

Abstract
Helicobacter pylori induces an infiltration of dendritic cells (DCs) into the infected gastric mucosa. Although DCs play an important role in the regulation of inflammation, the effects of H. pylori vacuolating cytotoxin (VacA) on DC maturation process have not yet been elucidated. The role of VacA in DC maturation following co-exposure to Escherichia coli lipopolysaccharide (LPS) was investigated. The treatment of immature DCs with LPS up-regulated the expression of surface molecules [e.g. CD40, CD80, CD86 and major histocompatibility complex (MHC) class II], as well as the production of cytokines [e.g. interleukin (IL)-1β, IL-12p70 and tumour necrosis gactor (TNF)-α] compared with those of unstimulated controls. Co-stimulation with H. pylori VacA significantly reduced the up-regulated DC maturation markers induced by LPS. In addition, VacA sustained the immature state of DCs with high endocytosis and low migratory capacity. The LPS-induced down-regulation of E2F1 expression in DCs was recovered by co-stimulation with VacA. Moreover, suppression of E2F1 by small interfering RNA resulted in a significant recovery of the inhibited DC maturation by VacA. In contrast, VacA did not affect nuclear factor (NF)-κB responses to LPS and the NF-κB signal was not associated with VacA-induced inhibition of DC maturation. These results suggest that the exposure of DCs to H. pylori VacA negatively regulates DC maturation via the restoration of E2F1. The immunomodulatory action of VacA on DCs may contribute to the ability of VacA-producing H. pylori to establish a persistent infection in the gastric mucosa.
Keywords: dendritic cells, E2F1, H. pylori vacuolating cytotoxin, maturation
Introduction
Helicobacter pylori is a Gram-negative bacterium that colonizes the mucus layer of the stomach and is the causative agent of diseases such as chronic gastritis, peptic ulcers, gastric cancers and gastric mucosa-associated lymphoid tissue (MALT) lymphoma [1]. Although the bacteria do not invade the gastric lamina propria, they can induce severe infiltrations of neutrophils and lymphocytes. In addition, many dendritic cells (DCs) are observed in human gastric mucosa infected with H. pylori[2].
The majority of H. pylori-infected patients show lifelong persistence of infection [3]. The inability to eliminate H. pylori may be due to immune-evasive strategies of the bacteria. Several mechanisms of immune evasion have been proposed, including antigenic and phase variations, modulation of adhesion molecules and immune inhibition by H. pylori vacuolating cytotoxin (VacA) [4–6]. Recently, a study demonstrated that adoptive transfer of H. pylori-pulsed DCs skewed regulatory T cells (Treg) and reduced the ratio of gastric interleukin (IL)-17/forkhead box P3 (FoxP3) mRNA expression [7]. These results suggest that DCs may play a role in the suppression of effective immunity against H. pylori infection.
DCs are potent antigen-presenting cells (APCs) [8]. In the resting state, the majority of immature DCs exist within peripheral tissues where they continuously incorporate antigens from the environment via endocytosis. Thus, contact with pathogens and/or proinflammatory mediators induces DCs to undergo a complex transformation process termed ‘DC maturation’. The remarkable features of DC maturation contain the up-regulation of surface co-stimulatory (e.g. CD40, CD80 and CD86) and major histocompatibility complex (MHC) molecules, the activation of lysosomal antigen-processing mechanisms, the production of cytokines and the acquisition of the capacity to migrate to secondary lymphoid organs [8–10]. Upon arriving at the secondary lymphoid organs, mature DCs present antigenic peptides to naive T cells using surface MHC molecules. The antigen presentation leads to the initiation of antigen-specific T cell immune responses. These processes are a unique to mature DCs. Therefore, controlling DC maturation is critical for regulation of adaptive immune responses [11].
Vacuolating cytotoxin (VacA) is one of the major virulence factors in the pathogenesis of H. pylori-related diseases [12,13]. VacA has a high potency for negatively regulating the proliferation of lymphocytes in vitro[14–19]. The bacterium H. pylori itself is reported to be a stimulus to the maturation of human monocyte-derived DCs [20], although the CagA protein of H. pylori inhibits the maturation of DCs [21]. Based on these data, there is a possibility that VacA may have an effect on the regulation of DC functions.
Nuclear factor (NF)-κB activation is known to be an important event underlying DC maturation [22,23]. Transcription factor E2F1 is also involved in the regulation of DC maturation [11]. Although H. pylori can activate NF-κB signalling pathways in several cells such as monocytes and gastric epithelial cells [24,25], little information is available on DC maturation in response to stimulation with H. pylori VacA. The goal of this study was to investigate the role of VacA in modulating the maturation process of mouse bone marrow (BM)-derived DCs. H. pylori VacA is shown herein to suppress DC maturation through a restoration of E2F1 signalling.
Materials and methods
Reagents
Murine recombinant granulocyte–macrophage colony-stimulating factor (GM-CSF), murine recombinant IL-4 and CCL21 (SLC) were obtained from PeproTech (Rocky Hill, NJ, USA). RPMI-1640, lipopolysaccharide (LPS)-free fetal bovine serum (FBS), antibiotics, l-glutamine and Trizol were obtained from Gibco BRL (Gaithersburg, MD, USA). Red blood cell (RBC) lysing buffer, 2-mercaptoethanol, LPS from Escherichia coli (serotype O55:B5) and fluorescein isothiocyanate (FITC)-conjugated dextran (40 000 MW) were provided by Sigma Chemical Co. (St Louis, MO, USA). FITC-conjugated monoclonal antibodies (mAbs) against murine CD11c, CD40, CD80, CD86 and MHC class II (I-A/I-E) were obtained from BD Pharmingen (San Diego, CA, USA). Antibodies against IκBα and actin were acquired from Cell Signaling Technology, Inc. (Beverly, MA, USA), and the antibody against E21b was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Hoechst 33258 [2′-(4-hydroxyphenyl)-5-(4-methyl-piperazinyl)-2,5-bi-1H-benzimidazole] was purchased from CalBiochem (San Diego, CA, USA).
Purification of H. pylori VacA
VacA-producing H. pylori strain 60190 (American Type Culture Collection, Manassas, VA, USA; ATCC 49503, CagA+, vacA s1a/m1) was grown in sulphite-free Brucella broth containing 5% FBS at 37°C under microaerophilic conditions. VacA was purified from broth culture supernatants according to our previously published procedures [26,27]. The purification method of VacA protein was kindly supported by Professor Patrice Boquet at Laboratoire de bacteriologie, Hopital de l'Archet 2, France [28]. Immediately before use in cells, the purified VacA protein was activated through the addition of 250 mM HCl until the pH reached 2·0 [29]. NH4Cl (5 mM) was also added to the medium to enhance the VacA activity [30].
Generation of BM-derived DCs and culture conditions
Femurs and tibias of specific pathogen-free C57BL/6 or NF-κB knock-out mice (4–12 weeks old; Jackson Laboratory, Bar Harbor, ME, USA) were removed and BM cells were obtained, as described previously [23]. Cells were depleted of RBCs using RBC lysing buffer and then cultured in ultra-low-adherence 24-well plates (Costar, Corning, NY, USA) in complete DC media. Cultures were fed every 2 days by removing and replacing half the supernatant with fresh media. RPMI-1640 media supplemented with 10% heat-inactivated FBS, 1% antibiotics, l-glutamine (2 mM), 2-mercaptoethanol (55 µM), murine recombinant GM-CSF (10 ng/ml) and murine recombinant IL-4 (10 ng/ml) was used. After 6 days of culture, DCs were harvested and stimulated with VacA or Escherichia coli LPS for the indicated times. All animal procedures were approved by Hanyang University College of Medicine. On day 6 of the culture, ∼80% of the harvested cells expressed CD11c assessed by flow cytometry.
DC2·4, an immature murine DC cell line, was cultured in RPMI-1640 supplemented with 10% FBS, 1% antibiotics, l-glutamine (2 mM), sodium pyruvate (1 mM) and non-essential amino acids (2 mM), and were grown at 37°C with 5% CO2, as described previously [31].
Immunophenotypic analysis for DC maturation
Cells were washed and resuspended in phosphate-buffered saline (PBS) containing 3% FBS and 0·09% NaN3. Cells were then incubated with a panel of FITC-conjugated mAbs. Isotype-matched antibodies were used as a background control. Cells were analysed with flow cytometry (FACSCalibur cytometer; Becton Dickinson, San Jose, CA, USA), including 20 000 cells per sample. The expression levels of each antigen were expressed as mean fluorescence intensity (MFI), as described previously [23].
Quantification of endocytosis
DCs (1 × 106) were pulsed with FITC-conjugated dextran (40 000 MW, 1 mg/ml) and were incubated for 60 min at 37°C or 4°C. Cold staining buffer was added to stop the reaction, after which the cells were washed three times at 4°C, stained with phycoerythrin (PE)-conjugated anti-CD11c antibody, and then analysed using flow cytometry. Non-specific binding of dextran to DCs was determined by incubating DCs with FITC-conjugated dextran at 4°C. This value was subtracted to determine the levels of specific binding, as described previously [32].
Migration assay
For an in vitro study, DCs (5 × 105) in serum-free medium were placed in a transwell migration chamber (Costar 3421) and allowed to migrate through a polycarbonate membrane (pore size, 5 mm) at 37°C, as described previously [11]. Briefly, secondary lymphoid-tissue chemokine CCL21 (SLC, 100 ng/ml) was added to the lower chamber to induce cell chemotaxis. After 6 h, the wells were removed and the cells at the top of the membrane were removed with a cotton swab. Cells at the bottom of the membrane were fixed with 3·7% formaldehyde, stained with the DNA dye Hoechst 33258 (5 µg/ml) and counted using epifluorescence microscopy (Axiophot, Carl Zeiss, Oberkochen, Germany).
Transfection and NF-κB-luciferase reporter assay
Small interfering RNA (siRNA) against E2F1 was constructed as described by Bukur et al. [33]. The specific oligonucleotide sequence for the E2F1 gene was 5′-CCU GGA GCA AGA AGC AGU AUU GCC A-3′. Negative control non-silencing RNA (NS-RNA) was purchased from Invitrogen. Transfection was performed according to the procedure described previously [23,27]. Briefly, DC2·4 cells were transfected in 24-well plates with HiPerFect transfection reagent (Qiagen, Valencia, CA, USA), according to the manufacturer's instructions. siRNAs were diluted in a final volume of 100 µl of OptiMEM medium, to which 6 µl of HiPerFect reagent was added, and the mixture was incubated for 15 min for complex formation. The transfection mixture was added dropwise to the culture, and the cells were incubated at 37°C in a 5% CO2 environment for 48 h until used in the indicated experiments.
The p2x NF-κB-, pβ-actin- and pRSV-β-galactosidase transcriptional reporters were kindly provided by Dr Martin F. Kagnoff of the University of California, San Diego [34]. Immature DCs were transfected at day 5 with 1·5 µg of plasmid DNA using FuGene6 transfection reagent (Roche, Mannheim, Germany). The transfected cells were incubated for 24 h at 37°C in a 5% CO2 incubator and were then treated with LPS and/or VacA. Luciferase activity was determined in accordance with the manufacturer's instructions (Tropix Inc., Bedford, MA, USA). Light release was quantitated for 10 s using a luminometer (MicroLumat Plus, Berthold GmbH & Co. KG, Bad Wildbad, Germany), as described previously [35].
Quantitative reverse transcription–polymerase chain reaction (RT–PCR) and enzyme-linked immunosorbent assay (ELISA)
Total cellular RNA was extracted using an acid guanidinium–phenol–chloroform method (Trizol). Quantitative RT–PCR using an internal standard was used to quantify mouse E2F1 and β-actin mRNA levels. Standard RNA for mouse E2F1 was generated by in vitro transcription using T7 RNA polymerase, as described previously [36] and encoded 5′ and 3′ priming sites in E2F1. PCR amplification consisted of 35 cycles of 1 min denaturation at 95°C, and 2·5 min annealing and extension at either 50°C (E2F1) or 72°C (β-actin). The primers were 5′-TAG CCC TGG GAA GAC CTC AT-3′ (sense) and 5′-CCC CAA AGT CAC AGT CAA AGA G-3′ (anti-sense) for E2F1 [11] and 5′-GTG GGC CGC TCT AGG CAC CAA-3′ (sense) and 5′-CTC TTT GAT GTC ACG CAC GAT TTC-3′ (anti-sense) for β-actin. The PCR product sizes for the target RNA were 368 base pairs (bp) (E2F1) and 540 bp (β-actin) and those of the standard RNA were 310 bp (E2F1) and 746 bp (β-actin).
The amounts of cytokines in triplicate cell culture supernatants were measured using commercially available ELISA kits (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's instructions. Prior to performing ELISA, supernatants were filtered through a 0·22-µm filter to remove any contaminants. To measure the levels of phospho-IκBα, an IκBα ELISA kit (Active Motif, Carlsbad, CA, USA) was used in accordance with the manufacturer's directions [24].
Electrophoretic mobility shift assay (EMSA)
Cells were harvested and nuclear extracts prepared as described previously [35]. Protein concentrations in the extracts were determined according to the Bradford assay (Bio-Rad, Hercules, CA, USA), and EMSA was performed according to the manufacturer's instructions (Promega, Madison, WI, USA). In brief, 5 µg of nuclear extract was incubated for 30 min at room temperature with a γ32P-labelled oligonucleotide probe (5′-AGT TGA GGG GAC TTT CCC AGG C-3′ for the NF-κB binding site). After incubation, both bound and free DNA were resolved on 5% polyacrylamide gels, as described previously [35,37].
Immunoblot analyses
DCs were washed with ice-cold PBS and lysed in 0·5 ml/well lysis buffer (150 mM NaCl, 20 mM Tris, pH 7·5, 0·1% Triton X-100, 1 mM phenylmethylsulphonyl fluoride (PMSF), 10 µg/ml aprotonin), as described previously [23]. Protein concentrations in the lysates were determined using the Bradford assay. Fifteen to 50 µg of protein/lane was size-fractionated on 6% polyacrylamide minigels (Mini-PROTEIN II; Bio-Rad) and transferred electrophoretically to a nitrocellulose membrane (0·1-µm pore size). The immunoreactive proteins using primary mAbs were visualized using goat anti-rabbit or anti-mouse secondary antibodies conjugated to horseradish peroxidase (Transduction Laboratories, Lexington, KY, USA), followed by enhanced chemiluminescence (ECL system; Amersham Life Sciences, Buckinghamshire, UK) and exposure to X-ray film (XAR5; Eastman Kodak Company, Rochester, NY, USA).
Statistical analyses
Data are presented as the mean ± standard deviation (s.d.) or mean ± standard error of the mean (s.e.m.). Wilcoxon's rank sum test was used for statistical analysis.
Results
H. pylori VacA reduces the maturation of E. coli LPS-treated DCs
Immature DCs were generated from murine BM cells by culturing with GM-CSF and IL-4 for six days. The generated DCs were incubated with VacA (10 µg/ml) in the presence or absence of E. coli LPS (200 ng/ml) for 24 h, and the effect of VacA on DC maturation was evaluated. As shown in Fig. 1, the addition of LPS significantly increased the number of cells with DC maturation markers, including CD40, CD80, CD86 and MHC class II, compared with that of the unstimulated control. In contrast, the number of cells with DC maturation markers was reduced significantly in the combined treatment with VacA and LPS compared with that of LPS alone. Stimulation of immature DCs with increasing concentrations of VacA led to a dose-dependent decrease in the surface expression of CD40 molecules. At VacA concentrations of 0·1, 1, 10 and 50 µg/ml, the co-stimulation with VacA and LPS (200 ng/ml) reduced the MFI of CD40 by 3%, 20%, 42% and 51%, respectively, relative to that of LPS-stimulated cells alone (mean value, n = 3). Significant suppression of CD40 expression was first apparent 24 h after co-stimulation with VacA and LPS and continued to suppress over the ensuing 48 h compared with LPS alone (Fig. 2a).
Fig. 1.
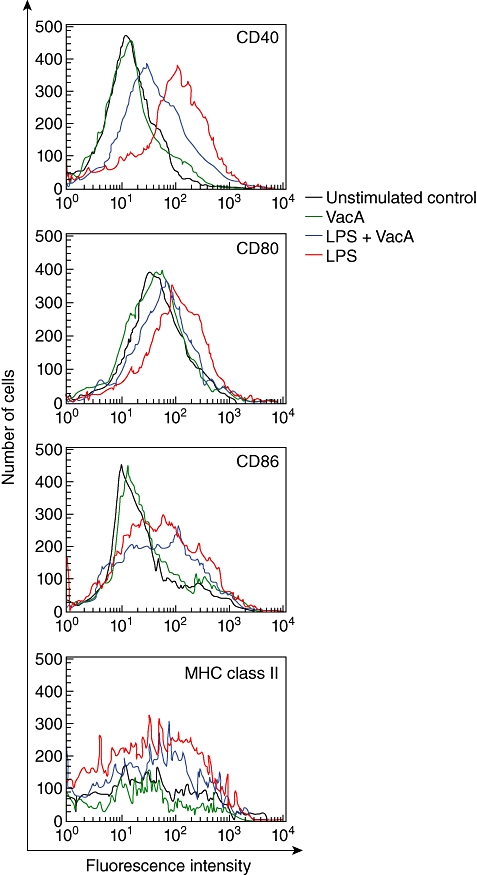
Helicobacter pylori VacA inhibits the maturation of lipopolysaccharide (LPS)-treated dendritic cells (DCs). DCs generated from C57BL/6 mice were incubated with vacuolating cytotoxin (VacA) (10 µg/ml) in the presence or absence of LPS (200 ng/ml) for 24 h. Cells were stained with monoclonal antibodies (mAbs) against surface molecules and were analysed using flow cytometry. Results are representative of five independent experiments. Black lines, unstimulated DCs; green lines, VacA-stimulated DCs; red lines, LPS-stimulated DCs; blue lines, VacA + LPS-stimulated DCs.
Fig. 2.

Effects of Helicobacter pylori VacA on lipopolysaccharide (LPS)-induced CD40 expression and cytokine production in dendritic cells (DCs). (a) Kinetics of CD40 expression on DCs treated with LPS and vacuolating cytotoxin (VacA). Murine DCs were incubated with VacA (10 µg/ml) in the presence or absence of LPS (200 ng/ml) for the indicated time-periods. Cells were stained with monoclonal antibodies against CD40 and were analysed using flow cytometry. The data represent the mean fluorescence intensity ± standard error of the mean (s.em.) (n = 3). *P < 0·05 versus LPS-stimulated DCs (b) DCs were incubated with VacA (10 µg/ml) in the presence or absence of LPS (200 ng/ml) for 24 h. Concentrations of each cytokine in the culture supernatants were determined using enzyme-linked immunosorbent assay (mean ± s.e.m., n = 5). *P < 0·05 versus LPS-stimulated DCs.
H. pylori VacA inhibits cytokine production in E. coli LPS-stimulated DCs
After DCs were incubated with LPS in the presence or absence of VacA, production of cytokines in the culture supernatants was measured using ELISA. LPS-treated DCs produced high amounts of IL-1β, IL-12p70 and TNF-α, and the combined treatment with LPS and VacA decreased cytokine production significantly compared with that of LPS alone (Fig. 2b).
H. pylori VacA sustains the immature state of DCs with high endocytosis and low migratory capacity
DC maturation is accompanied by the reduced capacity for antigen-uptake and enhanced migration [8,9]. To investigate the effects of VacA on antigen-uptake in LPS-treated DCs, a mannose receptor-mediated endocytosis assay was performed using FITC-conjugated dextran internalization. As shown in Fig. 3a, the cell population with CD11c+ and FITC–dextran+ (area R1) was higher in the untreated control than it was in the LPS-treated group. However, the combined treatment with LPS and VacA increased the numbers of CD11c+ and FITC–dextran+ cells compared with those of LPS alone. The quantitative data for area R1 obtained from several independent experiments are presented in Fig. 3b. To examine the effect of VacA on DC migratory capacity, DCs stimulated with VacA and/or LPS were examined with regard to migration in response to CCL21. A migration assay revealed that CCL21 increased the migration of LPS-treated DCs, and that the combined treatment with LPS and VacA reduced the migration of DCs significantly (Fig. 3c). These data suggest that VacA may sustain DCs in the immature state.
Fig. 3.

Effects Helicobacter pylori VacA on endocytosis and migration in dendritic cells (DCs). (a) Murine DCs were incubated with vacuolating cytotoxin (VacA) (10 µg/ml) in the presence or absence of lipopolysaccharide (LPS) (200 ng/ml) for 24 h. Fluorescein isothiocyanate (FITC)–dextran was analysed on CD11c-phycoerythrin (PE)-positive cells using flow cytometry. The results are representative of more than three independent experiments. (b) The percentage of CD11c+ and FITC–dextran+ cells. Data are expressed as the mean ± standard deviation of area R1 (n = 3). (c) DCs (5 × 105/well) were placed in a transwell migration chamber, with chemokine CCL21 (100 ng/ml) in the lower chamber, as described in the Materials and methods. Values represent the mean of migrated cells ± standard error of the mean of five independent experiments. *P < 0·05.
VacA attenuates the LPS-induced down-regulation of E2F1 activity in DCs
Because E2F1 is reported to be a critical regulator of DC maturation [11], we assessed whether E2F1 expression could be altered in the course of DC maturation due to treatment with LPS. The kinetics of E2F1 expression in DCs was examined using quantitative RT–PCR analysis with standard RNA, which showed that DCs treated with LPS exhibited a time-dependent change in E2F1 mRNA expression. Thus, E2F1 expression began to decrease in as little as 3 h after LPS treatment and continued to decrease over the next 24 h (Fig. 4a). In this system, the co-stimulation of DCs with VacA and LPS significantly attenuated the down-regulation of E2F1 at 12–24 h compared with that of LPS alone. The β-actin mRNA levels in stimulated and unstimulated DCs remained relatively constant throughout the same period (∼5 × 106 transcripts/µg of total RNA). Immunoblot analysis confirmed that the LPS-induced down-regulation of E2F1 expression at the protein level in DCs was recovered by the addition of VacA (Fig. 4b).
Fig. 4.
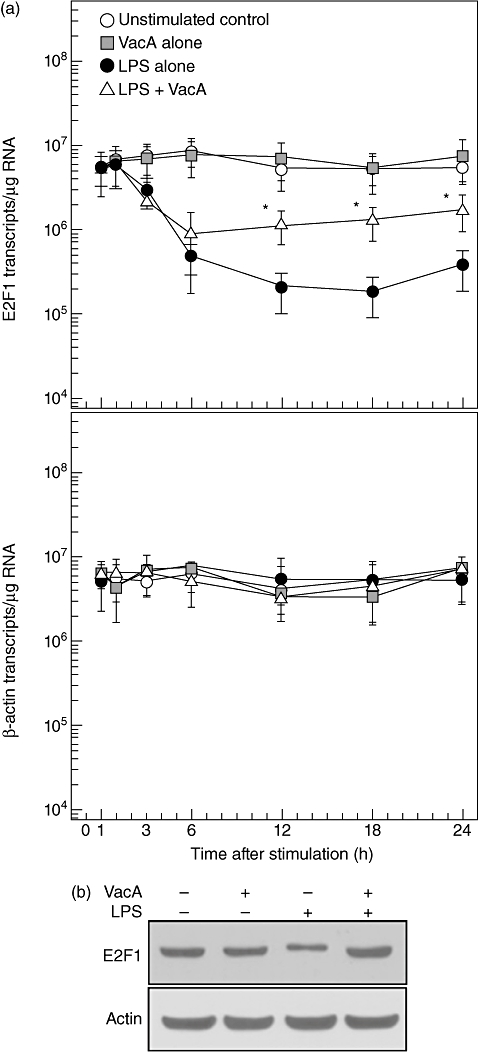
Effects of Helicobacter pylori VacA on E2F1 expression in the time course of lipopolysaccharide (LPS)-treated dendritic cells (DCs). (a) Murine BM-derived DCs were incubated with vacuolating cytotoxin (VacA) (10 µg/ml) in the presence or absence of LPS (200 ng/ml) for the indicated periods of time. Levels of E2F1 and β-actin mRNA were analysed using quantitative reverse transcription–polymerase chain reaction with each standard RNA. Values are expressed as the mean ± SD (n = 5). *P < 0·05 versus LPS alone. (b) The protein levels of E2F1 and actin in DCs treated with VacA (10 µg/ml) or LPS (200 ng/ml) for 12 h were detected using immunoblot analysis. The results are representative of more than three independent experiments.
Restoration of E2F1 activity by H. pylori VacA is associated with inhibition of LPS-induced DC maturation
We next asked whether the change of E2F1 expression could affect the maturation of DCs stimulated with LPS and VacA. In this experiment, siRNA against E2F1 and the DC2·4 cell line were used. DC2·4 cells transfected with E2F1 siRNA exhibited a significantly lower E2F1 protein level in a resting state (Fig. 5a). In this experimental system, co-stimulation with both LPS and VacA showed higher MFI values of surface molecules such as CD40 and CD80 in E2F1 siRNA-transfected cells compared to those in untransfected or non-silencing RNA (NS-RNA)-transfected cells (Fig. 5b). However, there was no significant difference in the MFI values between the LPS alone and LPS + VacA group in the E2F1 siRNA-transfected cells.
Fig. 5.
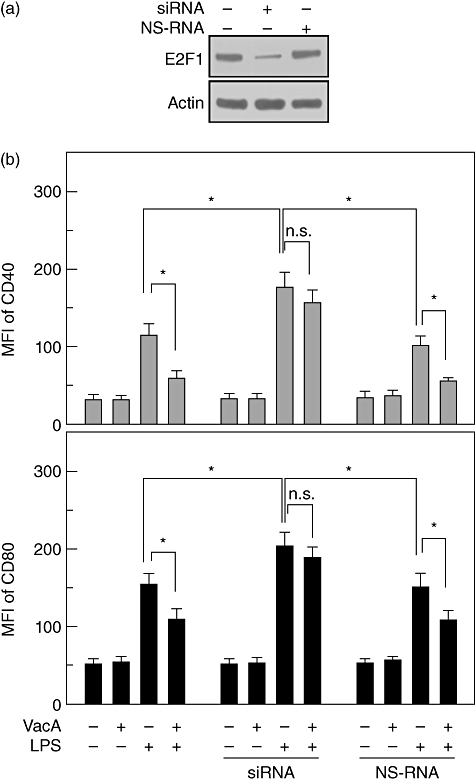
Effects of E2F1 suppression on surface molecule expression in DC2·4 cells stimulated with lipopolysaccharide (LPS) and vacuolating cytotoxin (VacA). (a) DC2·4 cells were transfected with siRNA against E2F1 or non-silencing RNA (NS-RNA), as described in the Materials and methods. The protein levels of E2F1 and actin were determined using immunoblot analysis. The results are representative of three independent experiments. (b) The transfected cells were treated with VacA (10 µg/ml) and/or LPS (200 ng/ml) for 24 h. Cells were stained with monoclonal antibodies against CD40 or CD80, and were analysed using flow cytometry. The data represent the mean fluorescence intensity ± standard error of the mean of five independent experiments. *P < 0·05; n.s.: non-significant.
Transfection with siRNA against E2F1 decreased the endocytotic capacity of DCs; however, significant differences in endocytosis were not observed between LPS alone and LPS + VacA in siRNA-transfected cells (Fig. 6a). In untransfected cells, a migration assay showed that CCL21 increased the migration of LPS-treated DCs, but that the addition of VacA reduced their migration significantly (Fig. 6b). In E2F1 siRNA-transfected cells, the migratory capacity of DCs due to co-stimulation with LPS and VacA was enhanced compared with that of untransfected cells. However, significant differences in migration were not shown between LPS alone and LPS + VacA in siRNA-transfected cells. Similar results were also observed with regard to cytokine production such as those of IL-12p70 and TNF-α (Fig. 6c). These results suggest that H. pylori VacA can attenuate the LPS-induced down-regulation of E2F1 activity, and that the restoration of E2F1 may play a role in the inhibition of LPS-induced DC maturation.
Fig. 6.
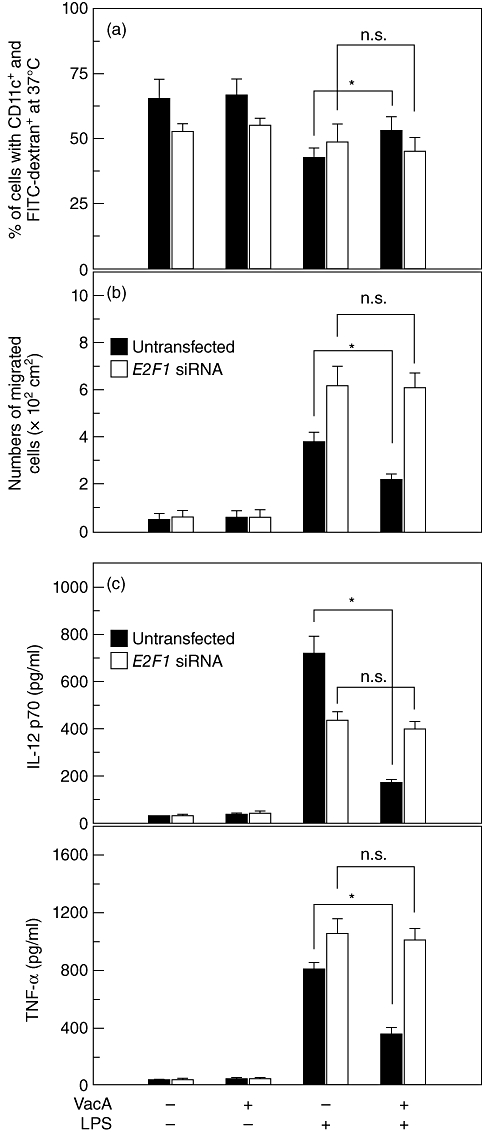
Effects of E2F1 suppression on endocytosis, migration and cytokine production in DC2·4 cells stimulated with lipopolysaccharide (LPS) and vacuolating cytotoxin (VacA). (a) DC2·4 cells were transfected with siRNA against E2F1 as described in the Materials and methods. The transfected or untransfected cells were stimulated with VacA (10 µg/ml) and LPS (200 ng/ml) for 24 h. Fluorescein isothiocyanate (FITC)–dextran was analysed on CD11c-phycoerythrin-positive cells using flow cytometry. Values represent the percentages of cells with CD11c+ and FITC–dextran+ at 37°C ± standard deviation of five independent experiments. The percentage of cells with CD11c+ and FITC–dextran+ at 4°C remained relatively constant in each experimental group (∼7%). (b) Cell culture conditions were identical to those in (a). DC2·4 cells (5 × 105/well) were placed in a transwell migration chamber, with chemokine CCL21 (100 ng/ml) in the lower chamber, as described in the Materials and methods. Values represent the mean of the migrated cells ± standard error of the mean (s.e.m.) of five independent experiments. (c) The transfected or untransfected cells were incubated with VacA (10 µg/ml) in the presence or absence of LPS (200 ng/ml) for 24 h. Concentrations of cytokines in the culture supernatants were determined using enzyme-linked immunosorbent assay (mean ± s.e.m., n = 5). *, P < 0·05; n.s.: non-significant.
NF-κB signals are not associated with the inhibition of DC maturation in response to VacA stimulation
NF-κB is a well-known transcriptional factor that plays an important role in DC maturation [23,38,39]. We determined whether H. pylori VacA affected activated NF-κB signals in DCs stimulated with E. coli LPS. As shown in Fig. 7a, there was a remarkable increase in DNA binding activity of NF-κB when DCs were treated with LPS. The combination of both LPS and VacA did not affect NF-κB response to stimulation with LPS alone. Consistent with this, degradation of IκBα by LPS was not recovered significantly after co-stimulation with VacA, as determined by immunoblot analysis. Levels of phospho-IκBα were measured using a commercial phospho-IκBα ELISA kit, and the results showed that the combined treatment with LPS and VacA did not change the levels of phospho-IκBα significantly, while LPS alone did increase these levels (Fig. 7b). To confirm these results, a reporter gene assay was performed. As shown in Fig. 7c, no significant difference in NF-κB activity was observed between LPS alone and LPS + VacA.
Fig. 7.

Helicobacter pylori vacuolating cytotoxin (VacA) does not affect nuclear factor (NF)-κB signals in lipopolysaccharide (LPS)-treated dendritic cells (DCs). (a) Murine bone marrow (BM)-derived DCs were incubated with VacA (10 µg/ml) in the presence of LPS (200 ng/ml) for 1 h. NF-κB DNA binding activity was assessed using electrophoretic mobility shift assay (EMSA). Immunoblots for concurrent IκBα and actin levels in cells under the same conditions are provided beneath each EMSA. The results are representative of more than five independent experiments. (b) IκBα phosphorylation in DCs treated with LPS or VacA for 1 h was measured using an IκBα enzyme-linked immunosorbent assay kit. Data are expressed as the mean fold induction ± standard error of the mean (s.e.m.) of phosphorylated IκBα activity relative to that of the untreated controls (n = 5). (c) Mouse BM-derived immature DCs at day 5 were transfected with 2× NF-κB-luciferase reporter plasmid as described in the Materials and methods. After 24 h, transfected cells were treated with VacA (10 µg/ml) in the presence of LPS (200 ng/ml) for 1 h. Data are expressed as mean fold induction ± s.e.m. of luciferase activity relative to that of the unstimulated controls (n = 5). The mean fold induction of β-actin reporter gene activity relative to that of the unstimulated controls remained relatively constant throughout each experiment; n.s.: non-significant.
We next determined whether suppression of NF-κB activity could influence DC maturation in VacA-treated immature DCs. For this experiment, BM-derived DCs from NF-κB wild-type and knock-out mice were used. In wild-type mice-derived DCs, increased MFI values of CD40 molecules due to treatment with LPS was reduced significantly by co-treatment with VacA. In knock-out mice-derived DCs, the MFI of CD40 was decreased by ∼ 53% compared with that of wild-type mice-derived DCs after exposure to LPS alone. In this experimental system, combined treatment with LPS and VacA significantly decreased the MFI of CD40 compared with that of LPS alone (Fig. 8a). Similar results for IL-12p70 production were noted following LPS and VacA stimulation of DCs derived from NF-κB knock-out mice (Fig. 8b). These results suggest that H. pylori VacA does not affect NF-κB responses to LPS, in which NF-κB signalling may not be associated with the inhibition of DC maturation in response to VacA stimulation.
Fig. 8.
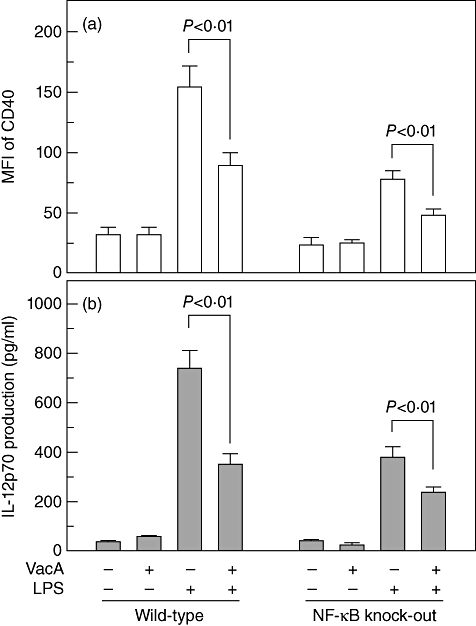
Comparative analysis of dendritic cell (DC) maturation in DCs derived from nuclear factor (NF)-κB wild-type and knock-out mice. (a) Immature DCs were derived from NF-κB wild-type and knock-out mice. DCs were treated with vacuolating cytotoxin (VacA) (10 µg/ml) and lipopolysaccharide (200 ng/ml) for 24 h. Cells were stained with monoclonal antibodies against CD40, and were analysed using flow cytometry. The data represent the mean fluorescence intensity ± standard error of the mean (s.e.m.) (n = 5). (b) Cell culture conditions were identical to those in (a). Concentrations of interleukin-12p70 in culture supernatants were determined using enzyme-linked immunosorbent assay (mean ± s.e.m., n = 5).
Discussion
During the DC maturation process induced by exogenous and endogenous stimulators, DCs display high levels of surface molecules, including CD40, CD80, CD86 and MHC [40]. DC maturation also promotes the acquisition of additional functional features such as high cytokine production, low antigen-uptake and high migratory capacity [8–10]. The present study demonstrated that the positive regulation of those surface molecules and their functional features by E. coli LPS decreased after co-exposure to H. pylori VacA. Moreover, the down-regulation of DC maturation by VacA was mediated via the restoration of E2F1 but not NF-κB signals. These results may indicate a possible mechanism of immune inhibition by VacA-producing H. pylori strains through the prevention of DC maturation, which precludes defensive immune activities against bacterial clearance. Considering that VacA has a high potency for inhibiting proliferation of lymphocytes in vitro[16–19], VacA-induced negative regulation of DC maturation may be a plausible mechanism. However, this hypothesis has not yet been verified, as this is the first report concerning the effects of purified H. pylori VacA protein on DC maturation via E2F1.
Mucosal CD11c+ DCs are located near the surface of normal gastric epithelium, and their numbers increase after H. pylori infection [7]. Wild-type H. pylori bacteria promoted the maturation of DCs, in which DC maturation was independent of the cag PAI and VacA statuses of H. pylori[20]. In contrast to the presence of whole bacteria, CagA protein of H. pylori negatively regulated the maturation functions of DCs [21]. The levels of CagA or VacA proteins that DCs encounter may be low when exposed to intact H. pylori. Thus, non-physiological levels of purified antigens are required to observe inhibitory effects on DC maturation in several studies, and the difference between virulent proteins and intact H. pylori may therefore lead to the disparity.
Although the transcription factor E2F1 is a promoter of S-phase transition in the cell cycle, E2F1 is an important regulator of DC maturation [11]. Our study showed that LPS, a stimulator of DC maturation, down-regulated E2F1 expression in DCs. In an LPS-treated state, suppression of E2F1 by siRNA resulted in the significant recovery of the down-regulated DC maturation with co-treatment of VacA. Thus, the DC maturation features caused by LPS treatment (e.g. increased expression of surface molecules, increased production of cytokines, low antigen-uptake function and high migratory capacity) were not decreased significantly after co-exposure to VacA in E2F1 siRNA-transfected cells, indicating that these results were due to the disappearance of the suppressive effect of E2F1. In contrast, the DC maturation features induced by LPS were reduced after co-exposure to VacA in untransfected cells. These results suggest that H. pylori VacA can attenuate the LPS-induced down-regulation of E2F1 activity, in which the restoration of E2F1 may play a role in the inhibition of DC maturation. Fang et al. [11] reported that blocking the E2F1 molecule can, by itself, induce a significant DC maturation, indicating that this is contrast with our results. Thus, in the present study, the increased cell maturation was not observed in unstimulated DCs that were transfected with E2F1 siRNA. Further study will be needed to evaluate clearly the roles of E2F1 suppression in DC maturation under unstimulated conditions.
It is not possible to make an assumption regarding the mechanism of how VacA is able to restore the LPS-induced down-regulation of E2F. The over-expression of E2F1 was demonstrated to inhibit the NF-κB-mediated transcription of MnSOD in murine fibroblasts by interacting with RelA, disrupting the RelA-p50 dimer and therefore inhibiting the DNA-binding activity of NF-κB [41]. In addition, the dependence of NF-κB activation for LPS-inducible E2F1 binding, as well as the physical association of E2F1 with NF-κB in THP-1 (human acute monocytic leukaemia cell line) cells, suggests that NF-κB recruits E2F1 to these LPS-responsive loci [42]. Thus, LPS-induced E2F1 suppression appears to lead to the relatively increase of DNA-binding activity of NF-κB. However, when considering that the stimulation with both LPS and VacA did not affect the NF-κB response to the challenge with LPS alone, VacA does not seem to affect the physical association of LPS-induced E2F1 with NF-κB. The precise role of E2F1 in the course of DC maturation remains to be established in VacA-stimulated DCs.
The activation of macrophages induced by LPS is accompanied by the recruitment of E2F1 to the promoters of many NF-κB-regulated genes [43]. In addition, DC maturation processes are driven by gene programmes triggered by transcription factors such as NF-κB [23,38,39]. Therefore, a possible relationship between NF-κB activation and E2F1 restoration during the DC maturation process is suggested. In a resting state, inactive NF-κB dimers are sequestered in the cytoplasm by inhibitory protein IκBs. Upon stimulation through Toll-like receptors (TLR) or cytokine receptors, IκB proteins are phosphorylated on specific serine residues by IκB kinase (IKK). Free NF-κB dimers then translocate to the nucleus where they can bind to several gene promoters. In the present study, co-stimulation of DCs with LPS increased the NF-κB DNA binding activity; however, the addition of VacA did not affect the increased NF-κB signals. Consistent with this, the combined treatment with LPS and VacA did not change the phospho-IκBα signals significantly, while LPS increased the levels of phospho-IκBα in DCs. Furthermore, the up-regulated DC maturation due to LPS was attenuated after co-exposure to VacA in both NF-κB wild-type mice- and knock-out mice-derived DCs. These results suggest that H. pylori VacA does not affect NF-κB responses to LPS, in which NF-κB signals seem to not be associated with the inhibition of DC maturation in response to VacA stimulation.
A major attribute of mature DCs is the synthesis and release of cytokines with important modulatory functions in inflammation and T cell differentiation. In particular, IL-12 plays an important role in the activities of natural killer cells and T lymphocytes. Thus, DCs treated with LPS produce IL-12 to regulate T helper type 1 (Th1) responses [44]. Considering that VacA inhibited the IL-12 expression in DCs, VacA seems to play a role in the negative regulation of Th1 responses.
As E2F1 plays a role in a suppressor of DC maturation, it is possible that E2F1 may act as an anti-inflammatory or immunosuppressive transcription factor. Consistent with this, a report demonstrated that E2F1 suppressed TNF-α-induced activation of human aortic endothelial cells by inhibiting NF-κB activation and translocation [45]. Therefore, restoration of E2F1 by VacA may lead to the inhibition of NF-κB signalling. The present study demonstrated that co-stimulation with VacA in DCs did not affect LPS-induced NF-κB activation, in which assays were performed 1 h after stimulation with LPS and VacA. Considering that the significant restoration of E2F1 transcripts first appeared 12 h after stimulation, the irrelevance of VacA to LPS-induced NF-κB activation may be due to the time interval. However, further study would be needed to clarify the relationship between E2F1 restoration and NF-κB-dependent proinflammatory cytokine gene activation pathway.
In summary, H. pylori VacA may play a negative role in DC maturation, in which the maturation process may be associated with the restoration of E2F1 signals. Based on these findings, we propose a possible mechanism of immune escape; the inhibition of DC maturation by VacA exposure may lead to distorted clearance of H. pylori, although many DCs exist in gastric mucosal layers infected with VacA-producing toxigenic H. pylori.
Acknowledgments
We thank Dr Patrice Boquet for VacA purification, Dr Martin F. Kagnoff for providing pβ-actin- and pRSV-β-galactosidase luciferase plasmids and standard β-actin RNA, Dr Tae Woo Kim for assistance with the DC2·4 cells and Jiyoung Kim, Hye Ri Park and Han-Jin Lee for the excellent technical help. This research was supported by a grant from the Basic Science Research Program through the National Research Foundation of Korea (NRF) (313–2008-2-E00154) and by a grant from the NRF of Korea Grant funded by the Korean Government (MEST) (MRC program no. 2010–0029507).
Disclosure
The authors declare no conflict of interest.
References
- 1.Polk DB, Peek RM., Jr Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer. 2010;10:403–14. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Necchi V, Manca R, Ricci V, Solcia E. Evidence for transepithelial dendritic cells in human H. pylori active gastritis. Helicobacter. 2009;14:208–22. doi: 10.1111/j.1523-5378.2009.00679.x. [DOI] [PubMed] [Google Scholar]
- 3.Hopkins RJ, Morris JG., Jr Helicobacter pylori: the missing link in perspective. Am J Med. 1994;97:265–77. doi: 10.1016/0002-9343(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 4.Cooke CL, Huff JL, Solnick JV. The role of genome diversity and immune evasion in persistent infection with Helicobacter pylori. FEMS Immunol Med Microbiol. 2005;45:11–23. doi: 10.1016/j.femsim.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Park H, Kim NI, Kim JM, et al. Expression of eotaxin in gastric epithelial cells stimulated with Helicobacter pylori vacuolating cytotoxin. J Bacteriol Virol. 2006;36:11–20. [Google Scholar]
- 6.Wilson KT, Crabtree JE. Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology. 2007;133:288–308. doi: 10.1053/j.gastro.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Kao JY, Zhang M, Miller MJ, et al. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology. 2010;138:1046–54. doi: 10.1053/j.gastro.2009.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 9.Mellman I. Antigen processing and presentation by dendritic cells: cell biological mechanisms. Adv Exp Med Biol. 2005;560:63–7. doi: 10.1007/0-387-24180-9_9. [DOI] [PubMed] [Google Scholar]
- 10.Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. 2005;5:617–28. doi: 10.1038/nri1670. [DOI] [PubMed] [Google Scholar]
- 11.Fang F, Wang Y, Li R, et al. Transcription factor E2F1 suppresses dendritic cell maturation. J Immunol. 2010;184:6084–91. doi: 10.4049/jimmunol.0902561. [DOI] [PubMed] [Google Scholar]
- 12.Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19:449–50. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu H, Yamaoka Y, Graham DY. Helicobacter pylori virulence factors: facts and fantasies. Curr Opin Gastroenterol. 2005;21:65365–9. doi: 10.1097/01.mog.0000181711.04529.d5. [DOI] [PubMed] [Google Scholar]
- 14.Algood HM, Torres VJ, Unutmaz D, Cover TL. Resistance of primary murine CD4+ T cells to Helicobacter pylori vacuolating cytotoxin. Infect Immun. 2007;75:334–41. doi: 10.1128/IAI.01063-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boncristiano M, Paccani SR, Barone S, et al. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J Exp Med. 2003;198:1887–97. doi: 10.1084/jem.20030621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science. 2003;301:1099–102. doi: 10.1126/science.1086871. [DOI] [PubMed] [Google Scholar]
- 17.Molinari M, Salio M, Galli C, et al. Selective inhibition of Ii-dependent antigen presentation by Helicobacter pylori toxin VacA. J Exp Med. 1998;187:135–40. doi: 10.1084/jem.187.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sundrud MS, Torres VJ, Unutmaz D, Cover TL. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc Natl Acad Sci USA. 2004;101:7727–32. doi: 10.1073/pnas.0401528101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torres VJ, VanCompernolle SE, Sundrud MS, Unutmaz D, Cover TL. Helicobacter pylori vacuolating cytotoxin inhibits activation-induced proliferation of human T and B lymphocyte subsets. J Immunol. 2007;179:5433–40. doi: 10.4049/jimmunol.179.8.5433. [DOI] [PubMed] [Google Scholar]
- 20.Kranzer K, Söllner L, Aigner M, et al. Impact of Helicobacter pylori virulence factors and compounds on activation and maturation of human dendritic cells. Infect Immun. 2005;73:4180–9. doi: 10.1128/IAI.73.7.4180-4189.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanaka H, Yoshida M, Nishiumi S, et al. The CagA protein of Helicobacter pylori suppresses the functions of dendritic cell in mice. Arch Biochem Biophys. 2010;498:35–42. doi: 10.1016/j.abb.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 22.Rescigno M, Martino M, Sutherland CL, Gold MR, Ricciardi-Castagnoli P. Dendritic cell survival and maturation are regulated by different signaling pathways. J Exp Med. 1998;188:2175–80. doi: 10.1084/jem.188.11.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JY, Kim H, Cha MY, et al. Clostridium difficile toxin A promotes dendritic cell maturation and chemokine CXCL2 expression through p38, IKK, and the NF-kappaB signaling pathway. J Mol Med. 2009;87:169–80. doi: 10.1007/s00109-008-0415-2. [DOI] [PubMed] [Google Scholar]
- 24.Kim JM, Kim JS, Kim YJ, et al. Conjugated linoleic acids produced by Lactobacillus dissociates IKK-gamma and Hsp90 complex in Helicobacter pylori-infected gastric epithelial cells. Lab Invest. 2008;88:541–52. doi: 10.1038/labinvest.2008.16. [DOI] [PubMed] [Google Scholar]
- 25.Sagaert X, Van Cutsem E, De Hertogh G, Geboes K, Tousseyn T. Gastric MALT lymphoma: a model of chronic inflammation-induced tumor development. Nat Rev Gastroenterol Hepatol. 2010;7:336–46. doi: 10.1038/nrgastro.2010.58. [DOI] [PubMed] [Google Scholar]
- 26.Kim JM, Kim JS, Lee JY, et al. Vacuolating cytotoxin in Helicobacter pylori water-soluble proteins upregulates chemokine expression in human eosinophils via Ca2+ influx, mitochondrial reactive oxygen intermediates, and NF-kappaB activation. Infect Immun. 2007;75:3373–81. doi: 10.1128/IAI.01940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim JM, Kim JS, Lee JY, et al. Dual effects of Helicobacter pylori vacuolating cytotoxin on human eosinophil apoptosis in early and late periods of stimulation. Eur J Immunol. 2010;40:1651–62. doi: 10.1002/eji.200939882. [DOI] [PubMed] [Google Scholar]
- 28.Gauthier NC, Monzo P, Kaddai V, Doye A, Ricci V, Boquet P. Helicobacter pylori VacA cytotoxin: a probe for a clathrin-independent and Cdc42-dependent pinocytic pathway routed to late endosomes. Mol Biol Cell. 2005;16:4852–66. doi: 10.1091/mbc.E05-05-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Bernard M, Papini E, de Filippis V, et al. Low pH activates the vacuolating toxin of Helicobacter pylori, which becomes acid and pepsin resistant. J Biol Chem. 1995;270:23937–40. doi: 10.1074/jbc.270.41.23937. [DOI] [PubMed] [Google Scholar]
- 30.Yamasaki E, Wada A, Kumatori A, et al. Helicobacter pylori vacuolating cytotoxin induces activation of the proapoptotic proteins Bax and Bak, leading to cytochrome c release and cell death, independent of vacuolation. J Biol Chem. 2006;281:11250–9. doi: 10.1074/jbc.M509404200. [DOI] [PubMed] [Google Scholar]
- 31.Kang TH, Bae HC, Kim SH, et al. Modification of dendritic cells with interferon-gamma-inducible protein-10 gene to enhance vaccine potency. J Gene Med. 2009;11:889–98. doi: 10.1002/jgm.1371. [DOI] [PubMed] [Google Scholar]
- 32.Shinoda K, Nakagawa K, Kosaka T, et al. Regulation of human dendritic cells by a novel specific nuclear factor-kappaB inhibitor, dehydroxymethylepoxyquinomicin. Hum Immunol. 2010;71:763–70. doi: 10.1016/j.humimm.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 33.Bukur J, Herrmann F, Handke D, Recktenwald C, Seliger B. Identification of E2F1 as an important transcription factor for the regulation of tapasin expression. J Biol Chem. 2010;285:30419–26. doi: 10.1074/jbc.M109.094284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elewaut D, DiDonato JA, Kim JM, Truong F, Eckmann L, Kagnoff MF. NF-kappaB is a central regulator of the intestinal epithelial cell innate immune response induced by infection with enteroinvasive bacteria. J Immunol. 1999;163:1457–66. [PubMed] [Google Scholar]
- 35.Kim JM, Oh YK, Kim YJ, Oh HB, Cho YJ. Polarized secretion of CXC chemokines by human intestinal epithelial cells in response to Bacteroides fragilis enterotoxin: NF-κB plays a major role in the regulation of IL-8 expression. Clin Exp Immunol. 2001;123:421–7. doi: 10.1046/j.1365-2249.2001.01462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eckmann L, Stenson WF, Savidge T, et al. Role of intestinal epithelial cells in the host secretory response to infection by invasive bacteria. J Clin Invest. 1997;100:296–309. doi: 10.1172/JCI119535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoon YM, Lee JY, Yoo D, et al. Bacteroides fragilis enterotoxin induces human beta-defensin-2 expression in intestinal epithelial cells via a mitogen-activated protein kinase/I kappaB kinase/NF-kappaB-dependent pathway. Infect Immun. 2010;78:2024–33. doi: 10.1128/IAI.00118-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bharadwaj AS, Agrawal DK. Transcription factors in the control of dendritic cell life cycle. Immunol Res. 2007;37:79–96. doi: 10.1007/BF02686091. [DOI] [PubMed] [Google Scholar]
- 39.Lyakh LA, Koski GK, Telford W, Gress RE, Cohen PA, Rice NR. Bacterial lipopolysaccharide, TNF-α, and calcium ionophore under serum free conditions promote rapid dendritic cell-like differentiation in CD14+ monocytes through distinct pathways that activate NK-κB. J Immunol. 2000;165:3647–55. doi: 10.4049/jimmunol.165.7.3647. [DOI] [PubMed] [Google Scholar]
- 40.Randolph GJ, Ochando J, Partida-Sánchez S. Migration of dendritic cell subsets and their precursors. Annu Rev Immunol. 2008;26:293–316. doi: 10.1146/annurev.immunol.26.021607.090254. [DOI] [PubMed] [Google Scholar]
- 41.Tanaka H, Matsumura I, Ezoe S, et al. E2F1 and c-Myc potentiate apoptosis through inhibition of NF-kappaB activity that facilitates MnSOD-mediated ROS elimination. Mol Cell. 2002;9:1017–29. doi: 10.1016/s1097-2765(02)00522-1. [DOI] [PubMed] [Google Scholar]
- 42.Lim CA, Yao F, Wong JJ, et al. Genome-wide mapping of RELA (p65) binding identifies E2F1 as a transcriptional activator recruited by NF-kappaB upon TLR4 activation. Mol Cell. 2007;27:622–35. doi: 10.1016/j.molcel.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 43.Lim CA, Yao F, Wong JJ, et al. Genome-wide mapping of RELA (p65) binding identifies E2F1 as a transcriptional activator recruited by NF-kappaB upon TLR4 activation. Mol Cell. 2007;27:622–35. doi: 10.1016/j.molcel.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 44.Langrish CL, McKenzie BS, Wilson NJ, de Waal Malefyt R, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev. 2004;202:96–105. doi: 10.1111/j.0105-2896.2004.00214.x. [DOI] [PubMed] [Google Scholar]
- 45.Chen M, Capps C, Willerson JT, Zoldhelyi P. E2F-1 regulates nuclear factor-kappaB activity and cell adhesion: potential antiinflammatory activity of the transcription factor E2F-1. Circulation. 2002;106:2707–13. doi: 10.1161/01.cir.0000038706.30661.86. [DOI] [PubMed] [Google Scholar]