

Abstract
A void in understanding primary biliary cirrhosis (PBC) is the absence of appropriate animal models. Our laboratory has studied a murine model of autoimmune cholangitis induced following immunization with 2-octynoic acid (2OA), an antigen identified following extensive quantitative structural activity relationship (QSAR) analysis, using human autoantibodies and three-dimensional analysis of the mitochondrial autoantigen, the E2 subunit of the pyruvate dehydrogenase complex (PDC-E2). Mice immunized with 2OA coupled to bovine serum albumin (BSA) develop anti-mitochondrial antibodies (AMAs) of the identical specificity as humans with PBC, and in addition develop inflammatory portal cell infiltrates in liver. However, the natural history of disease is less severe than in humans and does not include fibrosis. Data from human and autoimmune murine models suggest that environmental and/or infectious agents can exacerbate autoimmune reactions, and a model of PBC has been described in which polyinosinic-polycytidylic acid (poly I:C), a viral RNA mimetic and Toll-like receptor 3 (TLR-3) agonist induces low-titre AMAs and in mild portal infiltrates. We took advantage of our established model to determine whether immunization with 2OA-BSA coupled with poly I:C alters the disease process. Indeed, the addition of poly I:C produces a profound exacerbation of autoimmune cholangitis, including a significant increase in CD8+ infiltrating T cells, as well as a marked increase of proinflammatory cytokines. In addition, mice have evidence of fibrosis. These findings lend support to the concept that besides breakdown of self-tolerance, there is a requirement of a second ‘hit’ during the breakdown process that leads to disease which more faithfully mimics human PBC.
Keywords: liver fibrosis, poly I:C, primary biliary cirrhosis, TLRs, xenobiotics
Introduction
Primary biliary cirrhosis (PBC) is a progressive autoimmune liver disease characterized by immune-mediated destruction of intrahepatic small bile ducts [1]. Our laboratory has taken advantage of our extensive work on the quantitative structural activity relationship of the major mitochondrial autoantigen (PDC-E2) and, in particular, identification of molecular mimics of the inner lipoyl domain of PDC-E2 and the use of such mimics in inducing autoimmune cholangitis in mice [2–5]. This model has helped to define early events that lead to liver infiltrates and the production of anti-mitochondrial antibodies. It also provides a persuasive environmental argument for induction of human PBC. However, the natural history and severity of the disease in these mice is of relatively low intensity, and does not include fibrosis that is more typical of human PBC. It is well known that chemical and infectious agents modulate Toll-like receptors (TLRs) and can potentially amplify the inflammatory response [6–8]. These latter data prompted us to examine the role of one such agent in our murine model of human PBC.
Polyinosinic-polycytidylic acid (poly I:C) is a viral RNA mimetic that induces immune responses via TLR-3 [9,10]. Treatment with poly I:C increases the cytotoxic effect mediated by macrophages and natural killer (NK) cells [11,12] and activates T cells [13]. In murine liver, poly I:C treatment causes liver injury [14] which is interleukin (IL)-12-dependent [15]. PBC patients have an increased level of hepatic TLR-3 expression compared to liver tissues from control patients [16]. In addition, when stimulated by poly I:C, monocytoid cells from PBC patients produce increased proinflammatory cytokines compared with other TLR agonists [17]. Furthermore, long-term in vivo administration of mice with poly I:C can lead to a PBC-like disease, including the production of anti-mitochondrial antibodies (AMAs) associated with lymphocyte infiltrations around bile ducts [18,19]. However, the relationship between the mechanism by which poly I:C induces AMAs and liver pathology and the relevance of these findings to the pathogenesis of PBC remains unclear.
In the present study, we employed the 2OA xenobiotic-induced murine model of PBC to investigate the potential augmenting effect of poly I:C administration on the exacerbation of autoimmune cholangitis to test the ‘two-hit’ hypothesis of autoimmune disease. Indeed, results of our studies show that co-administration of poly I:C during immunization with 2OA led to a dramatic augmentation of autoimmune cholangitis. These mice demonstrate marked increases in inflammatory cytokine production associated with an increase in effector CD8+ T cell infiltration of liver. Interestingly, the fact that poly I:C administration did not influence titres of AMAs suggests that the exacerbation of PBC-like disease in these mice probably involved innate immunity. Indeed, poly I:C produced eosinophil infiltration and bridging fibrosis in the liver of 2OA-immunized mice. These data provide compelling evidence to conduct studies aimed at identification of additional natural environmental factor(s) that mimic the effect of poly I:C which can modulate the natural history of PBC, and thus provide useful targets for therapeutic intervention.
Materials and methods
Experimental protocol
Female C57BL/6J (B6) 5–6-week-old mice (n = 12 per group) were obtained from Jackson Labs (Bar Harbor, ME, USA) and maintained in ventilated cages under specific pathogen-free conditions at the animal facilities of the University of California at Davis. All studies were approved by the University of California at Davis Animal Care and Use Committee. The induction of autoimmune cholangitis employed the method that we have described in detail previously [2]. Essentially, a 2OA-bovine serum albumin (BSA) conjugate [100 µg/100 µl phosphate-buffered saline (PBS)] was injected intraperitoneally with complete Freund's adjuvant (CFA; Sigma-Aldrich, St Louis, MO, USA) containing 1 mg/ml of Mycobacterium tuberculosis strain H37Ra and was boosted after 2 weeks with 2OA-BSA and incomplete Freund's adjuvant (IFA; Sigma-Aldrich) after the first immunization. Additionally, mice received 100 ng of pertussis toxin (List Biological Laboratories, Campbell, CA, USA) at the time of and 2 days after the initial immunization with 2OA-BSA in CFA. Poly I:C (Sigma Chemical Co., St Louis, MO, USA) was injected intraperitoneally at a dose of 5 mg/kg once every 3 days from three days after the initial immunization with 2OA-BSA in CFA (2OA+poly I:C-treated mice). As controls, a comparable numbers of female B6 mice were immunized with 2OA-BSA alone (2OA-treated mice) and treated with poly I:C alone (poly I:C-treated mice) with the identical protocol. Sera was collected at weeks 2, 4 and 8 after the first immunization with 2OA-BSA, and stored at −80°C until use. Mice were killed 8 weeks after the first immunization (13–14 weeks of age), and the livers and spleens were collected for histological evaluation and flow cytometric analysis for cellular phenotypes.
Detection of anti-mitochondrial antibodies and cytokine levels
Serum titres of anti-PDC-E2, anti-E2 subunit of the branched-chain ketoacid dehydrogenase (BCKD-E2) and anti-E2 subunit of the 2-oxoglutarate dehydrogenase complex (OGDC-E2) autoantibodies were measured by enzyme-linked immunosorbent assay (ELISA), as described previously [2], using our standardized recombinant autoantigens [20]. Briefly, purified recombinant PDC-E2, BCKD-E2 or OGDC-E2 antigen at 5 µg/ml in carbonate buffer (pH 9·6) was coated onto 96-well plates at 4°C overnight, washed five times with PBS containing 0·05% Tween-20 (PBS-T) and blocked with 3% skimmed milk in PBS for 1 h before use. The thawed and diluted sera (1:250) with 3% skimmed milk in PBS were added to the wells and incubated at room temperature for 1 h, then washed five times with PBS-T. Horseradish peroxidase (HRP)-conjugated anti-mouse immunoglobulin (Ig)G, IgM and IgA (1:3000) (Zymed, San Diego, CA, USA) was added to each well and incubated for 1 h at room temperature. Immunoreactivity was determined by measuring the optical density at 450 nm with an Emax precision microplate reader (Molecular Devices, Sunnyvale, CA, USA) after incubation with tetramethylbenzidine (TMB) substrate (BD Biosciences, San Jose, CA, USA) for 15 min.
Cytokine concentrations
Serum interleukin (IL)-6, interferon (IFN)-γ and tumour necrosis factor (TNF)-α levels were measured with the mouse inflammatory cytometric bead array kit (BD Immunocytometry Systems, San Jose, CA, USA). Following preparation, samples were analysed using a fluorescence activated cell sorter (FACScan) flow cytometer (BD Immunocytometry System). Serum IL-12p40 was measured using a mouse IL-12/IL23 p40 allele-specific DuoSet ELISA development kit (R&D Systems, Minneapolis, MN, USA). Briefly, capture antibody at 4 µg/ml in PBS was coated onto 96-well plates at room temperature overnight, washed five times with PBS-T and blocked with 3% skimmed milk in PBS for 1 h before use. The thawed and diluted sera (1:4) with 3% skimmed milk in PBS were added to the wells and incubated for 2 h at room temperature, then washed five times with PBS-T. Detection antibody was added to each well and incubated for 2 h at room temperature. Streptavidin–HRP was added to each well and incubated for 20 min at room temperature. Immunoreactivity was determined by measuring the optical density at 450 and 540 nm with an Emax precision microplate reader (Molecular Devices) after incubation with TMB substrate (BD Biosciences) for 15 min. Subtraction of the readings at 540 nm from the readings at 450 nm was performed to correct for optical imperfections in the plate.
Cell preparation and flow cytometry
Livers were first perfused with PBS containing 0·2% BSA (0·2% BSA/PBS), passed through a 100-µm nylon cell strainer (BD Bioscience) and resuspended in 0·2% BSA/PBS. Hepatocytes were removed as pellets after centrifugation at 75 g for 1 min and the remainder of cells collected. The spleen was disrupted between two glass slides and suspended in 0·2% BSA/PBS. Lymphocytes were separated by centrifugation with Histopaque-1·077 (Sigma-Aldrich, St Louis, MO, USA). After centrifugation, cells at the interface were collected and washed with 0·2% BSA/PBS, and the viability of cells was confirmed by Trypan Blue dye (Invitrogen, Camarillo, CA, USA) exclusion. Cell numbers were determined by a haemacytometer. For flow cytometry, cell preparations were incubated with mAb for Fc receptor blocking (BioLegend, San Diego, CA, USA) and then incubated at 4°C with a combination of fluorochrome-conjugated antibodies, including fluorescein isothiocyanate (FITC)-conjugated anti-CD44 (BioLegend), phycoerythrin (PE)-conjugated anti-CD62L (BioLegend), peridinin chlorophyll/cyanin (PerCP/Cy)5·5-conjugated anti-NK1·1 (BioLegend), allophycocyanin (APC)-conjugated anti-T cell receptor (TCR)-β (eBioscience, San Diego, CA, USA) and APC-Cy7-conjugated anti-CD4 (BioLegend) for T cell phenotype analysis. For T cell activation analysis, cells were stained with FITC-conjugated anti-CD69 (BioLegend), PE-conjugated anti-NKG2D (eBioscience), PerCP/Cy5·5-conjugated anti-NK1·1 (BioLegend), APC-conjugated anti-TCR-β (eBioscience) and APC-Cy7-conjugated anti-CD4 (BioLegend). For myeloid precursor cell population analysis, cells were stained with AlexaFluor 488-conjugated anti-Gr-1 (BioLegend), PE-conjugated anti-CD11c (BD Biosciences), PE-Cy5-conjugated CD11b (BioLegend), APC-conjugated anti-B220 (Caltag, Burlingame, CA, USA) and APC Cy7-conjuagated anti-TCR-β (BioLegend). Multi-colour flow analyses were performed using a FACScan flow cytometer (BD Biosciences) upgraded by Cytec Development (Fremont, CA, USA) to allow for five-colour analysis. Acquired data were analysed with CellQuest software (BD Biosciences).
Liver histopathology
Sections of liver tissue were fixed immediately with 10% buffered formalin at room temperature for 2 days, then embedded in paraffin and cut into 4-µm sections for routine haematoxylin (DakoCytomation, Carpinteria, CA, USA) and eosin (American Master Tech Scientific, Lodi, CA, USA) (H&E) staining, and additional Azan and silver staining. Evaluation under light microscopy was performed on coded H&E-stained sections of the livers using a set of six indices by a ‘blinded’ pathologist (K. T.); indices included the degree of liver inflammation, presence of portal inflammation, bile duct damage, hepatic granulomas, eosinophil infiltration and fibrosis. Each section was scored as either 0 = no significant change, 1 = minimal, 2 = mild, 3 = moderate or 4 = severe pathology. Details of this scoring system have been described previously [21,22].
Expression of data and statistical analysis
The data are expressed as the mean ± standard error of the mean. Two-sample comparisons were performed with one-tailed unpaired t-test. The Mann–Whitney U-test was performed for histological evaluation. The Prism statistical package (GraphPad Software Inc, La Jolla, CA, USA) was used and P-values less than 0·05 were considered statistically significant.
Results
Poly I:C administration exacerbates autoimmune cholangitis in the liver of 2OA-immunized mice
Mice immunized with 2OA-BSA and co-treated with poly I:C (2OA+poly I:C-treated mice) demonstrate profound inflammatory changes in liver at 15–16 weeks of age (Fig. 1). In contrast, mice treated with poly I:C only (poly I:C-treated mice) did not show any histological changes in the liver (Fig. 1a,b). Livers from mice immunized with 2OA-BSA (i.e. 2OA-BSA alone and 2OA-BSA with poly I:C) had similar degrees of lymphocyte infiltration, granulomas and bile duct damage (Fig. 1a). Interestingly, eosinophil infiltration was found only in the portal tracts and hepatic parenchyma of the 2OA+poly I:C group. These pathological findings are similar to those observed during the early stage of human PBC. Hepatic bridging fibrosis, moreover, was observed only in the livers of the mice that were immunized with 2OA+poly I:C (three of the 11 mice in the group). This was confirmed with Azan and silver staining (Fig. 2b,c) and is consistent with the histology of the late-stage human PBC. As summarized in Table 1 these results, along with the fact that mice immunized with 2OA-BSA alone and those treated with both 2OA-BSA and poly I:C had similar degrees of lymphocyte infiltration, granulomas and bile duct damage in the livers, suggests that co-treatment with poly I:C exacerbates the cholangitis induced by 2OA-BSA immunization.
Fig. 1.
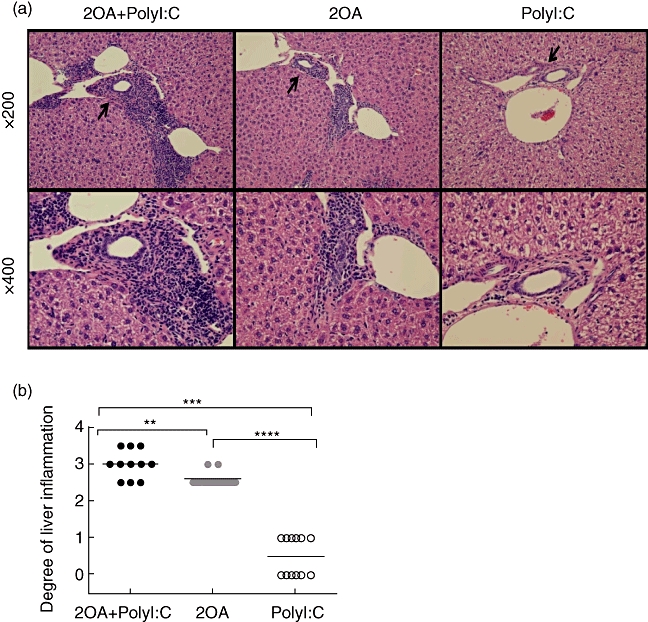
(a) Haematoxylin and eosin-stained tissue sections of livers from 2OA+polyinosinic-polycytidylic acid (poly I:C)-, 2OA-, poly I:C-treated mice at 8 weeks post-immunization. Infiltration of lymphocytes in portal tracts, particularly surrounding intralobular bile ducts (black arrow) were observed in the liver of both 2OA+poly I:C- and 2OA-treated mice, but none in poly I:C-treated mice. (b) The degree of liver inflammation is presented and compared for 2OA+poly I:C- (black dot; n = 11), 2OA- (grey dot; n = 11), poly I:C (white dot; n = 12)-treated mice at 8 weeks post-immunization. *P < 0·05; **P < 0·01; ***P < 0·001 (Mann–Whitney U-test).
Fig. 2.
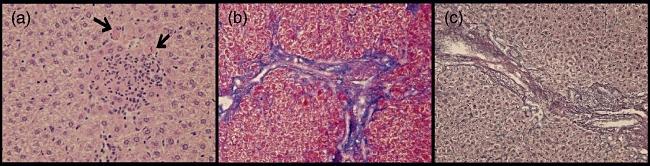
Microscopic examination of liver tissue from 2OA+polyinosinic-polycytidylic acid (poly I:C)-treated mice at 8 weeks post-immunization. Eosinophil infiltration (black arrow) was observed only in this group (haematoxylin and eosin × 400). Fibrosis formation was evaluated further with Azan (b, × 400) and silver staining (c, × 400) of liver sections from 2OA+poly I:C-treated mice at 8 weeks post- immunization.
Table 1.
Cholangitis in 2OA-immunized mice
| 2OA+poly I:C | 2OA | Poly I:C | |
|---|---|---|---|
| Serum autoantibodies | |||
| AMA | + | + | − |
| Liver histology | |||
| Portal inflammation | ++ | + | − |
| Eosinophil infiltration | + | + | − |
| Lymphocyte infiltration | + | + | − |
| Bile duct damage | + | + | − |
| Granulomas | + | + | − |
| Fibrosis | + | − | − |
AMA: anti-mitochondrial antibody; poly I:C: polyinosinic-polycytidylic acid; −: negative; +: positive; ++: more severe.
Co-treatment with poly I:C does not alter levels of AMA level induced in 2OA-immunized mice
Sera from 100% of mice immunized with 2OA-BSA [i.e. 2OA-BSA alone and 2OA-BSA with poly I:C (n = 11 each)] reacted with PDC-E2, BCOADC-E2 and OGDC-E2. In contrast, sera from none of the 11 control mice treated with poly I:C alone had any detectable reactivity against PDC-E2, BCOADC-E2 and OGDC-E2 (Fig. 3). We also found that the 2OA+poly I:C- and 2OA-treated mice developed similar levels of autoantibody against PDC-E2, BCOADC-E2 and OGDC-E2. These findings indicate that co-treatment with poly I:C does not influence AMA production by 2OA-BSA immunization (Fig. 3), despite the significant difference in liver pathology between the 2OA+poly I:C and 2OA groups (Fig. 1b). This finding is similar to what we observe in PBC patients, because AMA titres do not correlate with the severity of disease in human PBC [23].
Fig. 3.
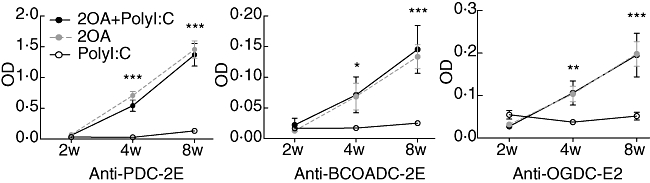
Quantification of serological anti-E2 subunit of the pyruvate dehydrogenase complex (PDC-E2), anti-branched chain 2-oxo acid dehydrogenase complex (BCOADC-E2) and anti-E2 subunit of the 2-oxoglutarate dehydrogenase complex (OGDC-E2) reactivity by enzyme-linked immunosorbent assay (ELISA) at 2, 4 and 8 weeks after immunization. *P < 0·05; **P < 0·01; ***P < 0·001.
Poly I:C increases inflammatory cytokine production in 2OA-immunized mice
Levels of each of the inflammatory cytokines measured (IFN-γ, TNF-α, IL-12p40 and IL-6) were increased significantly in the sera of the 2OA+poly I:C-treated mice compared with levels in the sera from mice immunized with 2OA alone (P = 0·0191, P = 0·0002, P = 0·0057 and P = 0·0209, respectively) and poly I:C alone (P = 0·0002, P < 0·0001, P < 0·0001 and P = 0·0017, respectively) (Fig. 4). Specifically, serum levels of the major T helper type 1 (Th1) cytokines (IFN-γ, TNF-α and IL-12p40) in the 2OA+poly I:C group were 40–60% higher than those in sera of the 2OA-BSA-alone immunized mice. Of importance was the finding that sera levels of two of the potent proinflammatory cytokines (IFN-γ and TNF-α) were six to seven times greater in the 2OA+poly I:C group than those in the poly I:C-alone group. These results suggest that poly I:C treatment in addition to 2OA-BSA immunization induces strong proinflammatory cytokines. In contrast, poly I:C treatment alone induced the lowest cytokine levels among the groups, emphasizing that poly I:C is not a strong inflammatory inducer by itself, but markedly enhances the inflammatory response when co-administered during 2OA-BSA immunization. These results suggests that 2OA-BSA immunization alone induces a Th1 skewed inflammatory response, and that poly I:C co-treatment markedly augments the Th1-skewed profile in such mice.
Fig. 4.
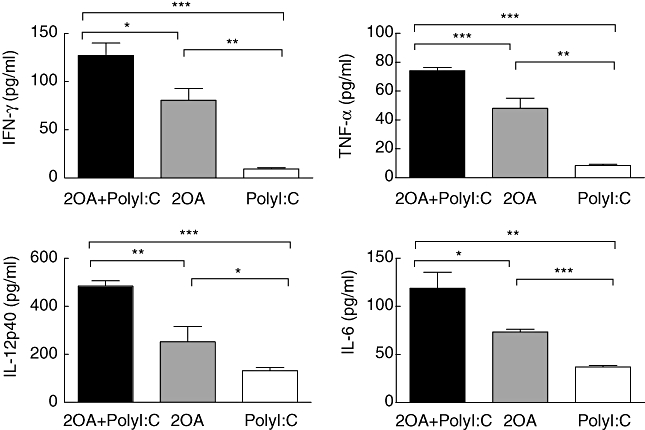
Serum concentration of interleukin (IL)-6, interferon (IFN)-γ, tumour necrosis factor (TNF)-α and IL-12p40 at 4 weeks after the immunization with 2-octynoic acid (2OA)-bovine serum albumin (BSA) conjugate with polyinosinic-polycytidylic acid (poly I:C) administration (n = 6) compared with 2OA-BSA alone (n = 4) and poly I:C alone (n = 6). *P < 0·05; **P < 0·01; ***P < 0·001.
Poly I:C increases the accumulation of effector CD8+ T cells in the liver of 2OA-immunized mice
Consistent with previous studies on human PBC and murine PBC models, the liver tissues of the 2OA-BSA-treated mice showed an infiltration of predominantly CD8+ T cells. Interestingly, the liver tissues of the 2OA+poly I:C-treated mice showed a significantly increased number of CD8+ T cells compared to mice immunized with 2OA-BSA alone (P = 0·0078) and/or poly I:C alone (P = 0·0004) (Fig. 5a,iii). These results indicate that poly I:C co-treatment with 2OA-BSA immunization can augment the trafficking and accumulation of cytotoxic CD8+ T cells in the liver. This is important, because autoreactive cytotoxic CD8+ T cells have been implicated in mediating the cytotoxicity of the intrahepatic bile ducts in PBC. In contrast, the number of CD4+ T cells in mice immunized with 2OA+poly I:C and mice immunized with 2OA alone was similar, but increased significantly compared to mice treated with poly I:C alone (P = 0·0137 and P = 0·0484, respectively) (Fig. 5a,ii). Although the liver tissues of mice immunized with 2OA alone had significantly higher effector memory CD8+ T cell (CD44+CD62L- population) frequency compared to mice treated with poly I:C alone (P = 0·0374), the frequency of such CD8+ T cells was increased even more in the liver of 2OA+poly I:C-immunized mice (P = 0·0103) (Fig. 5b,ii,iii), while a similar frequency was observed in effector memory CD4+ T cells (Fig. 5b,i,iii) for these groups. It is important to note that the infiltrating CD8+ T cells in the liver tissue of the 2OA+poly I:C-treated mice expressed CD69, a marker of activated cells. The frequency of CD69+ cells among hepatic CD8+ T cells was also increased significantly in 2OA+poly I:C immunized mice than mice immunized with 2-OA alone (P = 0·0007) (Fig. 5c,ii,iii), while a similar frequency was observed in CD4+ T cells (Fig. 5c,i,iii).
Fig. 5.
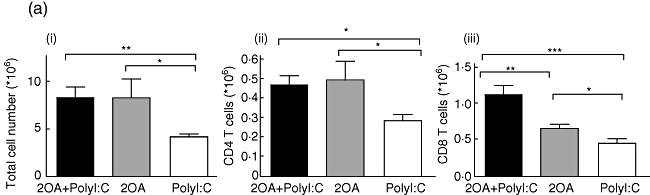
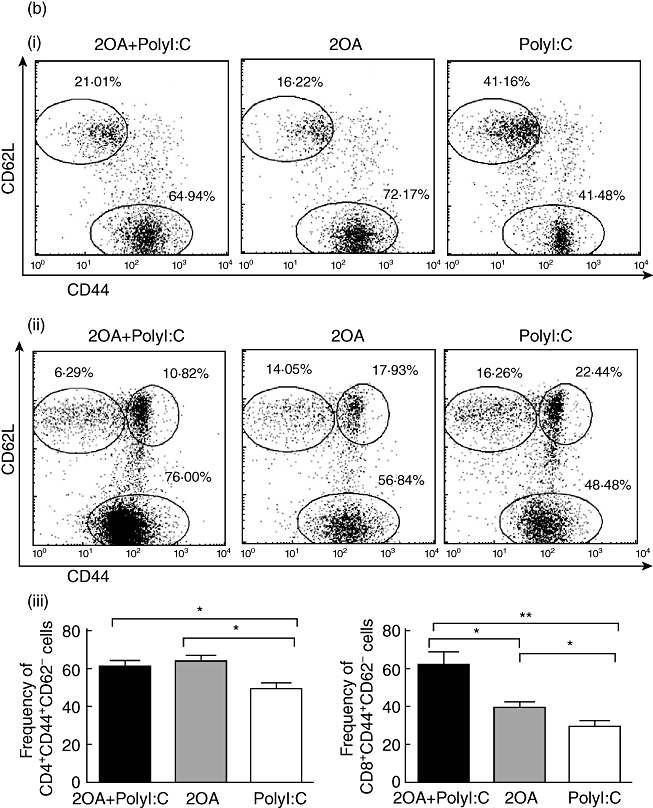
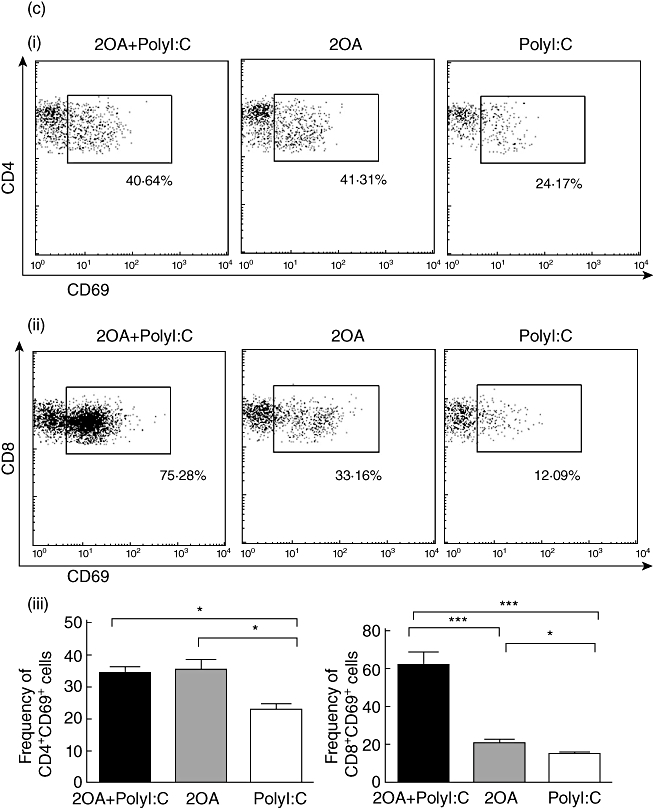
(a) The absolute numbers of total intrahepatic lymphocytes, hepatic CD4+ and CD8+ T cells were compared among 2OA+polyinosinic-polycytidylic acid (poly I:C) (n = 6), 2OA (n = 7), and poly I:C-treated mice (n = 8) at 8 weeks-post-immunization. *P < 0·05; **P < 0·01; ***P < 0·001. (b) Flow cytometric analysis of intrahepatic lymphocytes from 2OA+poly I:C (n = 6), 2OA (n = 7) and poly I:C-treated mice (n = 8) at 8 weeks post-immunization. Displayed in the dot-plots are cells gated on natural killer (NK)1·1- T cell receptor (TCR)-β+ T cell population. Numbers in the dot plots are percentages of cells in the specific gates out of the grated (i) CD4+ T cell population and (ii) CD8+ T cell population. The frequencies of (iii) CD4+CD44+CD62L- and CD8+CD44+CD62L- T cells were compared. *P < 0·05; **P < 0·01. (c) Flow cytometric analysis of intrahepatic lymphocytes from 2OA+poly I:C (n = 6), 2OA (n = 7) and poly I:C-treated mice (n = 8) at 8 weeks post-immunization. Displayed in the dot-plots are cells gated on NK1·1-TCR-β+ T cell population. Numbers in the dot-plots are percentages of cells in the specific gates of the gated (i) CD4+ T cell population and (ii) CD8+ T cell population. The frequencies of (iii) CD4+CD69+ and CD8+CD69+ T cells were compared. *P < 0·05; ***P < 0·001.
Poly I:C increases the accumulation of Gr-1low and Gr-1high CD11b+ cells in the liver of 2OA-immunized mice
The findings that myeloid cell subsets play an important role in the pathogenesis of a variety of murine autoimmune models [24–26] prompted us to include the analysis of these subsets. These studies thus included analysis of the frequencies of Gr-1low CD11b+ cells, which are comprised predominantly of monocytes and myeloid precursors, and Gr-1high CD11b+ cells, which are comprised primarily of granulocytes. As seen in Fig. 6, the frequency of Gr-1high CD11b+ cells, also known as ‘inflammatory’ monocytes [27,28], was increased significantly in the liver of 2OA+poly I:C-treated mice compared with values obtained in mice immunized with 2OA-BSA alone (P = 0·0291) and/or poly I:C alone (P = 0·0003). The frequency of Gr-1low CD11b+ cells, also known as ‘resident’ monocytes [27,28], was also increased significantly in the liver of 2OA+poly I:C-treated mice compared with mice immunized with 2OA-BSA alone (P = 0·0047) and/or poly I:C alone (P = 0·0474) (Fig. 6).
Fig. 6.
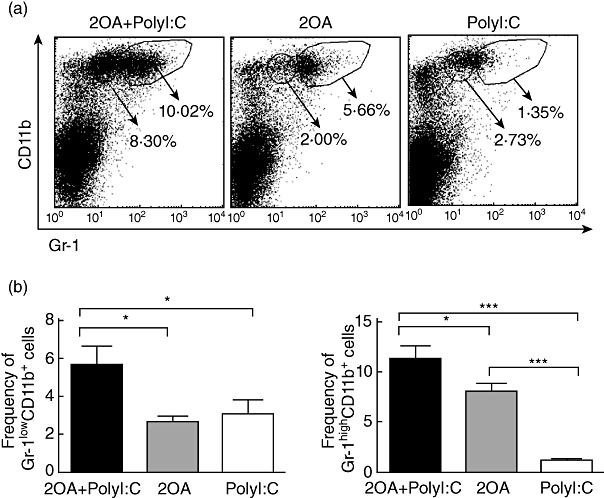
Flow cytometric analysis of intrahepatic lymphocytes from 2OA+polyinosinic-polycytidylic acid (poly I:C) (n = 6), 2OA (n = 7) and poly I:C-treated mice (n = 8) at 8 weeks-post-immunization. (a) Displayed in the dot plots are cells gated on the lymphocyte population. Numbers in the dot-plots are percentages of cells in the specific gates out of the grated lymphocyte population. The frequencies of (b) Gr-1lowCD11b+ and Gr-1highCD11b+ cells were compared. *P < 0·05; ***P < 0·001.
Poly I:C activates NK cells in the liver of 2OA-immunized mice
We first assessed the activation of NK cells in the liver of mice 8 weeks after immunization. As shown in Fig. 7, CD69 expression on NK cells was higher, but did not achieve statistical significance in the liver of 2OA+poly I:C-treated mice compared to 2OA-treated control. Of note, at 1 week after immunization, we detected significant elevation of NK cells activity in 2OA+poly I:C-treated mice, as evidenced by significant CD69 expression compared to 2OA-treated mice (Fig. 7).
Fig. 7.
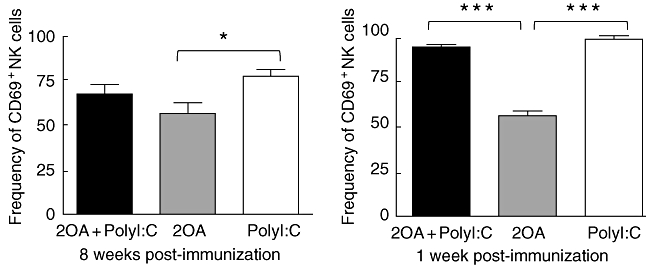
Activation of liver natural killer (NK) cells after 2 OA-immunization. At 1 and 8 weeks post-immunization, mice were killed and liver mononuclear cells were isolated and stained with monoclonal antibodies, as described. CD69 expression on NK1·1+ T cell receptor (TCR)-β- NK cells was analysed by flow cytometry. *P < 0·05; ***P < 0·001.
In addition, at 8 weeks post-immunization, both the frequency and absolute number of CD11c+ B220- conventional DC increased slightly; that of CD19+ B cells, CD11c+ B220- plasmacytoid DCs and NK1·1+TCR-β+ NK T cells decreased slightly in 2OA+poly I:C-treated mice, compared with 2OA-BSA-alone mice. However, these changes were not statistically different (data not shown).
Discussion
A number of studies have suggested that the TLR-3 pathway is critical in the pathogenesis of PBC [29–31]. Specifically, the liver from PBC patients has been shown to express markedly higher levels of TLR-3 than other liver diseases, i.e. autoimmune hepatitis (AIH) and chronic hepatitis C (CHC) [16]. We have demonstrated recently that peripheral monocytes of PBC patients produce more proinflammatory cytokines after poly I:C stimulation compared with other TLR agonists [17]. Furthermore, Okada et al. recently developed a murine PBC-like model induced by long-term poly I:C administration. Although the mice developed autoantibodies including AMAs in serum and lymphocyte infiltrations around bile ducts [18], the pathogenesis of TLR-3 in PBC is still unknown. The present study demonstrates that exposure to poly I:C treatment exacerbates murine autoimmune cholangitis induced by 2OA immunization in mice, including increased infiltration of effector CD8+ T cells and various myeloid cells (especially eosinophils), higher inflammatory cytokine production and fibrosis formation in the liver. These changes are very similar to that of early stage of the disease in human PBC. Our data suggest that TLR-3 activation is a critical factor in modulating the natural history of PBC.
In Okada's model, long-term administration (12–28 weeks) of poly I:C induced AMA production in mice [18]; however, the frequency of AMA-positive mice in the study was low (about 25%) at 4 and 8 weeks after the poly I:C treatment. In our model, 2OA immunization induced high levels of AMA in all mice, while poly I:C treatment alone did not cause any detectable AMAs at 4 and 8 weeks after the poly I:C treatment, which is similar to what was observed in Okada's model. However, the level of AMA did not increase significantly in sera of 2OA+poly I:C-treated mice compared to control 2OA-immunized mice. Although poly I:C cannot be excluded as a potential cause of induction of AMA and other autoantibodies, our data demonstrate that poly I:C treatment is not a main factor in the production of AMA in our murine PBC model.
AMA can be present in PBC patients long before the clinical disease [23]. Our results suggest that the transition from subclinical to clinical disease with overwhelming innate or bystander immune reactions may depend on exposure to viral infections that activate various TLRs, especially the TLR-3 pathway. Similar AMA titres between 2OA+poly I:C- and 2OA-alone immunized mice regardless of the difference in the liver pathology is consistent with the fact that AMA titres do not correlate with the severity of the disease in human PBC [32]. However, the role of poly I:C on induction of AMA could not be excluded in our animal model because we started treating mice with poly I:C 3 days after immunization. Indeed, long-term-treated mice with poly I:C caused AMA in sera of mice had been demonstrated [18]. It had been reported that poly I:C pretreatment inhibits concanavalin A (Con A)- and halothane-induced acute liver injury [11,33]. Further studies on the kinetics of their response will be evaluated in the future.
Interestingly, 2OA+poly I:C-treated mice demonstrated the presence of augmented autoimmune cholangitis, such as eosinophil infiltration and the presence of liver fibrosis, which are found characteristically in PBC patients [34,35], but rarely reported in murine PBC models. The induction of fibrosis in this model permits not only the elucidation of its induction but also has the potential to be useful in studies of intervention. In both human and animal studies, liver inflammation has been suggested as a requisite for the earliest stages of fibrosis [36,37], and several lymphoid subpopulations and cytokines clearly play a role in regulating this process. A Th1-skewed cytokine profile has been demonstrated in human PBC [38,39]. Therefore, an increase in Th1 cytokines such as IL-12 and IFN-γ in 2OA+poly I:C-treated mice compared with 2OA- and poly I:C-treated mice demonstrates that 2OA+poly I:C treatment provides the similar microenvironment as human PBC in mice.
In this study we demonstrate a significant increase of effector and activated CD8+ T cells in 2OA-treated mice, while poly I:C-treated mice did not show marked CD8+ T cell infiltration in liver. In contrast, poly I:C administration induced more effector CD8+ T cells accumulation in the liver of 2OA-immunized mice (2OA+poly I:C mice) compared with mice that did not receive poly I:C treatment (2OA-treated mice). These results suggest that poly I:C treatment alone does not induce the activation of CD8+ T cells but, rather, it elicits the trafficking and/or residence of cytotoxic effector CD8+ T cells into the liver. The increased CD8+ T cells may promote autoimmune cholangitis induced by the 2OA xenobiotic. These results are supported by our recent studies on the critical role of autoreactive CD8+ T cells and their role in the pathogenesis of cholangitis in cell transfer models [40,41]. It has been demonstrated that poly I:C can activate NK cells [12,42] via the TLR-3 pathway. In this study we found that NK cells were activated by treatment with poly I:C, as evidenced by high CD69 expression in 2 OA-immunized mice. Given that IFN-γ produced by NK cells is important for subsequent acquired immunity including T cell activation [43,44], the activated NK cells might result in more activation of cytotoxic CD8+ T cells found in our current model. However, how the effector CD8+ T cells migrate into the liver and damage the biliary epithelial cells in 2OA-immunized mice needs to be determined in future studies. In addition, poly I:C treatment enhanced granulocyte, especially eosinophil, cell infiltrates in the liver. The accumulation of inflammatory granulocytes by poly I:C administration may exacerbate the local hepatic inflammation and autoimmune cholangitis induced by 2OA-BSA in mice. These changes are significant, because in human PBC not only myeloperoxidase positive cells but also CD8+ T cells are implicated in the destruction of bile ducts cells and the pathogenesis of PBC [45–47].
We have demonstrated recently the importance of NK T cells in the liver pathology in the 2OA-BSA-induced murine model of PBC. In particular, the activation of invariant NK T cells by α-galactosylceramide (α-GalCer) administration exacerbated murine autoimmune cholangitis, including increased AMA production, portal inflammation and bile duct damage [22]. More importantly, significant fibrosis was induced by multiple administrations of α-GalCer. Although others have shown that poly I:C treatment could induce NK T cell accumulation in liver [48], we did not find a significant increase in the number of NK and NK T cells at 8 weeks after immunization. In contrast, a significant increase of cytotoxic CD8+ T cells, which is critical in adaptive autoimmunity, was detected in the liver. As in other autoimmune diseases, PBC is a multi-orchestrated immune response, and the profound importance of innate immune responses in the disease pathogenesis of PBC is evident in murine models [22,49]. The role of innate immune response in the pathogenesis of PBC is supported further by the observation that monocytes from patients with PBC are more sensitive to TLR ligands [17]. Collectively, our data suggests that the natural history of the disease, and perhaps the initiation from the asymptomatic PBC to symptomatic PBC, may depend on activation of innate immunity, especially through TLRs.
Disclosure
The authors have no financial conflicts of interest.
References
- 1.Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–73. doi: 10.1056/NEJMra043898. [DOI] [PubMed] [Google Scholar]
- 2.Wakabayashi K, Lian ZX, Leung PS, et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology. 2008;48:531–40. doi: 10.1002/hep.22390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wakabayashi K, Yoshida K, Leung PS, et al. Induction of autoimmune cholangitis in non-obese diabetic (NOD).1101 mice following a chemical xenobiotic immunization. Clin Exp Immunol. 2009;155:577–86. doi: 10.1111/j.1365-2249.2008.03837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ueno Y, Ambrosini YM, Moritoki Y, Ridgway WM, Gershwin ME. Murine models of autoimmune cholangitis. Curr Opin Gastroenterol. 2010;26:274–9. doi: 10.1097/MOG.0b013e32833755aa. [DOI] [PubMed] [Google Scholar]
- 5.Amano K, Leung PS, Rieger R, et al. Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid. J Immunol. 2005;174:5874–83. doi: 10.4049/jimmunol.174.9.5874. [DOI] [PubMed] [Google Scholar]
- 6.Long SA, Quan C, Van de Water J, et al. Immunoreactivity of organic mimeotopes of the E2 component of pyruvate dehydrogenase: connecting xenobiotics with primary biliary cirrhosis. J Immunol. 2001;167:2956–63. doi: 10.4049/jimmunol.167.5.2956. [DOI] [PubMed] [Google Scholar]
- 7.Selmi C, Gershwin ME. Bacteria and human autoimmunity: the case of primary biliary cirrhosis. Curr Opin Rheumatol. 2004;16:406–10. doi: 10.1097/01.bor.0000130538.76808.c2. [DOI] [PubMed] [Google Scholar]
- 8.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 9.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 10.Matsumoto M, Kikkawa S, Kohase M, Miyake K, Seya T. Establishment of a monoclonal antibody against human Toll-like receptor 3 that blocks double-stranded RNA-mediated signaling. Biochem Biophys Res Commun. 2002;293:1364–9. doi: 10.1016/S0006-291X(02)00380-7. [DOI] [PubMed] [Google Scholar]
- 11.Cheng L, You Q, Yin H, Holt M, Franklin C, Ju C. Effect of polyI:C cotreatment on halothane-induced liver injury in mice. Hepatology. 2009;49:215–26. doi: 10.1002/hep.22585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Xu J, Zhang W, Wei H, Tian Z. TLR3 ligand-induced accumulation of activated splenic natural killer cells into liver. Cell Mol Immunol. 2005;2:449–53. [PubMed] [Google Scholar]
- 13.Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med. 1999;189:521–30. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yin S, Gao B. Toll-like receptor 3 in liver diseases. Gastroenterol Res Pract. 2010;pii:750904. doi: 10.1155/2010/750904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong Z, Wei H, Sun R, Hu Z, Gao B, Tian Z. Involvement of natural killer cells in PolyI:C-induced liver injury. J Hepatol. 2004;41:966–73. doi: 10.1016/j.jhep.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 16.Takii Y, Nakamura M, Ito M, et al. Enhanced expression of type I interferon and toll-like receptor-3 in primary biliary cirrhosis. Lab Invest. 2005;85:908–20. doi: 10.1038/labinvest.3700285. [DOI] [PubMed] [Google Scholar]
- 17.Mao TK, Lian ZX, Selmi C, et al. Altered monocyte responses to defined TLR ligands in patients with primary biliary cirrhosis. Hepatology. 2005;42:802–8. doi: 10.1002/hep.20859. [DOI] [PubMed] [Google Scholar]
- 18.Okada C, Akbar SM, Horiike N, Onji M. Early development of primary biliary cirrhosis in female C57BL/6 mice because of poly I:C administration. Liver Int. 2005;25:595–603. doi: 10.1111/j.1478-3231.2005.01043.x. [DOI] [PubMed] [Google Scholar]
- 19.Jiang T, Han Z, Chen S, et al. Resistance to activation-induced cell death and elevated FLIPL expression of CD4+ T cells in a polyI:C-induced primary biliary cirrhosis mouse model. Clin Exp Med. 2009;9:269–76. doi: 10.1007/s10238-009-0052-2. [DOI] [PubMed] [Google Scholar]
- 20.Miyakawa H, Tanaka A, Kikuchi K, et al. Detection of antimitochondrial autoantibodies in immunofluorescent AMA-negative patients with primary biliary cirrhosis using recombinant autoantigens. Hepatology. 2001;34:243–8. doi: 10.1053/jhep.2001.26514. [DOI] [PubMed] [Google Scholar]
- 21.Tsuneyama K, Nose M, Nisihara M, Katayanagi K, Harada K, Nakanuma Y. Spontaneous occurrence of chronic non-suppurative destructive cholangitis and antimitochondrial autoantibodies in MRL/lpr mice: possible animal model for primary biliary cirrhosis. Pathol Int. 2001;51:418–24. doi: 10.1046/j.1440-1827.2001.01223.x. [DOI] [PubMed] [Google Scholar]
- 22.Wu SJ, Yang YH, Tsuneyama K, et al. Innate immunity and primary biliary cirrhosis: activated invariant natural killer T cells exacerbate murine autoimmune cholangitis and fibrosis. Hepatology. 2011;53:915–25. doi: 10.1002/hep.24113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishibashi H, Komori A, Shimoda S, Ambrosini YM, Gershwin ME, Nakamura M. Risk factors and prediction of long-term outcome in primary biliary cirrhosis. Intern Med. 2011;50:1–10. doi: 10.2169/internalmedicine.50.4462. [DOI] [PubMed] [Google Scholar]
- 24.Zhu B, Bando Y, Xiao S, et al. CD11b+Ly-6C(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J Immunol. 2007;179:5228–37. doi: 10.4049/jimmunol.179.8.5228. [DOI] [PubMed] [Google Scholar]
- 25.Haile LA, von Wasielewski R, Gamrekelashvili J, et al. Myeloid-derived suppressor cells in inflammatory bowel disease: a new immunoregulatory pathway. Gastroenterology. 2008;135:871–81. doi: 10.1053/j.gastro.2008.06.032. 81 e1-5. [DOI] [PubMed] [Google Scholar]
- 26.Hegde VL, Nagarkatti M, Nagarkatti PS. Cannabinoid receptor activation leads to massive mobilization of myeloid-derived suppressor cells with potent immunosuppressive properties. Eur J Immunol. 2010;40:3358–71. doi: 10.1002/eji.201040667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 28.Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–47. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobayashi Y, Murakami H, Akbar SM, Matsui H, Onji M. A novel and effective approach of developing aggressive experimental autoimmune gastritis in neonatal thymectomized BALB/c mouse by polyinosinic:polycytidylic acid. Clin Exp Immunol. 2004;136:423–31. doi: 10.1111/j.1365-2249.2004.02467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qu WM, Miyazaki T, Terada M, et al. A novel autoimmune pancreatitis model in MRL mice treated with polyinosinic:polycytidylic acid. Clin Exp Immunol. 2002;129:27–34. doi: 10.1046/j.1365-2249.2002.01881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren X, Zhou H, Li B, Su SB. Toll-like receptor 3 ligand polyinosinic:polycytidylic acid enhances autoimmune disease in a retinal autoimmunity model. Int Immunopharmacol. 2011;11:769–73. doi: 10.1016/j.intimp.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 32.Van Norstrand MD, Malinchoc M, Lindor KD, et al. Quantitative measurement of autoantibodies to recombinant mitochondrial antigens in patients with primary biliary cirrhosis: relationship of levels of autoantibodies to disease progression. Hepatology. 1997;25:6–11. doi: 10.1002/hep.510250103. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Sun R, Wei H, Dong Z, Gao B, Tian Z. Poly I:C prevents T cell-mediated hepatitis via an NK-dependent mechanism. J Hepatol. 2006;44:446–54. doi: 10.1016/j.jhep.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 34.Lee RG, Epstein O, Jauregui H, Sherlock S, Scheuer PJ. Granulomas in primary biliary cirrhosis: a prognostic feature. Gastroenterology. 1981;81:983–6. [PubMed] [Google Scholar]
- 35.Nakamura A, Yamazaki K, Suzuki K, Sato S. Increased portal tract infiltration of mast cells and eosinophils in primary biliary cirrhosis. Am J Gastroenterol. 1997;92:2245–9. [PubMed] [Google Scholar]
- 36.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–69. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Connolly MK, Bedrosian AS, Mallen-St Clair J, et al. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-alpha. J Clin Invest. 2009;119:3213–25. doi: 10.1172/JCI37581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harada K, Van de Water J, Leung PS, et al. In situ nucleic acid hybridization of cytokines in primary biliary cirrhosis: predominance of the Th1 subset. Hepatology. 1997;25:791–6. doi: 10.1002/hep.510250402. [DOI] [PubMed] [Google Scholar]
- 39.Nagano T, Yamamoto K, Matsumoto S, et al. Cytokine profile in the liver of primary biliary cirrhosis. J Clin Immunol. 1999;19:422–7. doi: 10.1023/a:1020511002025. [DOI] [PubMed] [Google Scholar]
- 40.Yang GX, Wu Y, Tsukamoto H, et al. CD8 T cells mediate direct biliary ductule damage in nonobese diabetic autoimmune biliary disease. J Immunol. 2011;186:1259–67. doi: 10.4049/jimmunol.1001597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang GX, Lian ZX, Chuang YH, et al. Adoptive transfer of CD8(+) T cells from transforming growth factor beta receptor type II (dominant negative form) induces autoimmune cholangitis in mice. Hepatology. 2008;47:1974–82. doi: 10.1002/hep.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miyake T, Kumagai Y, Kato H, et al. Poly I:C-induced activation of NK cells by CD8 alpha+ dendritic cells via the IPS-1 and TRIF-dependent pathways. J Immunol. 2009;183:2522–8. doi: 10.4049/jimmunol.0901500. [DOI] [PubMed] [Google Scholar]
- 43.Medzhitov R, Janeway CA., Jr Innate immunity: impact on the adaptive immune response. Curr Opin Immunol. 1997;9:4–9. doi: 10.1016/s0952-7915(97)80152-5. [DOI] [PubMed] [Google Scholar]
- 44.Strowig T, Brilot F, Munz C. Noncytotoxic functions of NK cells: direct pathogen restriction and assistance to adaptive immunity. J Immunol. 2008;180:7785–91. doi: 10.4049/jimmunol.180.12.7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu CT, Eiserich JP, Ansari AA, et al. Myeloperoxidase-positive inflammatory cells participate in bile duct damage in primary biliary cirrhosis through nitric oxide-mediated reactions. Hepatology. 2003;38:1018–25. doi: 10.1053/jhep.2003.50407. [DOI] [PubMed] [Google Scholar]
- 46.Kita H, Matsumura S, He XS, et al. Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest. 2002;109:1231–40. doi: 10.1172/JCI14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kita H, Lian ZX, Van de Water J, et al. Identification of HLA-A2-restricted CD8(+) cytotoxic T cell responses in primary biliary cirrhosis: T cell activation is augmented by immune complexes cross-presented by dendritic cells. J Exp Med. 2002;195:113–23. doi: 10.1084/jem.20010956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol. 2009;86:513–28. doi: 10.1189/jlb.0309135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chuang YH, Lian ZX, Yang GX, et al. Natural killer T cells exacerbate liver injury in a transforming growth factor beta receptor II dominant-negative mouse model of primary biliary cirrhosis. Hepatology. 2008;47:571–80. doi: 10.1002/hep.22052. [DOI] [PubMed] [Google Scholar]