

Abstract
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory lung disease; it is a leading cause of death and existing treatments have no proven disease-modifying effect. The mechanisms underlying this resistance are largely unknown, but suggest the presence of some self-maintaining pathogenic process, possibly initiated by cigarette smoking, that prevents the normal resolution of inflammation. We have previously reported increased production of proinflammatory cytokines and granzyme b by CD8+ T cells in COPD; costimulatory receptor/ligand interactions required include CD80:86/CD28, B7-1/CTLA4, 4-1BB/1BBL and OX40/OX40L. We hypothesized that a dysregulated expression/function of these molecules may play a role in inflammatory/autoimmune components of COPD. We analysed T cell co-stimulatory molecules in blood from 34 controls, 15 smokers and 48 COPD subjects. We assessed the potential functional relevance of CD8/CD28null cells in COPD by measuring their production of proinflammatory cytokines, co-stimulatory molecules, granzyme and perforin. A smoke-exposed murine model was applied to investigate the relative expression of CD8/CD28null T cells in blood, lung tissue and airway. CD8/CD28null cells were increased in both current- and ex-smoker COPD groups; these cells expressed significantly more interferon (IFN)-γ, OX40, 4-1BB, CTLA4, granzyme and perforin when stimulated than CD8/CD28+ T cells. There were no changes in CD4/CD28null T cells. In mice exposed to cigarette smoke for 12 weeks, CD8/CD28null T cells were significantly increased in the airway with a trend for an increase in lung tissue and blood. Increased production of proinflammatory cytokines and expression of alternative co-stimulatory molecules by CD8/CD28null T cells may play a role in inflammatory or autoimmune responses in COPD and identify therapeutic targets.
Keywords: CD8/CD28null T cells, COPD, co-stimulatory molecules, granzyme, smoking mouse model
Introduction
Chronic obstructive pulmonary disease (COPD) is a leading cause of death worldwide. Although some reduction in the prevalence of smoking has recently been achieved, the lag time of 20–50 years will mean that there will be no reduction in incidence of COPD in the near future. Existing treatments are largely symptomatic, and the only approved anti-inflammatory medication, corticosteroids, has no proven disease-modifying effect [1]. The mechanisms underlying this resistance are largely unknown, but suggest the presence of some self-maintaining pathogenic process, possibly initiated by cigarette smoking, that prevents the normal resolution of the inflammatory response. Several groups have shown that smoking induces an irreversible change in the transcriptome of airway cells, such that ex-smokers resemble current smokers more closely than never-smokers, even after many years of smoking cessation [2,3]. Although these findings have generated considerable interest, the real challenge is to determine how this altered state translates into disease pathogenesis. While there may be a direct pathogenic role for certain altered proteins, an indirect role via the recognition of altered self-antigens could plausibly be a mechanism of much broader relevance, as this would not depend directly on the functionality of altered proteins. Evidence for an autoimmune component of COPD has been presented by several groups [4–7]. In support of this, our group has reported increased production of T helper type 1 (Th1) proinflammatory cytokines interferon (IFN)-γ and tumour necrosis factor (TNF)-α by CD8+ T cells in peripheral blood in COPD [8]. We and others have also shown that smoking and COPD are associated with increased numbers of cytotoxic CD8+ T cells in blood and airway [9–11]. These cells were also shown to produce higher levels of the cytotoxic mediators granzyme b and perforin compared to healthy never smokers [12].
Major histocompatibility complex–T cell receptor (MHC–TCR) signalling is necessary but not sufficient to induce antigen-specific T cell activation and cytokine production. Additional receptor–ligand interactions are required to provide co-stimulatory signals; these include interactions of CD80:86 with CD28, CD40 with CD40L (CD154), B7-1 with cytotoxic T lymphocyte antigen 4 (CTLA4) (CD152) and members of the TNFR/TNF superfamily members (4-1BB (CD137)/1BBL and OX40 (CD134)/OX40L [13]. It is possible that dysregulated expression of some or all of these molecules may play a role in the chronic inflammatory response or autoimmune components of COPD, including the increased production of proinflammatory cytokines. In this regard, CD8/CD28null oligoclonal T cells have been reported in other autoimmune diseases including granulomatous interstitial lung diseases [14] and in the ageing immune system and its relationship to the development of COPD [15]. CD8/CD28null clones divide faster and live longer than CD8CD28+ T cells due to a shorter cell division cycle, higher resistance to apoptosis and a different response to regulatory cytokines [16]. However, the expression of CD8/CD28null cells in the airway compartment, or the expression of other co-stimulatory molecules, has not been assessed in COPD. We therefore analysed T cell co-stimulatory molecules (CD28, CD40L, CTLA4, 4-1BB and OX40) on CD4+ and CD8+ T cells from peripheral blood from healthy controls and smokers, and current and ex-smokers with COPD. Having found an increase in CD8/CD28null T cells in COPD, we further assessed the effects of cigarette smoke in the various lung compartments by measuring CD8/CD28null cells in blood, lung tissue and airway from mice exposed to cigarette smoke for 12 weeks. We then assessed the potential functional relevance of these CD8/CD28null cells in COPD by measuring their production of proinflammatory cytokines and cytotoxic mediators, granzyme b and perforin.
Materials and methods
Reagents
The following monoclonal antibodies (mAbs) and immunological reagents were employed: CD3 [phycoerythrin-cyanin 5 (PC5)] (Coulter/Immunotech, Marseille, France), CD8 [fluorescein isothiocyanate (FITC)] (BD Biosciences, San Jose, CA, USA), CD8 [peridinin chlorophyll (PerCP) Cy5·5; BD), CD4 and CD28 [phycoerythrin (PE)] (BD), CD28 (FITC) (BD), CD40L, CTLA4, 4-1BB and OX40 (PE; Coulter/Immunotech), granzyme b and perforin (PE; BD), a-IFN-γ (PE; BD), anti-mouse immunoglobulin control [immunoglobulin (Ig)G1] (FITC; BD)/IgG1a (PE; BD)/IgG1 (PerCP Cy5·5) (Coulter/Immunotech), FACSPerm and FACSlyse (BD), brefeldin A, ionomycin and phorbol myristate acetate (PMA; Sigma Aldrich, Sydney, Australia). For mouse studies, CD3 hamster anti-mouse (PE; AbD Serotec, Kidlington, UK), CD28 hamster anti-mouse (FITC; AbD Serotec) and CD8a rat anti-mouse (PerCP-Cy 5·5; BD) were employed.
Patient and control groups
COPD volunteers were recruited specifically for the study and informed consent obtained. There was no exacerbation of COPD for 6 weeks prior to study. Subjects with other co-existing lung disease or aged greater than 75 years were excluded. Ethics approval was obtained from the Royal Adelaide Hospital and the experiments were conducted with the understanding and the consent of each participant. COPD was diagnosed using the Global Initiative for COPD (GOLD) criteria with clinical correlation (mild COPD: FEV1/FVC < 70% but FEV1 ≥ 80% predicted; moderate COPD FEV1 50% ≤ 80% predicted, severe COPD FEV1 < 30% predicted) [17]. Eighteen of the COPD subjects were ex-smokers (at least 1 year) and 30 were current smokers. A further 15 healthy current smokers of at least 10 pack-years with no evidence of COPD or other lung disease were also recruited. Blood was also obtained from 34 never smokers (Table 1). These were healthy, recruited volunteers with no history of airways disease. All subjects underwent spirometry as part of their routine clinical assessment. Venous blood was collected into 10 U/ml preservative-free sodium heparin (DBL, Sydney, Australia) and maintained at 4°C until processing. All patients were submitted to the same protocol and analysis performed retrospectively.
Table 1.
Demographic data for the subjects tested
| n | Age (years) | FEV1 % pred | Pack years | Total WCC × 109/ml | Lymphocytes × 109/ml | CD8+ T cells (%CD3) | |
|---|---|---|---|---|---|---|---|
| Controls | 34 | 53 | 97 | 0 | 6·13 | 2·04 | 31 |
| (12) | (11) | (1·57) | (0·40) | (8) | |||
| Smokers | 15 | 54 | 93 | 57* | 6·34 | 2·42 | 32 |
| (12) | (10) | (32) | (2·37) | (0·83) | (9) | ||
| COPD | 30 | 59 | 57* | 57* | 7·48 | 2·55 | 38* |
| Curr-smokers | (7) | (20) | (28) | (2·21) | (1·06) | (13) | |
| COPD | 18 | 60 | 62* | 52* | 6·12 | 2·06 | 35 |
| Ex-smokers | (6) | (26) | (35) | (1·52) | (0·65) | (16) |
COPD: chronic obstructive pulmonary disease, FEV1: forced expiratory volume in 1 s, FVC: forced vital capacity, WCC: white cell count, Curr-smoker: current smoker.
P < 0·05 versus control. Data are presented as mean ± standard deviation.
Total and differential leucocyte counts
Blood differential cell counts were performed using a CELL DYN 4000 (Abbott Diagnostics, Sydney, Australia).
CD4 and CD8 cell counts
The percentages of CD3, CD4 and CD8 lymphocytes were calculated using flow cytometry. One hundred µl of peripheral blood were stained with appropriately diluted fluorescently conjugated monoclonal as described previously [8,10,12]. Briefly, samples were analysed by gating using forward-scatter (FSC) versus side-scatter (SSC) to exclude platelets and debris. Control staining of leucocytes was performed on each sample and background readings of < 2% were obtained. A minimum of 10 000 events were acquired for analysis.
Expression of granzyme b and perforin
The percentages of CD8+ and CD4+ T cells expressing CD28, granzyme b and perforin were calculated using flow cytometry, as described previously [12].
Expression of co-stimulatory molecules
Because the expression of co-stimulatory molecules or CD8/CD28null cells has not been assessed previously in the airway compartment, we analysed T cell co-stimulatory molecules (CD28, CD40L, CTLA4, 4-1BB and OX40) on CD4+ and CD8+ T cells from peripheral blood from healthy controls and smokers, and current and ex-smokers with COPD. The percentages of CD8+ and CD4+ T cells expressing CD28 were calculated using flow cytometry, as described previously [8,10,12]. For CD40L, CTLA4, 4-1BB and OX40, 1-ml aliquots of blood (diluted 1:2 with RPMI-1640 medium) were placed in 10 ml sterile conical polyvinyl chloride (PVC) tubes (Johns Professional Products, Sydney, NSW, Australia). Phorbol myristate (25 ng/ml) and ionomycin (1 mg/ml) were added to stimulate the T cells. Brefeldin A (10 mg/ml) was added to prevent the shedding of the co-stimulatory molecules from the T cell surface. The tubes were incubated in a humidified 5%CO2/95% air atmosphere at 37°C. At 16 h 100 ml 20 mm ethylenediamine tetraacetic acid/phosphate-buffered saline (EDTA/PBS) was added to the culture tubes, which were vortexed vigorously for 20 s to remove adherent cells. Three hundred µl aliquots of cells were treated with 2 ml FACSLyse for 10 min. Cells were centrifuged, supernatant discarded and 500 ml FACSPerm added for 10 min. Two ml 0·5% bovine serum albumin (BSA) (Sigma) in IsoFlow (Beckman Coulter) was then added and the tubes were centrifuged at 300 g for 5 min. After decanting supernatant, Fc receptors were blocked with 10 ml human immunoglobulin (Intragam, CSL, Parkville, Victoria, Australia) for 10 min at room temperature. Five µl of appropriately diluted anti-CD8 (BD), anti-CD3 PC5 (Coulter/Immunotech) and PE-conjugated monoclonal antibodies to CD40L, CTLA4, 4-1BB, OX40 or isotype control were added for 15 min in the dark at room temperature. Cells were washed and events acquired and analysed as described above.
Expression of CD4/CD28null and CD8/CD28null T cells in a mouse smoke-exposure model
Having found an increase in CD8/CD28null T cells in COPD patients, we further assessed the effects of cigarette smoke in the various lung compartments by measuring CD8/CD28null cells in peripheral blood, lung tissue and airway from mice exposed to cigarette smoke for 12 weeks. Methods of smoke exposure, sample preparation and measurements have been described previously [18,19]. Ethics approval was granted by the Institute of Medical and Veterinary Science Ethics Committee. Briefly, mice were exposed to nine cigarettes per day (three cigarettes per hour, three times per day, 5 days per week for 12 weeks). There were six mice in the control group (‘sham’-exposed mice, exposed to room air only) and 10 smoke-exposed mice. At the conclusion of the experimental time–course mice were weighed, killed and bronchoalveolar lavage (BAL), blood and lung tissue collected from separate animals, as we have reported previously [18,19]. A ‘Medimachine’ tissue disaggregator (BD) was used to prepare cell suspensions from lung tissue [18,19]. Total cell counts were performed in BAL and disaggregated lung tissue using a modified Neubauer haemocytometer. Blood was obtained by terminal bleeding by cardiac puncture and collected into microtainer tubes with lithium heparin (Greiner Bio-One, Stonehouse, UK).
Macrophages were removed by adherence to plastic and the remaining fluid used for analysis of T cells. The percentages of CD4, CD8, CD4/CD28null and CD8/CD28null T cells were assessed using flow cytometry as described above (using specific mouse mAbs).
Production of cytokines, granzymes and expression of co-stimulatory molecules by CD8/CD28null T cells
To assess the potential functional relevance of increased CD8/CD28null cells in COPD, we measured their production of proinflammatory cytokines and cytotoxic mediators, granzyme b and perforin, as described above and as reported previously [8]. Briefly, following red cell lysis, permeabilization and blocking Fc receptors, 5 µl of appropriately diluted mAbs to CD3 (Alexa 647), CD8 (PerCP), anti-28 FITC and PE-conjugated monoclonal antibodies to IFN-γ, granzyme b, perforin, CD40L, CTLA4, 4-1BB and OX40 or isotype control were added for 15 min in the dark at room temperature. Events were acquired and analysed as described above.
Statistical analysis
Data from ex-vivo human studies was analysed using analysis of variance (anova) with Dunnett's post-hoc test. A normal data distribution was confirmed using kurtosis and skewness. For remaining analyses, the non-parametric Mann–Whitney U-test was applied to analyse the data. Analyses were performed using spss software. P values < 0·05 were considered significant.
Results
CD4+ and CD8+ T cell counts and expression of co-stimulatory molecules
Lymphocyte numbers ranged from 1·3–5·4 × 109/l and there were no significant differences between groups. The percentage of CD8+ T cells were significantly higher in peripheral blood from current smoker COPD subjects compared to controls (current smoking COPD subjects median 38% (range 15–64%) versus controls median 31% (range 11–44%).
Expression of co-stimulatory molecules by T cells
A significant increase in CD8/CD28null T cells in blood from current and ex-smokers with COPD versus controls was noted (Fig. 1). There was a non-significant trend for an increase in CD8/CD28null T cells in blood from healthy smokers (P = 0·11). There were no significant changes in CD4/CD28null T cells between the groups (Fig. 1). Overall, there were no significant correlations between age and the presence of CD8/CD28null cells (−0·199; Pearson's correlation; P = 0·069) and no significant correlations within the COPD group.
Fig. 1.
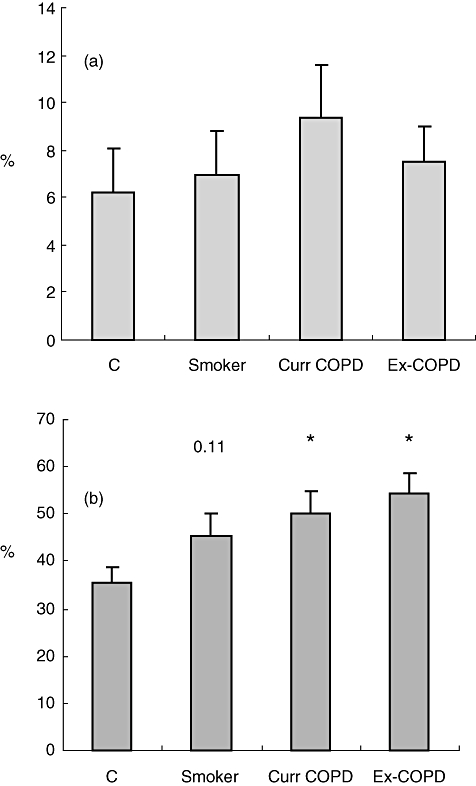
Expression of (a) CD4/ CD28null and (b) CD8/CD28null T cells from peripheral blood from controls (c), healthy smokers (Smoker), current smokers with chronic obstructive pulmonary disease (COPD) (Curr COPD) and ex-smokers with COPD (Ex-COPD). Note significantly increased percentage of CD8/CD28null T cells in blood from both COPD groups and a trend for an increase in healthy smokers. There were no changes in CD4/CD28null T cells. Histograms present mean ± standard deviation.
For both CD4 and CD8 T cells, expression of 4-1BB was increased in both COPD groups (P < 0·05) and a trend for smokers (P = 0·055) versus controls; CTLA4 was increased on CD4+ T cells from all groups (P < 0·05) and on CD8+ T cells from smoker (P < 0·05) and a trend for current smoker COPD (P = 0·07) groups. OX40 was increased significantly on CD8+ cells from smoker and current smoker COPD groups (Fig. 2). There were no significant changes in expression of CD40L (Fig. 2).
Fig. 2.
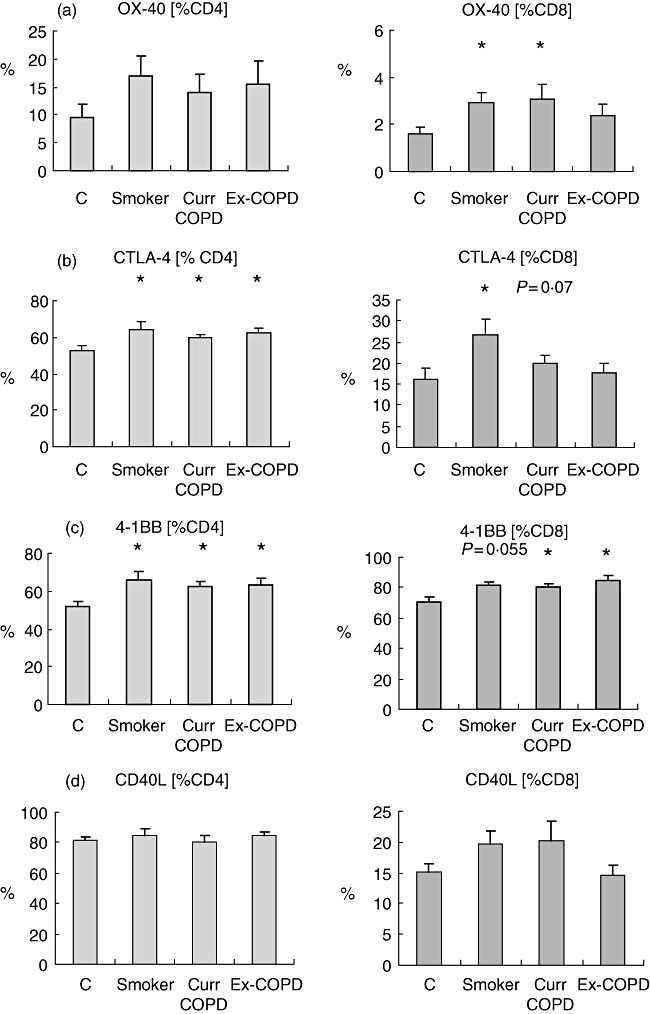
Expression of (a) OX40, (b) cytotoxic T lymphocyte antigen 4 (CTLA4), (c) 4-1BB and (d) CD40L by CD4 and CD8 T cells from phorbol mysristate acetate (PMA)-stimulated peripheral blood from controls (C), healthy smokers (Smoker), current smokers with chronic obstructive pulmonary disease (COPD) (Curr COPD) and ex-smokers with COPD (Ex-COPD). Note that for both CD4 and CD8 T cells, expression of 4-1BB was increased significantly in both COPD groups versus controls, with a trend for an increase in healthy smokers. CTLA4 was increased significantly on CD4+ T cells from all groups and on CD8+ T cells from smokers, with a trend for an increase in current-smoker COPD subjects. OX40 was increased significantly on CD8+ cells from smoker and current-smoker COPD groups. Histograms present mean ± standard deviation. *Significant (P < 0·05 increase versus controls).
Production of cytokines and granzymes by CD8/CD28null T cells
CD8/CD28null cells expressed significantly more IFN-γ, granzyme and perforin than CD8/CD28+ T cells (Fig. 3). CD4/CD28+ cells expressed significantly more IL-2 than CD4/CD28null or CD8/CD28+ or CD8/CD28null T cells (P < 0·05) (Fig. 3).
Fig. 3.
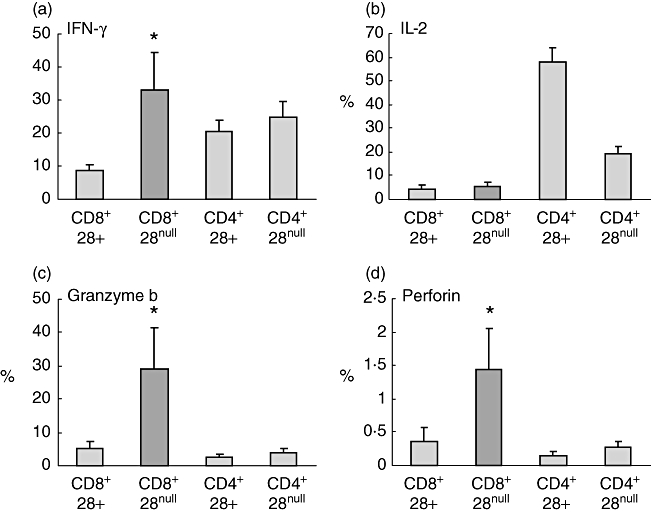
Histograms showing production of interferon (IFN)-γ, interleukin (IL)-2, granzyme b and perforin by CD8/CD28null T cells (dark shading) following stimulation of peripheral blood with phorbol myristate acetate (PMA) (n = 5 separate experiments; five never smoker control subjects). Note increased production of IFN-γ, granzyme b and perforin by CD8/CD28null T cells in contrast to increased expression of interleukin (IL)-2 by CD4+CD28+ T cells. Histograms present mean ± standard error of the mean. *Significant (P < 0·05 increase versus CD8+CD28+ T cells).
Expression of co-stimulatory molecules by CD8/CD28null T cells
CD8/CD28null cells expressed significantly more OX40, 4-1BB, and CTLA4 when stimulated than CD8/CD28+ T cells (Fig. 4). Representative plots showing gating strategy to identify CD8+ and CD8- (CD4+) T cells and increased expression of IFN-γ, granzyme b and OX40 by CD8/CD28null T cells in contrast to increased expression of IL-2 by CD4/CD28+ T cells are presented in Fig. 5.
Fig. 4.
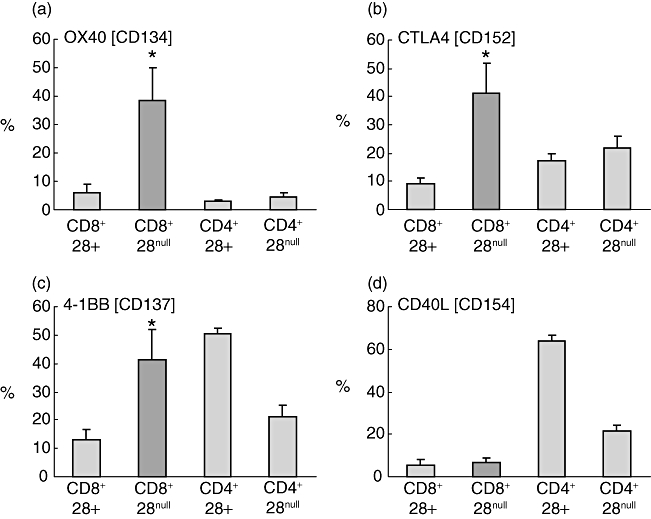
Histograms showing production of (a) OX40, (b) cytotoxic T lymphocyte antigen 4 (CTLA4), (c) 4-1BB and (d) CD40L by CD8/CD28null T cells (dark shading) following stimulation of peripheral blood with PMA (n = 5 separate experiments; five never smoker control subjects). Note increased production of OX40, CTLA4 and 4-1BB by CD8/CD28null T cells in contrast to increased expression of CD40L by CD4+CD28+ T cells. Histograms present mean ± standard error of the mean. *Significant (P < 0·05 increase versus CD8+CD28+ T cells).
Fig. 5.
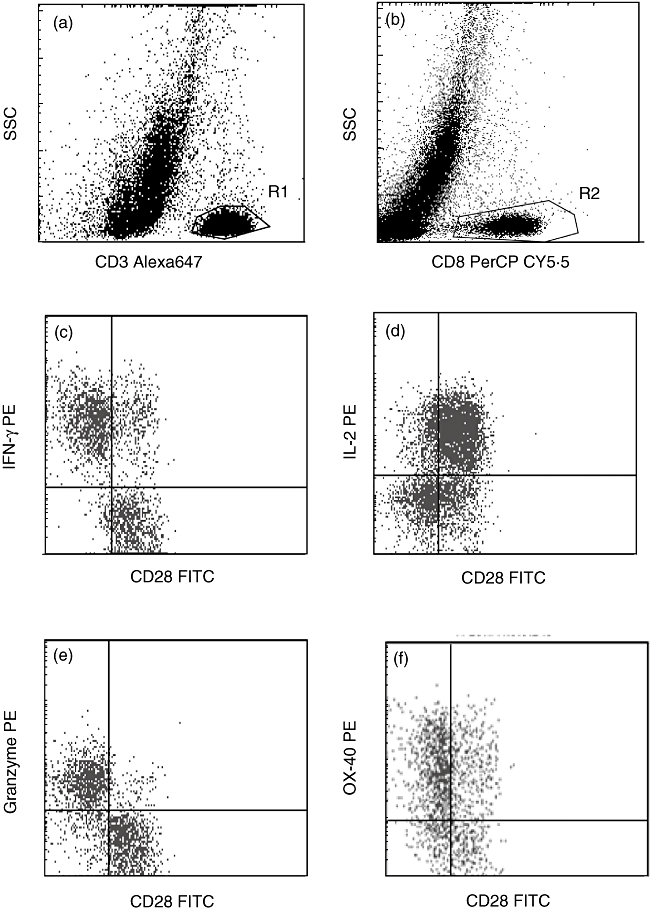
Representative flow cytometry dot-plots showing gating strategy. (a,b) CD8+ T cells were identified as cells in region 1 (R1) (CD3+ low side-scatter (SSC) events) and region 2 (R2) (CD8+ low SSC events) and CD4 T cells as events in R1 and not R2. Production of (c) interferon (IFN)-γ, (d) interleukin (IL)-2, (e) granzyme b and (f) OX40 by CD8/CD28+ and CD8/CD28null T cells and CD8-/CD28+ (CD4+) and CD8-/CD28null (CD4+) T cells. Note increased expression of IFN-γ, granzyme b and OX40 by CD8/CD28null T cells in contrast to increased expression of IL-2 by CD4+CD28+ T cells.
Expression of CD8/CD28null T cells in a mouse smoke-exposure model
Exposure of mice to cigarette smoke for 3 weeks resulted in no significant increases in leucocyte counts in BAL or lung tissue. [lung tissue: smoke-exposed 4·4 × 105/ml ± standard error of the mean (s.e.m.) 0·34 × 105/ml versus control 3·6 × 105/ml ± s.e.m. 1·9 × 105/ml P = 0·22; BAL: smoke-exposed 0·46 × 105/ml ± s.e.m. × 0·05 105/ml versus control 0·34 × 105/ml ± s.e.m. × 0·05 105/ml P = 0·18].
There were no significant differences in the percentage of CD8 T cells in any compartment following 12 weeks smoke exposure (not shown).
In mice exposed to cigarette smoke for 12 weeks, there was a significant increase in CD8/CD28null T cells in the airway (BAL) (P < 0·05) and a trend for an increase in lung tissue (P = 0·063) and in blood (P = 0·057) (Fig. 6).
Fig. 6.
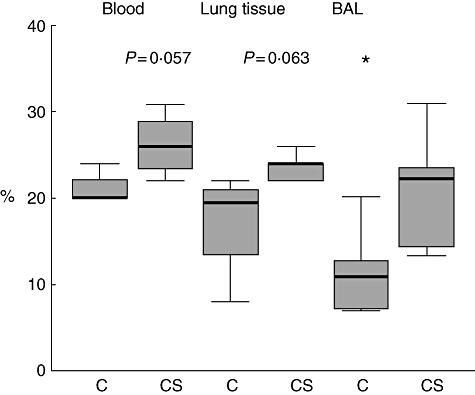
CD8+CD28null T cells in blood, lung tissue and bronchoalveolar lavage (BAL) from mice exposed to cigarette smoke for 12 weeks. C, Untreated mice, CS: cigarette smoke-treated mice. *Significant (P < 0·05) increase in BAL CD8+CD28null T cells versus control mice. Box plots present median ± 25th and 75th percentiles (solid box), with the 10th and 90th percentiles shown by whiskers outside the box.
Discussion
The present study shows that CD8/CD28null T cells are increased significantly in the peripheral blood of both current and ex- smoking COPD subjects, with a trend for an increase in the blood of healthy smokers. These cells are functionally active and produce increased levels of the proinflammatory cytokine IFN-γ, and the cytotoxic mediators granzyme b and perforin. We further show that one mechanism for the increased production of the proinflammatory mediators by the CD8/CD28null T cells may be an increased expression of alternative co-stimulatory molecules OX40, CTLA4 and 4-1BB. COPD is associated with damage throughout the lungs from the large airways to the alveolar walls, due to ineffective repair processes in the face of ongoing noxious insults. Once COPD disease is established, cessation of smoking does little to reduce physiological functioning or susceptibility to acute deterioration as a result of new insults such as infection. Understanding the basis of ongoing inflammation and individual susceptibility to COPD is critical to the development of new treatments for this disease.
The direct effect of cigarette smoke on CD8/CD28null T cells was shown using a smoking mouse model. Although the numbers of mice were very small, 12 weeks smoke exposure induced a significant increase in the percentage of CD8/CD28null T cells in the airway and a trend for increased expression in blood and lung tissue. This suggests an initial, direct effect of cigarette smoke in the airway. We have shown previously that cessation of smoking once COPD disease is established does not stop the persistence of defective efferocytosis and tissue damage in the airways or chronic local and systemic inflammation [1,20,21]. The mechanisms underlying this abnormal resolution of the inflammatory response are largely unknown, but our present data suggest that cigarette smoking may initiate a self-maintaining pathogenic process that inhibits resolution. Interestingly, in the group of COPD subjects who had ceased smoking, there was an increase in CD8+28null T cells and a significant increase in expression of 4-1BB by CD8+ T cells in the peripheral blood. This suggests that these clones may be related to COPD disease rather than smoking per se, and are likely to be particularly important in the pathogenesis of COPD disease. We propose that therapeutic targeting of these molecules may suppress an autoimmune reaction and reduce future morbidity in COPD.
The potential role of an autoimmune component of COPD in these mechanisms has been explored by several groups [4–7]. These studies have provided evidence to support a loss of tolerance to self-antigens, usually peptides, or developments of immunity to foreign epitopes that cross-react with self-antigens in COPD. Cigarette smoke may play a role, as it has been shown to generate new epitopes by oxidizing existing proteins in COPD [2,3]. Any resultant autoantibodies have the potential to exert pathogenic effects in the disease via several mechanisms that include the increased production of proinflammatory cytokines by Th1 T cells and increased proliferation of CD8+ cytotoxic T cells. We and others have identified the presence of some of these effects in COPD. We have found an increased infiltration of activated T cells, and particularly CD8+ cytotoxic T cells; these cells have been correlated positively with airflow limitation and disease progression in COPD [8–12]. Consistent with our present findings, an increased percentage of oligoclonal CD8/CD28null clonally expanded T cells has been reported in other autoimmune states. One study reported significant correlations between the percentage of peripheral blood CD8/CD28null T cells and disease severity in sarcoidosis [14]. However, we found no significant correlations between CD8+/CD28null T cells and FEV1 in our COPD patient cohort.
Loss of CD28 expression on T cells has been reported previously for sarcoidosis and COPD; however, the studies were focused on expansion of CD4+/CD28null T cells and did not investigate the important CD8+ T cell subset or investigate alternative co-stimulatory molecules in COPD [22–24]. Although levels of CD4+/CD28null were lower in our COPD patients than healthy controls, the difference did not reach statistical significance. In contrast, the significant increase in CD8+/CD28null T cells in COPD was accompanied by their increased production of proinflammatory cytokines and granzyme. Taken together, these data suggest that CD8/CD28null T cells may play an important role in the dysregulated immune responses in COPD. This is consistent with the increased numbers of cytotoxic T cells in COPD and the known effector cell activity of these cytotoxic cells that arises as a result of production of cytokines including IFN-γ and granzymes [8].
Cross-talk between CD28 and the co-localized 4-1BB/OX40 is considered to be important in efficient co-stimulation signalling. However, our results suggest that 4-1BB/OX40 and CTLA4 may act independently of CD28 and that their increased expression may be important for immune modulation in COPD, and have particular relevance in the autoimmune and inflammatory components of the disease. This was evident by our findings that stimulated CD8/CD28null T cells from patients with COPD were able to express significantly higher levels of these co-stimulatory molecules supporting the presence of alternative and increased co-stimulation in the absence of CD28 signalling in COPD. Further, our findings of increased production of proinflammatory cytokines and granzyme by the CD8+/CD28null T cells in COPD suggest a strong co-stimulatory capacity of these cells, despite the apparent lack of co-stimulation by the CD28 pathway.
Although COPD is primarily a disease that affects the airway, it is being recognized increasingly as a systemic disease [25,26]. Lymphocytes have been shown to traffic from the bloodstream to the bronchoalveolar space and visa versa [27], and our previous investigations have shown that several of the changes noted in the airways in COPD are reflected in the peripheral blood [8,12], supporting this view. We now show that changes in co-stimulatory molecule expression by CD8+ T cells were detected in peripheral blood T cells in both healthy smokers and current smokers with COPD, further confirming the systemic nature of the disease. The biological relevance of the differences noted in this study requires further study; it may be that there are larger differences in cells from the airway and lung tissue. Although we showed differences in the airway of non-smoking versus smoking mice, a future extension of these studies will be to investigate both the CD8+/CD28null cells and the expression of the various co-stimulatory molecules in mice with established emphysema.
In conclusion, our data showed increased expression of proinflammatory cytokines, granzyme/perforin and alternative co-stimulatory molecules by CD8/CD28null T cells in COPD that may play a role in inflammatory responses or an autoimmune component in this disease.
Acknowledgments
This study was supported by the Australian Lung Foundation, National Health and Medical Research Council (NHMRC) Career Development Award and NHMRC Practitioner Fellowship.
Disclosure
The authors have no disclosures.
References
- 1.Hodge S, Hodge G, Holmes M, Reynolds PN. Increased apoptosis in the airways in COPD persists after smoking cessation. Eur Respir J. 2005;25:447–54. doi: 10.1183/09031936.05.00077604. [DOI] [PubMed] [Google Scholar]
- 2.Eggleton P, Haigh R, Winyard PG. Consequence of neo-antigenicity of the ‘altered self’. Rheumatology (Oxf) 2008;47:567–71. doi: 10.1093/rheumatology/ken014. [DOI] [PubMed] [Google Scholar]
- 3.Agustí A, MacNee W, Donaldson K, Cosio M. Hypothesis: does COPD have an autoimmune component? Thorax. 2003;58:832–4. doi: 10.1136/thorax.58.10.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Strate BW, Postma DS, Brandsma CA, et al. Cigarette smoke-induced emphysema: a role for the B cell? Am J Respir Crit Care Med. 2006;173:751–8. doi: 10.1164/rccm.200504-594OC. [DOI] [PubMed] [Google Scholar]
- 5.Feghali-Bostwick CA, Gadgil AS, Otterbein LE, et al. Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:156–63. doi: 10.1164/rccm.200701-014OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stefanska AM, Walsh PT. Chronic obstructive pulmonary disease: evidence for an autoimmune component. Cell Mol Immunol. 2009;6:81–6. doi: 10.1038/cmi.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee SH, Goswami S, Grudo A, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. 2007;13:567–9. doi: 10.1038/nm1583. [DOI] [PubMed] [Google Scholar]
- 8.Hodge G, Nairn J, Holmes M, Reynolds P, Hodge S. Increased intracellular T helper type 1 proinflammatory cytokine production in peripheral blood, bronchoalveolar lavage and intraepithelial T cells of chronic obstructive pulmonary disease subjects. Clin Exp Immunol. 2007;150:22–9. doi: 10.1111/j.1365-2249.2007.03451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saetta M, Di Stefano A, Turato G, et al. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157:822–6. doi: 10.1164/ajrccm.157.3.9709027. [DOI] [PubMed] [Google Scholar]
- 10.Hodge SJ, Hodge GL, Reynolds PN, Scicchitano R, Holmes M. Increased production of TGF-beta and apoptosis of T lymphocytes isolated from peripheral blood in COPD. Am J Physiol Lung Cell Mol Physiol. 2003;285:L492–L499. doi: 10.1152/ajplung.00428.2002. [DOI] [PubMed] [Google Scholar]
- 11.de Jong JW, van der Belt-Gritter B, Koeter GH, Postma DS. Peripheral blood lymphocyte cell subsets in subjects with chronic obstructive pulmonary disease: association with smoking, IgE and lung function. Respir Med. 1997;91:67–76. doi: 10.1016/s0954-6111(97)90070-6. [DOI] [PubMed] [Google Scholar]
- 12.Hodge S, Hodge G, Nairn J, Holmes M, Reynolds PN. Increased airway granzyme b and perforin in current and ex-smoking COPD subjects. COPD. 2006;3:1–9. doi: 10.1080/15412550600976868. [DOI] [PubMed] [Google Scholar]
- 13.Guinan EC, Gribben JG, Bousiottis VA, Freeman GJ, Nadler LM. Pivotal role of the B7:CD28 pathway in transplantation tolerance and tumor immunity. Blood. 1994;84:3261–82. [PubMed] [Google Scholar]
- 14.Heron M, Claessen AM, Grutters JC, van den Bosch JM. T cell activation profiles in different granulomatous interstitial lung diseases – a role for CD8+CD28(null) cells? Clin Exp Immunol. 2010;160:256–65. doi: 10.1111/j.1365-2249.2009.04076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma G, Hanania NA, Shim YM. The aging immune system and its relationship to the development of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:573–80. doi: 10.1513/pats.200904-022RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arosa FA. CD8+CD28− T cells: certainties and uncertainties of a prevalent human T-cell subset. Immunol Cell Biol. 2002;80:1–13. doi: 10.1046/j.1440-1711.2002.01057.x. [DOI] [PubMed] [Google Scholar]
- 17.Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/ WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med. 2001;163:1256–76. doi: 10.1164/ajrccm.163.5.2101039. [DOI] [PubMed] [Google Scholar]
- 18.Hodge S, Matthews G, Dean M, et al. Is there a therapeutic role for mannose binding lectin in cigarette smoke-induced lung inflammation? Evidence from a murine model. Am J Respir Cell Mol Biol. 2010;42:235–42. doi: 10.1165/rcmb.2008-0486OC. [DOI] [PubMed] [Google Scholar]
- 19.Hodge S, Matthews G, Mukaro V, et al. Cigarette smoke-induced changes to alveolar macrophage phenotype and function is improved by treatment with procysteine. Am J Respir Cell Mol Biol. 2011;44:673–81. doi: 10.1165/rcmb.2009-0459OC. [DOI] [PubMed] [Google Scholar]
- 20.Hodge S, Hodge G, Flower R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with COPD are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. 2003;81:289–96. doi: 10.1046/j.1440-1711.2003.t01-1-01170.x. [DOI] [PubMed] [Google Scholar]
- 21.Hodge S, Hodge G, Ahern J, Jersman H, Holmes M, Reynolds PN. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2007;37:748–55. doi: 10.1165/rcmb.2007-0025OC. [DOI] [PubMed] [Google Scholar]
- 22.Lambers C, Hacker S, Posch M, et al. T cell senescence and contraction of T cell repertoire diversity in patients with chronic obstructive pulmonary disease. Clin Exp Immunol. 2009;155:466–75. doi: 10.1111/j.1365-2249.2008.03835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts SD, Kohli LL, Wood KL, Wilkes DS, Knox KS. CD4+CD28−T cells are expanded in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2005;22:13–19. [PubMed] [Google Scholar]
- 24.Katchar K, Wahlström J, Eklund A, Grunewald J. Highly activated T-cell receptor AV2S3(+) CD4(+) lung T-cell expansions in pulmonary sarcoidosis. Am J Respir Crit Care Med. 2001;163:1540–5. doi: 10.1164/ajrccm.163.7.2005028. [DOI] [PubMed] [Google Scholar]
- 25.Wouters EF. Chronic obstructive pulmonary disease. 5: Systemic effects of COPD. Thorax. 2002;57:1067–70. doi: 10.1136/thorax.57.12.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oudijk EJ, Lammers JW, Koenderman L. Systemic inflammation in chronic obstructive pulmonary disease. Eur Respir J Suppl. 2003;46:5s–13s. doi: 10.1183/09031936.03.00004603a. [DOI] [PubMed] [Google Scholar]
- 27.Lehmann C, Wilkening A, Leiber D, et al. Lymphocytes in the bronchoalveolar space reenter the lung tissue by means of the alveolar epithelium, migrate to regional lymph nodes, and subsequently rejoin the systemic immune system. Anat Rec. 2001;264:229–36. doi: 10.1002/ar.1163. [DOI] [PubMed] [Google Scholar]