dermatological autoimmune disorders as demonstrated in IVIG-responsive epidermolysis bullosa acquisita (EBA). A case report study of a patient with EBA who was switched to SCIG (0·9 g/kg/month) showed improved clinical outcome [31]. Successful treatment of MMN and EBA suggests that SCIG use can be explored in many other conditions where IVIG is effective.
Table 2.
Overview of results from studies using subcutaneous immunoglobulin (SCIG) and intravenous immunoglobulin (IVIG) for replacement therapy and immunomodulation in multi-focal motor neuropathy (MMN)
| Number of patients | Study design SCIG switch | Primary end-point | Result | Number continuing with SCIG* | Study |
|---|---|---|---|---|---|
| 10 | 50% dose (n = 5) | Muscle strength | 50% SCIG < IVIG | 0/4 | Eftimov et al., 2009 [29] |
| 100% dose (n = 5) | 100% SCIG = IVIG | 4/5 | |||
| 9 | 100% dose | Muscle strength | SCIG = IVIG | 5/9 | Harbo et al., 2009 [28] |
| SCIG–IVIG cross-over | |||||
| 8 | 100% dose | Muscle strength | SCIG = IVIG | 7/7 | Misbah et al., [30] |
Patients who completed the study and for whom data were analysed.
A recent retrospective study offers insight into new ways to improve convenience in SCIG administration. Infusion with a syringe and butterfly needle (rapid push) was compared with the usual pump administration. The rapid push method involves more frequent subcutaneous administration of smaller doses compared to weekly SCIG. Of 104 patients with PI who had either no previous IgG therapy or had been on IVIG, 74 patients used rapid push administration and 29 used a pump to infuse a 16% SCIG IgG formulation. Patients using rapid push underwent an average of 3·1 infusions per week, and those using pump an average of 2·9 infusions per week. Rapid push was found to be an efficacious alternative, as no difference in mean serum IgG levels was observed between the two different administration methods [32]. Additionally, serum IgG levels achieved with either route of SCIG infusion were higher than those achieved with the previous IVIG therapy, due probably to the frequent administration of smaller doses and the slow transition of IgG into the vascular space. Rapid push infusion thus offers a suitable alternative, for example, when a pump is not available or when high infusion volumes per injection site are not tolerated.
The Oxford Immunology Department uses SCIG for replacement therapy and immunomodulation in primary antibody deficiencies, autoimmune neuropathies and EBA, and has kept records on patients treated with SCIG since 1992. The indication, typical monthly SCIG doses used, serum levels achieved and duration of therapy are outlined in Table 3 (Misbah, unpublished). Results show that the mean dose used for PI is 0·57 g/kg/month and for immunomodulation, 1·2 g/kg/month. Resulting serum IgG levels were lower for immune replacement therapy (10·2 g/l) than for immunomodulation (18·6 g/l). The broad range of steady state serum IgG levels achieved in PI patients with SCIG reflects individual clinical responses. An important part of the successful therapy is the support of nursing staff, who introduce patients using SCIG to the Oxford home therapy training programme. The course involves six in-clinic training sessions and covers all practical aspects of infusing, theory and practice, such as venipuncture technique and blood sampling, priming of the SCIG administration set, controlling the infusion rate, recording details of infusion, safe disposal of equipment used and recognition of adverse reactions and actions to be taken. Patients are asked to take an examination before they may begin infusing at home.
Table 3.
Use of subcutaneous immunoglobulin (SCIG) for replacement therapy and immunomodulation at the Oxford Immunology Department
| Indication | Patients (number) | Dose g/kg/month, mean (range) | Serum IgG g/l, mean (range) | Therapy years, mean (range) |
|---|---|---|---|---|
| Primary antibody deficiency | 96 | 0·57 (0·1–2·2) | 10·2 (4·2–22) | 4·7 (0–17) |
| Autoimmune neuropathies | 17 | 1·2 (0·5–3·3) | 18·6 (13·8–28·5) | 1·4 (0–4) |
| Epidermolysis bullosa acquisita | 1 | 0·9 | 15·0 (13·8–17·7) | 6·0 |
As physicians become more experienced in the use and benefits of SCIG administration, and as patients exert their preferred choice of administration and with the availability of 20% solutions, the use of SCIG for patients with PI and autoimmune neuropathies is expected to increase. Enhancing absorption of IgG using hyaluronidase may facilitate infusion of higher doses required for immunomodulation. Monitoring of clinical outcomes in each patient will allow dose adjustments for achieving optimal results.
With the increasing use of SCIG therapy, the terminology associated traditionally with IVIG may have to be attuned. The term ‘trough serum IgG level’, which has been defined as the lowest serum IgG level prior to the next IVIG infusion, is inappropriate in SCIG therapy because of the stable serum IgG levels (lack of ‘peaks’ and ‘troughs’). Therefore, it would be more appropriate to refer to steady state IgG levels.
Fc receptors in IgG therapy
Molecular investigations of the FCGR genes show that functional single nucleotide polymorphisms and copy number variation occur and may impact the ability of IgG to modulate inflammatory responses [33–37]. Thus, better understanding of the clinical impact of FCGR gene variation and related Fc receptors for IgG (FcγRs) raises the possibility of therapy individualization for optimal benefit. A spectrum of diseases involves inflammatory cells known to express FcγRs. Based on their binding affinity for monomeric IgG, human FcγRs can be subdivided into high-affinity receptors (type I, CD64) and low-affinity receptors (type II, CD32; and type III, CD16). The genes encoding the low-affinity FcγRs (FCGR2A, FCGR2B, FCGR2C, FCGR3A and FCGR3B) are located on chromosome 1q23–24. The expression and activation status of FcγRs vary among immune cells.
Many of the FcγR-encoding genes show variation in SNPs, which may determine the IgG binding characteristics of the various FcγRs. The impact of genetic variation is not known for all receptors, but some functional FcγR polymorphisms have been characterized (Fig. 4, reviewed in [38]). The best-known SNP variant is R131H in the FcγRIIa, whereby an arginine at position 131 changes to histidine, which facilitates binding to IgG2 and enables phagocytosis of IgG2-coated particles. Homozygous carriers of arginine at this position may experience increased risk of infection, whereas those homozygous for histidine may be at higher risk for autoimmune disorders. A SNP (I232T) in the transmembrane area of the inhibitory FcγRIIb may impact the receptor's inhibitory activity. FcγRIIIa may express either a valine or a phenylalanine at position 158 (V158F). The V158 allotype has a higher affinity for IgG1 and IgG3 subclasses compared to 158F. In another example, the human neutrophil antigen (NA) is present on FcγRIIIb and expresses two allotypes (NA1 and NA2) which impact receptor binding. NA1 shows higher binding and phagocytosis of IgG1- and IgG3-coated particles and higher affinity for IgG3 in comparison to the NA2 allotype.
Fig. 4.
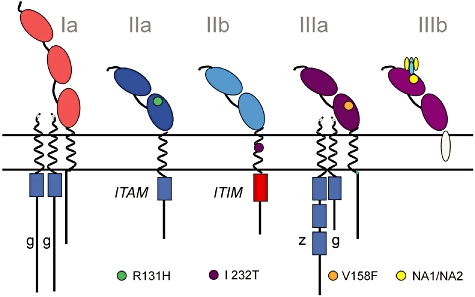
Single nucleotide polymorphisms (SNPs) in various human Fcγ receptors (FcγR). Different types of single nucleotide polymorphisms (SNPs) are known to have functional implications related to the affinity of the FcγR to IgG binding and/or signalling capacity. With respect to the SNP in the intramembranous domain of the inhibitory FcγRIIb, the infrequent FcγRIIb-232T variant is believed to give a stronger negative signal via the immunoreceptor tyrosine-based inhibitory motif (ITIM). FcγRIIa-H131 binds human IgG2, whereas FcγRIIa-R131 does not. In FcγRIIIa, FcγRIIIa-V158 has a higher affinity for IgG1 and IgG3 than FcγRIIIa-F158. Although some of the FcγRs do not contain the immunoreceptor tyrosine-based activating motif (ITAM), these receptors are associated with other proteins for signalling. Allelic variation in FcγRIIIb is composed of differences in four amino acids, referred to as human neutrophil antigen 1 [HNA1a (NA1) and HNA1b (NA2)]; FcγRIIIb-HNA1a internalizes IgG1- or IgG3-opsonized particles more efficiently than does FcγRIIIb-HNA1b.
In addition to SNPs, copy number variation (CNV) is now also being recognized as an important factor of variation. Gene dosage effects may occur as a functional consequence of CNV. Recently, an association between a low copy number of FCGR3B and glomerulonephritis in systemic lupus erythematosus (SLE) has been reported [33,34]. The low gene copy number correlates with reduced FcγRIIIb expression and is likely to contribute to the impaired clearance of immune complexes, a feature of SLE [33].
Recent studies identifying CNV in the human genome suggest that large areas at chromosome 1q23–24 exhibit a high degree of variation in gene copy number [39]. Indeed, FCGR3A, FCGR2C and FCGR3B show CNV at variable degrees of co-segregation, while FCGR2A and FCGR2B do not show CNV [36,37,40,41]. CNV may thus be an indicator for interindividual differences, including differential responsiveness to infection or predisposition to autoimmune disease as a result of unbalanced immunity [34].
The Multiplex Ligation-dependent Probe Amplification (MLPA) method was used to study FCGRs in a cohort of patients with idiopathic thrombocytopenic purpura (ITP) versus a control group of healthy volunteers [35]. Both control and ITP groups showed no variation in FCGR2A and FCGR2B. MLPA showed that FCGR2C, FCGR3A and FCGR3B CNV are present in the normal population. CNV was not associated with susceptibility to ITP in this cohort. A stop codon in exon 3 of FCGR2C suggests that it is a pseudogene (Table 4). A SNP at this site changes the region to an open reading frame (ORF). In healthy volunteers, STOP allele frequency was found to be 91·2% of all alleles and ORF frequency was 8·8%. The FCGR2C–ORF allele frequency was significantly higher in the ITP population (18·9%; P = 0·004). Also, significantly more ITP patients harboured ORF SNPs (34·5%) compared to healthy controls (18·0%; P = 0·009). Further investigations demonstrated that FCGR2C harbouring an ORF encodes a surface expressed FcγRIIc on natural killer (NK) cells (Fig. 5). Furthermore, NK cells with FcγRIIc can mediate antibody-dependent cellular cytotoxicity (ADCC) to antibody-coated targets, demonstrating that FcγRIIc acts as an activating IgG receptor.
Table 4.
Stop codon versus open reading frame (ORF) in exon 3 of FCGR2C in healthy controls and patients with ITP
| FCGR2C exon 3 | Number (%) of patients | |
|---|---|---|
| SNP | Healthy controls (n = 100) | ITP (n = 116) |
| STOP | 6 (6) | 4 (3·4) |
| STOP/STOP | 66 (66) | 68 (58·6) |
| STOP/STOP/STOP | 10 (10) | 4 (3·4) |
| ORF | 1 (1) | 2 (1·7) |
| ORF/ORF | 0 (0) | 3 (2·6) |
| ORF/STOP | 16 (16) | 32 (27·6) |
| ORF/ORF/STOP | 0 (0) | 1 (0·9) |
| ORF/STOP/STOP | 1 (1) | 2 (1·7)* |
| Allele frequency (%) | ||
| Allele frequency | Healthy controls | ITP |
| STOP | 186 (91·2) | 198 (81·1) |
| ORF | 18 (8·8) | 44 (18·9)† |
ORF single nucleotide polymorphisms (SNPs) healthy versus idiopathic thrombocytopenic purpura (ITP); P = 0·009.
ORF allele frequency healthy versus ITP; P = 0·004.
Fig. 5.
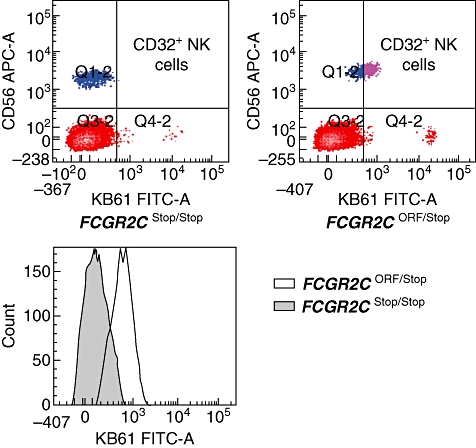
FcγRIIc expression on natural killer (NK) cells. FcγRIIc expression is observed on the FCGR2C-open reading frame (ORF) and not on the FCGR2C-STOP genotype. Control counts for each genotype are also shown. FcγRII expression on NK cells is limited to the FCGR2C-ORF genotype. Blood cells were incubated with CD56 and pan-CD32 (monoclonal antibody clone KB61). Lymphocytes were gated on the basis of their forward-scatter/side-scatter pattern. NK-cell population was determined as CD56-positive lymphocytes.
IVIG-induced anaphylaxis in a patient with CVID has been shown to be probably related to variation in FCGR genes (Kuijpers, unpublished data). A Caucasian female was diagnosed with CVID. She had recurrent infections and chronic Giardia lamblia-related diarrhoea. After the start of IVIG, the patient complained of abdominal pain, a generalized rash, tachypnoea and tachycardia with a fall in blood pressure, followed by chills and fever. IVIG infusion was stopped and anti-histamines (clemastin, 2 mg), steroids (DAF, 25 mg) and NaCl 0·9% (500 ml) were administered intravenously. Blood cultures remained sterile, concentrations of serum tryptase and complement activation products were not increased; however, elevated elastase was detected. IgG–anti-IgA complexes are not always clinically relevant and are no longer tested for routinely prior to infusion. In this case, due to the anaphylaxis, preinfusion serum samples were analysed and showed the presence of anti-IgA antibodies of the IgG1 subclass. Investigation of FCGR2 revealed a novel splice variant in exon 6 of FcγRIIa that is characterized by normal mRNA and protein expression, and represents a potential gain-of-function variant through elongation of the cytoplasmic tail. The expression of this splice variant has been found in eight individuals, including one patient with CVID, three with vasculitis of whom one developed insulin-dependent diabetes type 1 and in one healthy control. FcγRIIa-mediated hyper-reactivity may be proposed as a mechanism to explain severe anaphylactic reaction to IVIG. More CVID patient serum samples are required to fully characterize the clinical response.
Thus, FCGR2C represents a gene with variable expression that is highly relevant for immunity, probably contributing to susceptibility and severity of infections and autoimmune disease. A balance between inhibitory (FcγRIIb) and activating FcγRs (FcγRIIa, FcγRIIcorf, FcγRIIIa, FcγRIIIb) is important for immune reactivity.
High-dose IVIG treatment is thought to exert an immunomodulatory effect by numerous mechanisms, including engagement of the inhibitory FcγRIIb receptor and/or by saturation of the neonatal Fc receptor, FcRn. FcRn is a human leucocyte antigen (HLA) class I-related receptor that transports IgG antibodies within and across a diverse array of different cell types. Through this transport, FcRn serves multiple roles throughout adult life that extend well beyond its previously defined function of transcytosing IgG molecules from mother to offspring. The influence of FcRn on the tissue distribution of IgG has not yet been clarified [42]. The role of FcRn includes the maintenance of serum IgG and albumin levels and the delivery of antigen in the form of immune complexes to degradative compartments within cells. The FcRn–IgG interaction is strictly pH-dependent, with a maximum at pH 6, and becomes undetectable as near neutral pH is approached, a feature that is essential for efficient transport. IgG transport between the blood and interstitial compartments may proceed by convection through paracellular pores in the vascular endothelium, or via FcRn-mediated transcytosis across vascular endosomal cells. Because of the redundancy of the transport systems, high-dose IVIG may help to block FcRn resulting in the enhanced clearance of pathogenic autoantibodies, but will never be able to block it completely, as indicated in several experimental studies to date [42].
Although improving the binding of IgG to FcRn in vitro generally translates to an improved serum IgG half-life in vivo, this is not always the case. Recombinant therapeutics genetically engineered to contain IgG fragments with the CH2–CH3 domain that binds to FcRn can have significantly prolonged half-life due to protection of catabolism through FcRn binding. However, increased binding affinity to the FcRn does not appear to be proportional to the half-life extension. For example, when comparing variants of Herceptin antibody (an ERBB2-specific human IgG1 against mammary tumour cells) with a threefold increase in FcRn binding at acidic pH and another variant with a 12-fold increased binding at acidic pH and also enhanced binding at more neutral pH, both antibodies exhibited similar half-lives when tested in a humanized FcRn transgenic mouse model [43]. Increased binding may enhance degradation of IgG under neutral conditions. Clearly, there is an obvious need to have a better understanding of FcRn in the exact regulation of IgG-mediated responses and half-life in vivo.
Conclusions: applying the lessons of today
Research in immunoglobulin therapy with IVIG or SCIG has shown that therapy targets and treatment options evolve in parallel. Achieving good clinical outcomes to enable a state of health as found in immunocompetent individuals is achievable with the use of 0·4–0·6 g/kg/month for many patients with PI, although some patients may require higher doses. For patients with autoimmune neuropathies, an empirically derived starting dose of 2 g/kg is used frequently in the acute setting as in Guillain–Barré syndrome. For maintenance treatment, evidence from a recent randomized placebo-controlled trial in chronic inflammatory demyelinating neuropathy suggests that a dose of 1 g/kg every 3 weeks is sufficient to maintain strength [44]. Indications for review of immunoglobulin doses in patients with PI and autoimmune neuropathies are summarized in Table 5.
Table 5.
Indications for review of doses
| Primary immunodeficiency | Autoimmune neuropathies |
|---|---|
| Frequent infections despite achievement of trough or steady state IgG levels within the reference range | Progressive muscle weakness following achievement of stable disease |
| Failure to achieve adequate trough or steady state IgG levels despite use of currently recommended doses of immunoglobulin | Frequent dips in muscle strength before the next infusion of IVIG |
| Sudden deterioration in muscle power following an infection |
Ig: immunoglobulin; IVIG: intravenous immunoglobulin.
Immunoglobulin doses based on actual body weight have hitherto been derived empirically, yet the lack of diffusion of IgG into adipose tissue questions the validity of doses based on body weight in obese patients. The absence of correlation between trough IgG levels and annual dose of IgG in relation to body size (height, weight or body mass index) [45] suggests that dosing based on ideal body weight maybe equally effective, but this hypothesis remains to be proved.
While the questions physicians face are challenging and continually keep pace with progress itself, the future for patients in need of immunoglobulin therapies appears brighter than ever before. Through understanding the needs, specifics and clinical outcomes of patients in need of immune replacement therapy or immunomodulation, the application of IgG therapy can be improved by focusing upon the metrics derived from the patients themselves. The administration route, regimen and diversity of applications for IgG preparations are continually being optimized. A deeper understanding of immunoglobulin molecules, gene variability and its impact on the susceptibility of patients with certain gene patterns to IgG therapy may allow pharmacogenetic prediction of individual IgG dose requirements for patients and redefining IgG therapy.
Acknowledgments
The authors are grateful for the help of Mary Lucas for data collation, Helen Chapel, Jennifer Lortan and Smita Patel for inclusion of data on their patients, and nursing colleagues Janet Burton, Nicola Salome-Bentley and Carol Ross are gratefully acknowledged.
Disclosure
Dr Misbah has received honoraria for advisory board membership on immunoglobulin therapy from CSL Behring, Baxter and Biotest; Dr Kuijpers, honoraria for advisory activities of Sanquin; Dr van der Heijden, support from Sanquin for his scientific work as PhD student; Dr Grimbacher is a member of the IgPro20 Steering Committee and Advisory Boards, honoraria for presentations from Baxter and Grifols; Dr Orange, consultant fees from CSL Behring, Baxter Biosciences, IBT Reference Laboratories, and Octapharma research grants review committee. No other potential conflicts of interest were reported.
References
- 1.Yong PL, Boyle J, Ballow M, et al. Use of intravenous immunoglobulin and adjunctive therapies in the treatment of primary immunodeficiencies. A working group report of and study by the Primary Immunodeficiency Committee of the American Academy of Allergy Asthma and Immunology. Clin Immunol. 2010;135:255–63. doi: 10.1016/j.clim.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Hagan JB, Fasano MB, Spector S, et al. Efficacy and safety of a new 20% immunoglobulin preparation for subcutaneous administration, IgPro20, in patients with primary immunodeficiency. J Clin Immunol. 2010;30:734–45. doi: 10.1007/s10875-010-9423-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ochs HD, Gupta S, Kiessling P, Nicolay U, Berger M. Safety and efficacy of self-administered subcutaneous immunoglobulin in patients with primary immunodeficiency diseases. J Clin Immunol. 2006;26:265–73. doi: 10.1007/s10875-006-9021-7. [DOI] [PubMed] [Google Scholar]
- 4.Orange JS, Grossman WJ, Navickis RJ, Wilkes MM. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: a meta-analysis of clinical studies. Clin Immunol. 2010;137:21–30. doi: 10.1016/j.clim.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 5.Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol. 2010;125:1354–60. doi: 10.1016/j.jaci.2010.02.040. [DOI] [PubMed] [Google Scholar]
- 6.Halliday E, Winkelstein J, Webster AD. Enteroviral infections in primary immunodeficiency (PID): a survey of morbidity and mortality. J Infect. 2003;46:1–8. doi: 10.1053/jinf.2002.1066. [DOI] [PubMed] [Google Scholar]
- 7.Jolles S, Bernatowska E, de Gracia J, et al. Efficacy and safety of Hizentra® in patients with primary immunodeficiency after a dose-equivalent switch from intravenous or subcutaneous replacement therapy. Clin Immunol. 2011 doi: 10.1016/j.clim.2011.06.002. doi: 10.1016/j.clim.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Tarzi MD, Grigoriadou S, Carr SB, Kuitert LM, Longhurst HJ. Clinical immunology review series: an approach to the management of pulmonary disease in primary antibody deficiency. Clin Exp Immunol. 2009;155:147–55. doi: 10.1111/j.1365-2249.2008.03851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wasserman RL, Melamed I, Nelson RP, Jr, et al. Pharmacokinetics of subcutaneous IgPro20 in patients with primary immunodeficiency. Clin Pharmacokinetics. 2011;50:405–14. doi: 10.2165/11587030-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 10.Aghamohammadi A, Moin M, Farhoudi A, et al. Efficacy of intravenous immunoglobulin on the prevention of pneumonia in patients with agammaglobulinemia. FEMS Immunol Med Microbiol. 2004;40:113–18. doi: 10.1016/S0928-8244(03)00304-3. [DOI] [PubMed] [Google Scholar]
- 11.Foy HM, Cooney MK, Allan I, Kenny GE. Rates of pneumonia during influenza epidemics in Seattle, 1964 to 1975. JAMA. 1979;241:253–8. [PubMed] [Google Scholar]
- 12.Jokinen C, Heiskanen L, Juvonen H, et al. Incidence of community-acquired pneumonia in the population of four municipalities in eastern Finland. Am J Epidemiol. 1993;137:977–88. doi: 10.1093/oxfordjournals.aje.a116770. [DOI] [PubMed] [Google Scholar]
- 13.Woodhead MA, Macfarlane JT, McCracken JS, Rose DH, Finch RG. Prospective study of the aetiology and outcome of pneumonia in the community. Lancet. 1987;1:671–4. doi: 10.1016/s0140-6736(87)90430-2. [DOI] [PubMed] [Google Scholar]
- 14.Eades-Perner AM, Gathmann B, Knerr V, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2004−06. Clin Exp Immunol. 2007;147:306–12. doi: 10.1111/j.1365-2249.2006.03292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roifman CM, Schroeder H, Berger M, et al. Comparison of the efficacy of IGIV-C, 10% (caprylate/chromatography) and IGIV-SD, 10% as replacement therapy in primary immune deficiency. A randomized double-blind trial. Int Immunopharmacol. 2003;3:1325–33. doi: 10.1016/s1567-5769(03)00134-6. [DOI] [PubMed] [Google Scholar]
- 16.de Gracia J, Vendrell M, Alvarez A, et al. Immunoglobulin therapy to control lung damage in patients with common variable immunodeficiency. Int Immunopharmacol. 2004;4:745–53. doi: 10.1016/j.intimp.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 17.Mazer BD, Gelfand EW. An open-label study of high-dose intravenous immunoglobulin in severe childhood asthma. J Allergy Clin Immunol. 1991;87:976–83. doi: 10.1016/0091-6749(91)90420-s. [DOI] [PubMed] [Google Scholar]
- 18.Kainulainen L, Varpula M, Liippo K, Svedstrom E, Nikoskelainen J, Ruuskanen O. Pulmonary abnormalities in patients with primary hypogammaglobulinemia. J Allergy Clin Immunol. 1999;104:1031–6. doi: 10.1016/s0091-6749(99)70085-0. [DOI] [PubMed] [Google Scholar]
- 19.Thickett KM, Kumararatne DS, Banerjee AK, Dudley R, Stableforth DE. Common variable immune deficiency: respiratory manifestations, pulmonary function and high-resolution CT scan findings. QJM. 2002;95:655–62. doi: 10.1093/qjmed/95.10.655. [DOI] [PubMed] [Google Scholar]
- 20.Martinez Garcia MA, Hernandez F, de Rojas MD, et al. Respiratory disorders in common variable immunodeficiency. Respir Med. 2001;95:191–5. doi: 10.1053/rmed.2000.1020. [DOI] [PubMed] [Google Scholar]
- 21.Smith RP, Lipworth BJ. C-reactive protein in simple community-acquired pneumonia. Chest. 1995;107:1028–31. doi: 10.1378/chest.107.4.1028. [DOI] [PubMed] [Google Scholar]
- 22.Corsonello A, Pedone C, Battaglia S, Paglino G, Bellia V, Incalzi RA. C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) as inflammation markers in elderly patients with stable chronic obstructive pulmonary disease (COPD) Arch Gerontol Geriatr. 2010 doi: 10.1016/j.archger.2010.10.015. doi: 10.1016/j.archger.2010.10.015. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 23.Miedema KG, de Bont ES, Elferink RF, et al. The diagnostic value of CRP, IL-8, PCT, and sTREM-1 in the detection of bacterial infections in pediatric oncology patients with febrile neutropenia. Support Care Cancer. 2010 doi: 10.1007/s00520-010-0987-6. doi: 10.1007/s00520-010-0987-6. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chapel HM, Spickett GP, Ericson D, Engl W, Eibl MM, Bjorkander J. The comparison of the efficacy and safety of intravenous versus subcutaneous immunoglobulin replacement therapy. J Clin Immunol. 2000;20:94–100. doi: 10.1023/a:1006678312925. [DOI] [PubMed] [Google Scholar]
- 25.Gardulf A, Nicolay U, Asensio O, et al. Rapid subcutaneous IgG replacement therapy is effective and safe in children and adults with primary immunodeficiencies – a prospective, multi-national study. J Clin Immunol. 2006;26:177–85. doi: 10.1007/s10875-006-9002-x. [DOI] [PubMed] [Google Scholar]
- 26.Berger M. Subcutaneous immunoglobulin replacement in primary immunodeficiencies. Clin Immunol. 2004;112:1–7. doi: 10.1016/j.clim.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 27.Thepot S, Malphettes M, Gardeur A, et al. Immunoglobulin dosage and switch from intravenous to subcutaneous immunoglobulin replacement therapy in patients with primary hypogammaglobulinemia: decreasing dosage does not alter serum IgG levels. J Clin Immunol. 2010;30:602–6. doi: 10.1007/s10875-010-9417-2. [DOI] [PubMed] [Google Scholar]
- 28.Harbo T, Andersen H, Hess A, Hansen K, Sindrup SH, Jakobsen J. Subcutaneous versus intravenous immunoglobulin in multifocal motor neuropathy: a randomized, single-blinded cross-over trial. Eur J Neurol. 2009;16:631–8. doi: 10.1111/j.1468-1331.2009.02568.x. [DOI] [PubMed] [Google Scholar]
- 29.Eftimov F, Vermeulen M, de Haan RJ, van den Berg LH, van Schaik IN. Subcutaneous immunoglobulin therapy for multifocal motor neuropathy. J Peripher Nerv Syst. 2009;14:93–100. doi: 10.1111/j.1529-8027.2009.00218.x. [DOI] [PubMed] [Google Scholar]
- 30.Misbah S, Baumann A, Fazio R, et al. A smooth transition protocol for patients with multifocal motor neuropathy going from intravenous to subcutaneous immunoglobulin therapy: an open-label proof-of-concept study. J Peripher Nerv Syst. 2011;16:92–7. doi: 10.1111/j.1529-8027.2011.00330.x. [DOI] [PubMed] [Google Scholar]
- 31.Tayal U, Burton J, Dash C, Wojnarowska F, Chapel H. Subcutaneous immunoglobulin therapy for immunomodulation in a patient with severe epidermolysis bullosa acquisita. Clin Immunol. 2008;129:518–19. doi: 10.1016/j.clim.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Shapiro R. Subcutaneous immunoglobulin therapy by rapid push is preferred to infusion by pump: a retrospective analysis. J Clin Immunol. 2010;30:301–7. doi: 10.1007/s10875-009-9352-2. [DOI] [PubMed] [Google Scholar]
- 33.Willcocks LC, Lyons PA, Clatworthy MR, et al. Copy number of FCGR3B, which is associated with systemic lupus erythematosus, correlates with protein expression and immune complex uptake. J Exp Med. 2008;205:1573–82. doi: 10.1084/jem.20072413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aitman TJ, Dong R, Vyse TJ, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–5. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 35.Breunis WB, van Mirre E, Bruin M, et al. Copy number variation of the activating FCGR2C gene predisposes to idiopathic thrombocytopenic purpura. Blood. 2008;111:1029–38. doi: 10.1182/blood-2007-03-079913. [DOI] [PubMed] [Google Scholar]
- 36.de Haas M, Kleijer M, van Zwieten R, Roos D, von dem Borne AE. Neutrophil Fc gamma RIIIb deficiency, nature, and clinical consequences: a study of 21 individuals from 14 families. Blood. 1995;86:2403–13. [PubMed] [Google Scholar]
- 37.Koene HR, Kleijer M, Roos D, de Haas M, von dem Borne AE. Fc gamma RIIIB gene duplication: evidence for presence and expression of three distinct Fc gamma RIIIB genes in NA(1+,2+)SH(+) individuals. Blood. 1998;91:673–9. [PubMed] [Google Scholar]
- 38.Bournazos S, Woof JM, Hart SP, Dransfield I. Functional and clinical consequences of Fc receptor polymorphic and copy number variants. Clin Exp Immunol. 2009;157:244–54. doi: 10.1111/j.1365-2249.2009.03980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Breunis WB, van Mirre E, Geissler J, et al. Copy number variation at the FCGR locus includes FCGR3A, FCGR2C and FCGR3B but not FCGR2A and FCGR2B. Hum Mutat. 2009;30:E640–50. doi: 10.1002/humu.20997. [DOI] [PubMed] [Google Scholar]
- 41.Huizinga TW, Kuijpers RW, Kleijer M, et al. Maternal genomic neutrophil FcRIII deficiency leading to neonatal isoimmune neutropenia. Blood. 1990;76:1927–32. [PubMed] [Google Scholar]
- 42.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. 2007. pp. 715–25. [DOI] [PubMed]
- 43.Petkova SB, Akilesh S, Sproule TJ, et al. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. 2006;18:1759–69. doi: 10.1093/intimm/dxl110. [DOI] [PubMed] [Google Scholar]
- 44.Hughes RA, Donofrio P, Bril V, et al. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol. 2008;7:136–44. doi: 10.1016/S1474-4422(07)70329-0. [DOI] [PubMed] [Google Scholar]
- 45.Khan S, Grimbacher B, Boecking C, et al. Serum trough IgG level and annual intravenous immunoglobulin dose are not related to body size in patients on regular replacement therapy. Drug Metab Lett. 2011;5:132–6. doi: 10.2174/187231211795305302. [DOI] [PubMed] [Google Scholar]