

Abstract
Because invariant natural killer T (iNK T) cells link innate and adaptive immunity, the structure-dependent design of iNK T cell agonists may have therapeutic value as vaccines for many indications, including autoimmune disease. Previously, we showed that treatment of non-obese diabetic (NOD) mice with the iNK T cell activating prototypic glycolipid α-galactosylceramide (α-GalCer) protects them from type 1 diabetes (T1D). However, α-GalCer is a strong agonist that can hyperactivate iNK T cells, elicit several side effects and has shown only limited success in clinical trials. Here, we used a structure-guided design approach to identify an iNK T cell agonist that optimally protects from T1D with minimal side effects. Analyses of the kinetics and function of a panel of synthetic α-GalCer fatty acyl chain derivatives (C8:0-C16:0) were performed in NOD mice. C16:0 elicited the highest protection from insulitis and T1D, which was associated with a higher frequency and survival of iNK T cells and enhanced activity of tolerogenic dendritic cells (DCs) in draining pancreatic lymph nodes (PLN), inability to transactivate NK cells and a more rapid kinetics of induction and recovery of iNK T cells from anergy. We conclude that the length and structure of the acyl chain of α-GalCer regulates the level of protection against T1D in mice, and propose that the extent of this protection depends on the relative capacity of the acyl chain to accommodate an endogenous spacer lipid of appropriate length and structure. Thus, our findings with the α-GalCer C16:0 derivative suggest strongly that it be considered as a lead glycolipid candidate in clinical trials of T1D.
Keywords: dendritic cells, glycolipid agonist, immunoregulation, iNK T cells, Type 1 diabetes
Introduction
Invariant natural killer T (iNK T) cells comprise a non-conventional subset of T cells that express a canonical invariant T cell receptor (TCR)-α-chain encoded by Vα14-Jα18 in mice, which together with a limited repertoire of TCR-β chains (Vβ2, Vβ7, Vβ8·2) mediates their recognition of glycolipid antigens presented by CD1d molecules on antigen-presenting cells (APCs) [1–3]. Because iNK T cells regulate immune responses, deficiencies in iNK T cells may yield adverse effects in the host and elicit immune dysregulation and disease onset. For example, numerical and functional deficiencies in iNK T cells develop in islets during progression to autoimmune type 1 diabetes (T1D) in NOD mice [3–8]. Although this result is debatable in human patients with T1D [9–11], iNK T cell activation with a suitable agonist may conceivably have therapeutic value for individuals at risk for T1D.
After activation with the prototypic α-galactosylceramide C26:0 (α-GalCer) glycosphingolipid agonist, iNK T cells produce T helper type 1 (Th1)- and Th2-type cytokines that promote cell-mediated immunity or induce immune tolerance, respectively. We and others have reported [4–7] that treatment of NOD mice with a multi-dose (MD) protocol of α-GalCer protects them from T1D in an interleukin (IL)-4-dependent manner that augments the recruitment of tolerogenic dendritic cells (DCs) to the draining pancreatic lymph nodes (PLN) [12]. These tolerogenic DCs can suppress effector T cells responsible for islet β cell death, and also stimulate Th2 and regulatory T cells (Treg) cells to suppress immune responses and protect against T1D [4–7,13–17].
α-GalCer is not only safe, tolerated and not hepatotoxic in humans, but also has beneficial effects in protection from autoimmune diseases [18] and cancer [19–22]. Nevertheless, the therapeutic use of α-GalCer has remained questionable because, in mice, α-GalCer can elicit adverse side effects including hepatocyte damage [23], exacerbation of atherosclerosis [24], increased rate of spontaneous abortion [25] and enhanced allergic responses [26]. After an initial response to α-GalCer, activated iNK T cells become anergic and respond poorly upon restimulation [27]. While anergic iNK T cells can induce tolerogenic DCs that mediate protection from T1D, chronic administration of α-GalCer triggers a long-lasting anergy accompanied by significantly reduced iNK T cell frequencies in the PLN. Thus, the potential long-term therapeutic use of α-GalCer raises several concerns [18].
To circumvent complications associated with α-GalCer therapy, several α-GalCer derivatives have been synthesized in attempts to elicit immune responses that protect from T1D without adverse side effects. When either side chain of α-GalCer is shortened, the rate of lipid dissociation from human CD1d is increased, indicating that the length of the sphingosine chain influences the threshold of iNK T cell activation [28]. Additionally, the glycolipid OCH, which has shorter fatty acyl and sphingosine side chains than α-GalCer, selectively stimulates iNK T cells to produce fewer Th1 and more Th2 cytokines than α-GalCer [29]. The extent of unsaturation in the acyl chain also determines the strength and duration of iNK T cell activation and protection from T1D, as evidenced by our demonstration that the C20:2 Th2-biased N-acyl variant of α-GalCer protects from T1D [30–32].
In the current study, we used a structure-guided approach to design a more optimal iNK T cell agonist(s) for the prevention of T1D [33] by the characterization of a panel of synthetic α-GalCer derivatives that differ incrementally by only two carbon atoms in their acyl chain length (C8:0, C10:0, C12:0, C14:0 and C16:0). The longer (C14:0, C16:0) acyl chain derivatives provided significantly greater protection of NOD mice from T1D than the shorter (≤C12:00) derivatives. Compared to α-GalCer, C16:0 not only stimulated higher protection from insulitis and T1D but also elicited an increased iNK T cell frequency and enhanced activity of tolerogenic DCs in the PLN. The latter responses were not observed for C8:0. These analyses reveal that the acyl chain length of α-GalCer regulates protection from T1D and provide further insight into the structural design and mechanism of action required for an α-GalCer-based agonist to protect optimally against T1D.
Materials and methods
Mice
NOD/Del, NOD.CD1d–/– and C57BL/6 mice were bred in house or purchased from The Jackson Laboratory (Bar Harbor, ME, USA). The incidence of T1D among the female NOD mice in our colony is ≥80% by 30 weeks of age. All mice were maintained in a specific pathogen-free facility in the Animal Care and Veterinary Services at the University of Western Ontario according to the Canadian Council for Animal Care guidelines.
Monitoring for diabetes
Mice were monitored beginning at 10 weeks of age for hyperglycaemia, as described [32].
Glycolipids and antibodies
KRN7000 (α-GalCer, C26:0/C18:0) and its fatty acyl chain derivatives (C8:0-C16:0) were synthesized, solubilized at 1 mg/ml in dimethylsulphoxide (DMSO) and stored as aliquots at −20°C until use [34]. The control vehicle was 2% DMSO in phosphate-buffered saline (PBS). Allophycocyanin (APC)-conjugated PBS-57-loaded and -unloaded CD1d tetramers for staining mouse iNK T cells were provided by the NIH Tetramer Core Facility (Emory University, Atlanta, GA, USA) [32]. Fluorescein isothiocyanate (FITC)-conjugated anti-TCR-β (H57-597), anti-B220 (RA3-6B2), anti-Pan NK cells (DX5), anti-Siglec H (eBio440c), anti-CD11c (N418), anti-CD4 (GK1·5), anti-CD8 (53–6·7) anti-CD3ε (eBio500A2); phycoerythrin (PE)-conjugated anti-CD69 (H1·2F3), anti-I-Ad (AMS-32·1), anti-CD86 (Gl-1), anti-CD80 (16-10A1), anti-CD40L (MR1), anti-IL-4 (11B11), anti-interferon (IFN)-γ (XMG1·2), anti-IL-12 (C17·8), anti-IL-10 (JES5-16E3), anti-CD11b (M1/70), anti-immunoglobulin (Ig)G2b (eB149/1OH5); peridinin chlorophyll (PerCP)-conjugated anti-CD8α (53–2·1), anti-CD4 (RM4-5), anti-CD3ε (145-2C11); purified CD16/32 (93), and APC-conjugated anti-CD11c (N418), anti-CD8 (53–6·7) and IgG (eBio299Arm) monoclonal antibodies (mAbs) were purchased from eBiosciences (San Diego, CA, USA) or BD Biosciences (Mississauga, ON, Canada).
In vivo treatment with glycolipid
Mice were injected intraperitoneally (i.p.) with glycolipid (4 µg/dose) using a single dose or MD treatment (every other day for 3 weeks) [32].
Cell isolation and flow cytometry
Splenic and PLN lymphocyte suspensions were prepared [32] and cells (1 × 106) were incubated with Fc-block (CD16/32, eBiosciences) for 15 min before staining (30 min, 4°C) with fluorescent mAbs diluted in buffer [PBS + 2% fetal bovine serum (FBS) + 0·1% sodium azide]. Cells were then washed and fixed in 2% paraformaldehyde. Flow cytometry was performed using FACSCalibur and CellQuest software (BD Biosciences) or FACSCanto II and FACSDiva software. Analyses were conducted using FlowJo software (Treestar, Ashland, OR, USA). Intracellular cytokine (IL-4, IFN-γ, IL-12) staining was performed using a BD Cytofix/Cytoperm buffer set (BD Biosciences) [32].
In vitro analysis of cell proliferation and cytokine secretion
Spleen- and PLN-derived lymphocytes (5 × 105 cells for proliferation analysis, 5 × 106 cells for cytokine analysis) were cultured (72 h, 37°C) in triplicate with glycolipid (100 ng/ml) or vehicle. To assay proliferation, cells were pulsed with [3H]-thymidine (1 µCi/well, Perkin Elmer) for the last 18 h of culture and harvested, and incorporated radioactivity was quantitated using a 1450 Microbeta scintillation counter (Perkin Elmer, Woodbridge, ON, Canada). All cultures were maintained in RPMI-1640 medium supplemented with 10% heat-inactivated FBS (Hyclone, Scarborough, ON, Canada), 1 mm sodium pyruvate, 10 mm HEPES, 100 U/ml penicillin, 100 µg/ml streptomycin, 2 mm l-glutamine and 0·05 mm 2-mercaptoethanol (ME) (Invitrogen Life Technologies, Burlington, ON, Canada). A standard sandwich enzyme-linked immunosorbent assay (ELISA) was performed to analyse mouse cytokines in culture supernatants using paired antibody kits for IL-4, IL-10, IL-2 and IFN-γ (BD Biosciences). Streptavidin–horseradish peroxidase (HRP) conjugate and development solution for BD OptiEIA Reagent Set A (BD Biosciences) were used for signal detection. Plates were read at a dual wavelength of 450/570 nm using a Benchmark Microplate Reader (Bio-Rad Laboratories, Mississauga, ON, Canada).
Histological analysis
Pancreata were fixed (24 h in 10% neutral buffered formalin), transferred to 70% ethanol for an additional 24 h, embedded in paraffin, sectioned (5–6 µm/section) and placed on microscope slides by the Molecular Pathology Facility (Robarts Research Institute). Tissue sections were stained with haematoxylin and eosin. Scoring was performed blindly by examining ≥ 50 pancreatic islets per mouse from three different sections [32]. Islets were graded as: (0) normal islet; (1) < 25% (peri-insulitis); (2) 25–50% infiltration (mild insulitis); and (3) > 50% islet infiltration (severe insulitis).
Statistical analysis
Results are expressed as mean ± standard error of the mean (s.e.m.). Statistical analyses were performed using the Student's t-test or log-rank test (for T1D incidence) (Prism version 4·0; GraphPad, San Diego, CA, USA). Differences were considered statistically significant at P values ≤ 0·05.
Results
Length of the α-GalCer fatty acyl chain correlates with the efficiency of iNK T cell-mediated protection from T1D and insulitis
α-GalCer and its synthetic fatty acyl chain derivatives (Fig. 1a) were assayed after MD treatment for protection of NOD mice from T1D and insulitis. While > 90% of vehicle-treated mice developed T1D by 30 weeks of age, only ∼40% of α-GalCer-treated mice were diabetic at this age (Fig. 1b), as reported [4,5,32]. The highest incidence and most rapid onset of T1D (100% at 27 weeks of age) occurred in C8:0-treated mice (Fig. 1b). C10:0 and C12:0 treatments resulted in an intermediate (50–60%) incidence of T1D. For C14:0 and C16:0, the onset was delayed (21 and 25 weeks of age, respectively) and the incidence of T1D was reduced (40% and 10%, respectively). Treatment with α-GalCer or C16:0, but not C8:0, protected NOD mice from destructive insulitis at 15 weeks of age (Fig. 1c,d). Thus, among the α-GalCer derivatives tested, C16:0 yielded the greatest protection from insulitis and T1D.
Fig. 1.
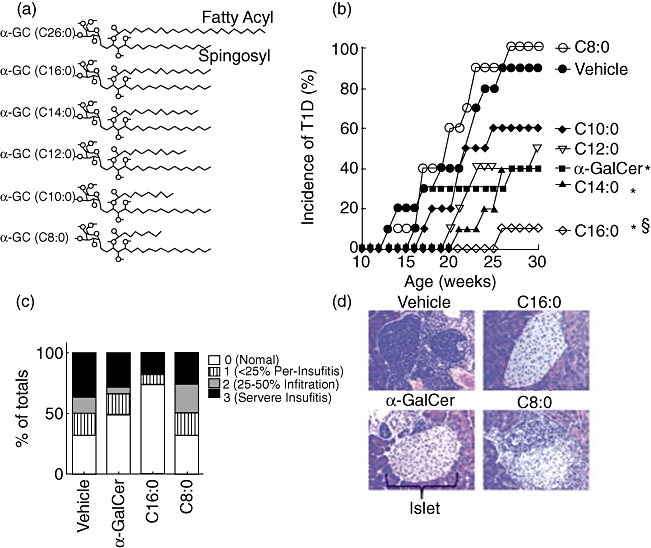
Glycolipid-induced protection against the spontaneous development of Type 1 diabetes (T1D). (a) Structure of α-galactosylceramide (α-GC) and its derivatives. The α-GC fatty acyl and sphingosyl side chains are shown. The carbon length of the fatty acyl chain of α-GC and each derivative is indicated in parentheses. Data in (b,c) are from one of two reproducible experiments. (b) Female non-obese diabetic (NOD) mice (n = 10, 6–8-week-old) were injected intraperitoneally using a multi-dose (MD) protocol in which mice were injected every other day for 3 weeks with 4 µg of glycolipid or vehicle. The mice were monitored for hyperglycaemia by measurement of blood glucose levels (BGL) twice weekly. Mice were considered diabetic when they had two consecutive BGL readings above 11·1 mmol/l. An asterisk (*) indicates significant reduction in incidence of T1D compared to vehicle control (P ≤ 0·05) calculated using a log-rank test. §Significant reduction compared to α-GalCer. (c) Histological analysis of pancreas. Female NOD mice (n = 5, 3–5-week-old) were treated with glycolipid using an MD protocol, killed at 15 weeks of age, and their pancreas were placed in cassettes and incubated at room temperature in neutral buffered formalin (10%, v/v) for 24 h. Tissues were transferred to 70% ethanol for an additional 24 h, then paraffin-embedded, sectioned and stained with haematoxylin and eosin (H&E). Each slide was analysed (in a blinded manner) and islet infiltration was scored using the following scale: 0 = no infiltration, 1 = < 25% infiltration, 2 = 25–50% infiltration and 3 = severe (> 50%) infiltration. Asterisks indicate a value significantly greater than vehicle control and C8:0 treatment group (P ≤ 0·05). (d) Example of a typical pancreatic islet obtained from a mouse in each treatment group described in Fig. 1c, magnified ×400.
α-GalCer derivatives differ in their stimulation of iNK T cell proliferation and cytokine secretion in vitro
The acyl side chain length dictates the Th-bias of an iNK T cell response in BALB/c and C57BL/6 (B6) mice [34]. To confirm this finding and extend it to NOD mice, we tested whether our α-GalCer derivatives stimulate BALB/c, B6, NOD and NOD.CD1d–/– spleen- and PLN-derived lymphocytes for proliferation and cytokine secretion. Proliferation and cytokine production were not detected for NOD.CD1d–/– splenocytes, demonstrating the CD1d dependency and role of iNK T cells in these responses (Fig. 2a,b). NOD lymphocyte proliferative responses were much weaker than those of BALB/c and B6 lymphocytes (Fig. 2a). The latter result is due probably to the decreased number and function of iNK T cells in the spleen of NOD mice relative to that in the spleen of BALB/c and B6 mice [35]. As NOD splenocytes yielded greater proliferative responses than PLN-derived lymphocytes, subsequent in vitro studies were conducted using NOD splenocytes. In all four mouse strains, C16:0 and C14:0 stimulated the lowest proliferative responses, while equivalent responses were obtained with C10:0, C8:0 and α-GalCer. For NOD splenocytes, C16:0 stimulated a much lower response than did C8:0 or α-GalCer (P ≤ 0·05).
Fig. 2.
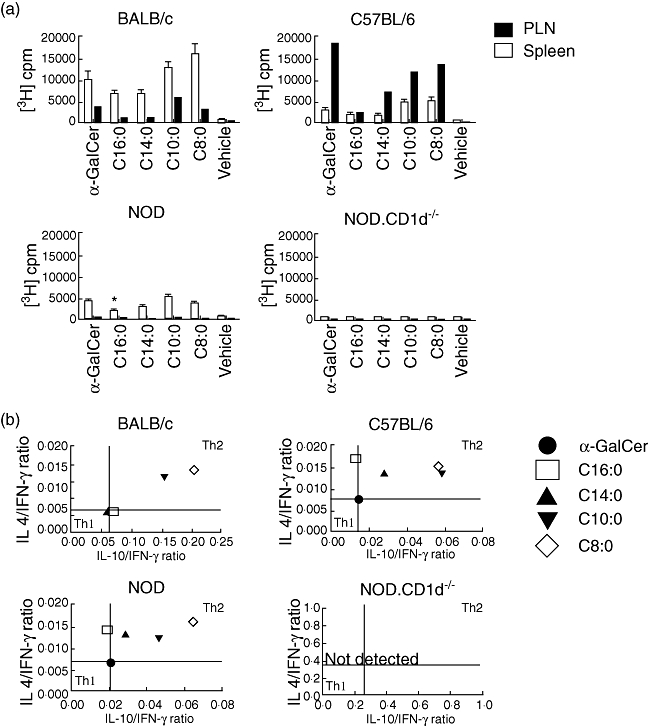
Glycolipid activation of splenocytes and lymphocytes from various mouse strains. Data from one of three independent and reproducible experiments are shown. (a) Splenocytes (5 × 105 cells) and pancreatic lymph node (PLN) lymphocytes (5 × 105 cells) from female C57BL/6 (B6), non-obese diabetic (NOD), BALB/c or NOD.CD1d–/– mice (n = 5, 3–5-week-old) were treated with ammonium chloride (ACK) lysis buffer to eliminate erythrocytes, cultured (72 h, 37°C) with glycolipid (100 ng/ml) in a 96-well tissue culture plate, and [3H]-thymidine (1 mCi) was added for the final 18 h of culture before harvesting the cells and quantitating their thymidine incorporation. Asterisks indicate a value significantly lower than the α-galactosylceramide (α-GalCer) and C8:0 treatment groups. (b) Supernatants of cultures of splenocytes obtained from the same mice as in (a) were collected after 72 h and analysed by enzyme-linked immunosorbent assay for the presence of interleukin (IL)-4, IL-10 and interferon (IFN)-γ. The mean values of the individual cytokine levels assayed at this time were compared to determine the IL-4/IFN-γ and IL-10/IFN-γ ratios. T helper type 1 (Th1)- and Th2-like cytokine responses are shown in the respective quadrants.
To determine whether the acyl chain length influences the Th bias of an α-GalCer derivative, levels of secreted IL-4, IL-10 and IFN-γ were assayed after stimulation of splenocytes (Fig. 2b). IL-10 was analysed as it is an anti-inflammatory cytokine that mediates immunosuppression, and we wished to identify an α-GalCer derivative that is anti-inflammatory and immunosuppressive. This reasoning is consistent with several reports showing that IL-10, when expressed in the pancreas, mediates elevated Treg activity and protection from T1D [36]. Th1- or Th2-biased cytokine profiles were defined by comparison to the α-GalCer-stimulated response. For BALB/c splenocytes, the C14:0, C16:0 and α-GalCer responses were equivalent, whereas the C8:0 and C10:0 responses were Th2-biased, with C8:0 being the most Th2-biased. B6 splenocytes yielded a Th2-biased response to each derivative, with C8:0 and C10:0 generating the highest Th2 responses. Similarly, the four derivatives elicited Th2-biased responses for NOD splenocytes, with the C8:0 response being the most Th2-biased. With the exception of CD1d–/– mice, the cytokine responses of splenocytes from the other mouse strains became more Th2-biased as the acyl chain length decreased. Normally, an in vivo shift of this nature is associated with protection from T1D in NOD mice [12,37]. Importantly, the observations that C8:0 is the most Th2-biased iNK T cell agonist in vitro and that it does not protect against T1D in vivo suggest that an in vitro Th2-biased response after a single stimulation is not an accurate biomarker for the predictive capacity of a given glycolipid to protect from T1D in NOD mice.
C16:0 and C8:0 do not promote iNK T cell expansion and differentially stimulate the early activation and cytokine production of iNK T cells in vivo
As C8:0 and C16:0 elicited the greatest difference in their incidence of T1D, severity of insulitis, lymphocyte proliferation and Th-bias, we reasoned that restriction of our additional functional analyses to these derivatives in NOD mice would be more informative about the properties of an iNK T cell agonist required to maximally protect from T1D. Only α-GalCer stimulated NOD iNK T cell expansion in vivo in the spleen and PLN, with the peak expansion (five- to sixfold > vehicle control) evident at 72 h in both tissues (Fig. 3a,b). To examine whether C16:0 and C8:0 modulate iNK T cell activation in vivo, CD69 expression induced by these derivatives on iNK T cells was assayed. α-GalCer, C16:0 and C8:0 each up-regulated (P < 0·05) CD69 expression on NOD splenic iNK T cells, and the same was true for α-GalCer- and C8:0-, but not C16:0-stimulated PLN iNK T cells (Fig. 4a). The percentage of CD69+ iNK T cells stimulated by C16:0 was two- to threefold less than that stimulated by α-GalCer or C8:0. While exposure to α-GalCer and C8:0 in vivo rapidly (by 2 h) activated a high percentage of iNK T cells producing intracellular IFN-γ (Fig. 4b), a significantly lower percentage (P < 0·05) of intracellular IFN-γ-producing iNK T cells was stimulated by C16:0. In contrast, intracellular IL-4 production in iNK T cells was activated significantly in vivo by α-GalCer but neither C16:0 nor C8:0. Thus, C16:0-induced protection from T1D does not appear to be mediated by the in vivo stimulation of the early activation, expansion or polarization of iNK T cells to a Th2-type response.
Fig. 3.
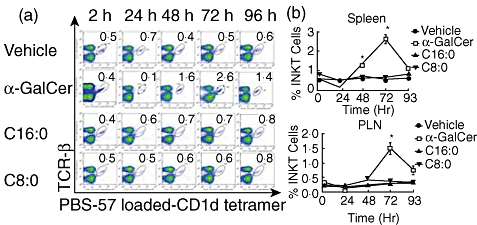
In vivo expansion of invariant natural killer T (iNK T) cells. Female non-obese diabetic (NOD) mice (n = 5, 4–6-week-old) were injected with glycolipid (4 µg) or vehicle intraperitoneally and killed 2, 24, 48, 72 or 96 h later. Spleens and pancreatic lymph nodes (PLN) were removed, placed into single cell suspension, counted, stained for iNK T cells using T cell receptor (TCR)-β and phosphate-buffered saline (PBS)-57-loaded CD1d tetramers and analysed by flow cytometry. (a) Fluorescence activated cell sorter (FACS) plots of splenic iNK T cells (gated) assayed from one mouse per treatment at each time-point after injection. After gating on live cells, iNK T cells were gated as PBS-57-loaded CD1d tetramer-positive and TCR-β-positive cells. The numbers in the right-hand corner of each FACS plot indicate the percentages of iNK T cells. (b) Cumulative data from FACS analyses of splenocytes and lymphocytes at each time-point post-injection. Data from one of two independent and reproducible experiments are presented as mean ± standard error of the mean. Asterisks (*) indicate significant significance (P ≤ 0·05) relative to control values.
Fig. 4.
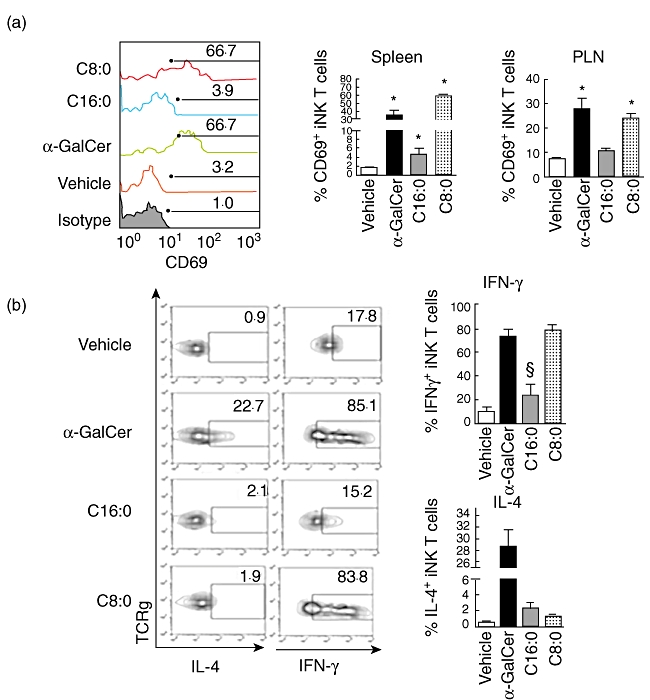
Early activation of invariant natural killer T (iNK T) cells in vivo. Female non-obese diabetic (NOD) mice (n = 5, 6-week-old) were injected intraperitoneally with glycolipid (4 µg) or vehicle control. Spleens and pancreatic lymph nodes (PLN) were harvested 2 h later and placed into single cell suspension free of erythrocytes. Cells were stained with anti-T cell receptor (TCR)-β, CD1d-tetramer [phosphate-buffered saline (PBS)-57-loaded or -unloaded] and anti-CD69. The cells were fixed and permeabilized, then stained intracellularly with anti-interleukin (IL)-4 and anti-interferon (IFN)-γ. (a) CD69 surface expression on iNK T cells. Left panel: representative fluorescence activated cell sorter (FACS) plot for each treatment group. Right panel: mean ± standard error of the mean (s.e.m.) surface CD69 expression for each treatment group. Numbers show percentages of CD69+ iNK T cells. Asterisks (*) indicate statistical significance (P ≤ 0·05) relative to vehicle control values. Representative data shown were obtained from one of the five mice analysed. (b) Intracellular cytokine data from cells in (a). Left panel: representative FACS plots for each treatment group. Right panel: average (mean ± s.e.m.) percentage of IL-4+ or IFN-γ+ iNK T cells for each treatment group. Gating was based on isotype controls. §Values are significantly (P ≤ 0·05) less than those obtained in the α-galactosylceramide (α-GalCer) and C8:0 treatment groups. Representative data averaged from five mice per experiment in one of three independent and reproducible experiments are shown.
C16:0- and C8:0-activated iNK T cells are reduced in their ability to transactivate leucocytes
α-GalCer-stimulated iNK T cells secrete cytokines that may transactivate the responses of other bystander immune cells [18,38]. These cytokines promote IFN-γ secretion by NK cells, antibody production by B cells and T cell activation upon interaction with APCs. To determine whether C16:0 and C8:0 stimulate lymphocyte transactivation responses differentially, we quantified the level of CD69 expression on NOD NK, B and T cells at 6 h post-injection. Only α-GalCer-stimulated iNK T cells transactivated these various responses in the spleen and PLN (Fig. 5). While C8:0 stimulated significant NK cell activation in the PLN but not the spleen, C16:0 did not activate NK cells appreciably in either of these tissues. These findings suggest that, compared to α-GalCer, the decreased capacity of C16:0 to transactivate NK cells is associated with its ability to stimulate increased protection of NOD mice against T1D. This reasoning is compatible with reports that upon activation, NK cells secrete significant amounts of IFN-γ, which can elicit islet beta cell destruction and the onset of T1D [39]. Conversely, inhibition of NK cell activity affords protection from T1D. Thus, our observation that C16:0 stimulates less transactivation of NK cells (Fig. 5) and IFN-γ secretion (Fig. 4b) in the PLN than either α-GalCer or C8:0 may explain partially why C16:0 yields more efficient protection from T1D.
Fig. 5.
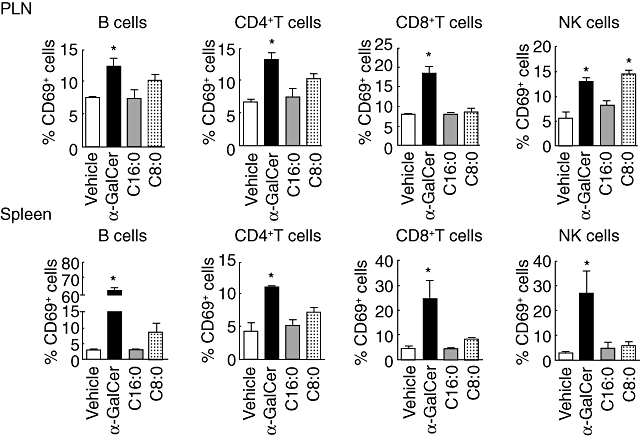
Analysis of lymphocyte transactivation. Female non-obese diabetic (NOD) mice (n = 5, 3–6-week-old) were injected intraperitoneally with glycolipid (4 µg) or vehicle control, killed 6 h later and spleens and pancreatic lymph nodes (PLN) were harvested. The presence of the early activation marker CD69 on the surface of B, T and natural killer (NK) cells was analysed by flow cytometry. Each bar represents the mean ± standard error of the mean of each treatment group. Representative data from one of two independent and reproducible experiments are shown. Asterisks (*) indicate statistical significance (P ≤ 0·05) greater than vehicle control values.
C16:0 modulates the function of DC subsets
Cytokines produced by α-GalCer-stimulated iNK T cells can regulate the phenotype and function of interacting APCs [3,40,41]. We therefore investigated the effect of treatment of NOD mice with α-GalCer, C16:0 or C8:0 on cytokine production by various DC subsets. At 6 h and 24 h post-treatment, α-GalCer and C8:0, but not C16:0, stimulated significant production of IL-12 in CD8α+ myeloid DCs (mDCs) (Fig. 6a, upper panels). In contrast, an increase in IL-10 production was noted for α-GalCer-, C16:0- and C8:0-stimulated CD8α+ mDCs at 6 h but not 24 h post-treatment (Fig. 6a, lower panels). As IL-12 production by DCs stimulates IFN-γ secretion by iNK T cells, these data suggest that the diminished production of IL-12 by proinflammatory CD8α+ mDCs elicited by C16:0 relative to C8:0 may mediate the greater protection from T1D obtained with C16:0. Further support for this notion is provided by our finding that treatment with α-GalCer and C16:0, but not C8:0, significantly (P ≤ 0·05) reduces the frequency of CD8α+ mDCs in the PLN (Fig. 6b). Importantly, our results that C16:0 elicits much lower levels of both IL-12 and IFN-γ production than α-GalCer may explain partly why C16:0 is considerably more protective from T1D than α-GalCer.
Fig. 6.
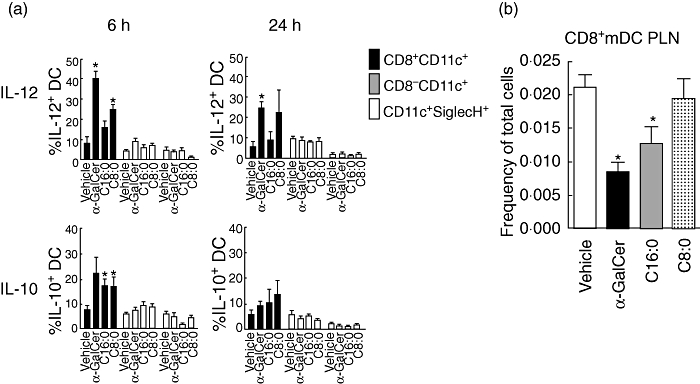
Modulation of dendritic cell (DC) function by α-galactosylceramide (α-GalCer) derivatives. (a) Production of interleukin (IL)-12 and IL-10 in splenic DC subsets after a single dose of glycolipid. Female non-obese diabetic (NOD) mice (n = 3, 5–7-week-old) were injected intraperitoneally with 4 µg of glycolipid or vehicle and killed 6 or 24 h later. DCs were analysed by fluorescence activated cell sorter (FACS) after separation into three distinct populations (CD11C+CD8+ mDC, CD11C+CD8– mDC, and CD11cintSiglec H+ plasmacytoid DC). Once separated, the percentages of IL-12+ or IL-10+ DC in each subset were determined and plotted as the mean ± standard error of the mean. Asterisks (*) indicate statistical significance (P ≤ 0·05) greater than vehicle control values. Data from one of three reproducible experiments are shown. (b) PLN were harvested from NOD mice treated with vehicle or the various glycolipids in a multi-dose protocol, and lymphocyte suspensions were fluorescently stained for the CD8α, CD11chi and Sieglec H surface markers before analysis by flow cytometry. mDCs were gated in the CD11chiSiglecH– cell subset, which was divided further into the CD8+ and CD8– subsets. *Values significantly less than (P ≤ 0·05) vehicle control. Data are from one of two representative and reproducible experiments.
C16:0-activated iNK T cells enter into and recover from anergy more rapidly than do C8:0-activated iNK T cells
Glycolipid-activated iNK T cells become anergic and respond poorly upon restimulation [27]. However, if iNK T cells remain anergic for a long duration, this may impair their survival and activity [18]. To investigate whether C16:0 and C8:0 differ in their induced kinetics of entry into and recovery of iNK T cells from anergy, iNK T cells from NOD mice treated previously (MD) with α-GalCer, C16:0 or C8:0 were restimulated in vitro following 1, 3, 7 or 28 days of rest and assayed for their proliferative and cytokine secretion (IL-2, IFN-γ) responses. At 1 day post-treatment, iNK T cells from C16:0-treated mice were hyporesponsive to restimulation, whereas the amount of IL-2 secreted by iNK T cells from C8:0- and α-GalCer-treated mice was equivalent to that secreted by iNK T cells from vehicle-treated mice (Fig. 7a). Similar results were observed for the cell proliferation and IFN-γ responses (our unpublished observations). While iNK T cells from C16:0- and α-GalCer-treated mice were both anergic at 3 days post-treatment, iNK T cells from C16:0- but not α-GalCer-treated mice recovered from anergy by 7 days post-treatment. α-GalCer experienced iNK T cells did not recover from anergy until 28 days post-treatment. Note that C8:0-treated iNK T cells were not anergic at any time post-treatment. Thus, iNK T cells activated by C16:0 enter into and exit from anergy more rapidly than iNK T cells stimulated with α-GalCer or C8:0. Accordingly, the kinetics of induction of and recovery from anergy of iNK T cells may be an important factor that controls the ability of a given sphingoglycolipid agonist to protect against T1D.
Fig. 7.
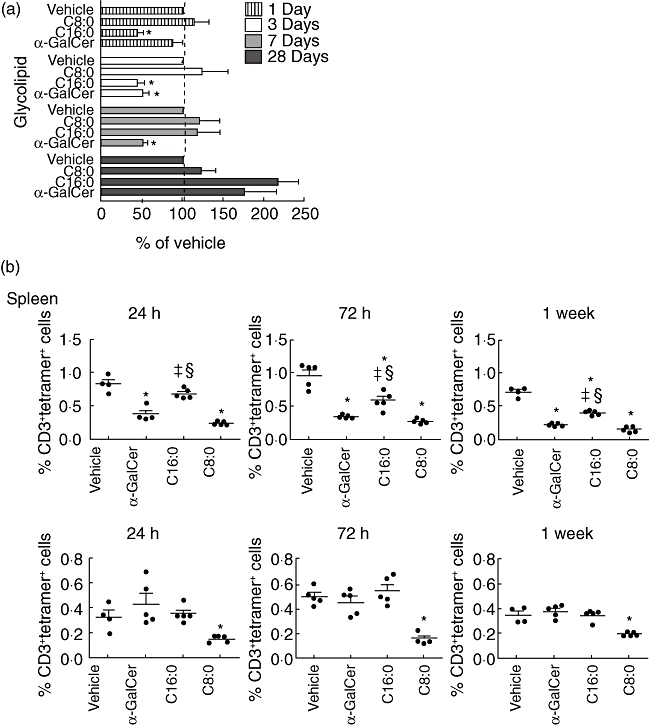
(a) Kinetics of invariant natural killer T (iNK T) cell entry into and exit from anergy varies with different glycolipids. Female non-obese diabetic (NOD) mice (n = 5, 3–5-week-old) were injected intraperitoneally every other day for 3 weeks with 4 µg glycolipid or vehicle, and were then rested for 1, 3, 7 or 28 days. At each time-point, the mice were killed and spleens were harvested. Splenocytes (5 × 106 cells) free of erythrocytes were restimulated in vitro with the same glycolipid (100 ng/ml) used in vivo or vehicle control for 72 h at 37°C. The amount of interleukin (IL)-2 in the supernatant was determined by enzyme-linked immunosorbent assay. The amount of IL-2 secreted for α-galactosylceramide (α-GalCer) or its derivative was converted into a percentage of the amount of IL-2 secreted in the vehicle in vivo/glycolipid in vitro control treatment. Asterisks (*) indicates values significantly lower than vehicle control (P ≤ 0·05). Data are from one of two reproducible experiments. (b) Frequency of iNK T cells varies after multi-dose treatment with different glycolipids. Female NOD mice (n = 5, 3–5-week-old) were treated every other day for 3 weeks with glycolipid (4 µg) or vehicle, and were then rested for 24 h, 72 h or 1 week. At this time, spleens and pancreatic lymph nodes (PLN) were harvested and placed into single-cell suspension free of erythrocytes. Cells were stained with anti-CD3-peridinin chlorophyll (PerCP) and phosphate-buffered saline (PBS)-57-loaded CD1d-tetramer to analyse the frequency of iNK T cells. An asterisk (*) indicates statistical significance (P ≤ 0·05) less than vehicle control values. §Values significantly greater than C8:0 values; ‡values significantly greater than α-GalCer values. Data are from one of three reproducible experiments.
C16:0 and C8:0 regulate iNK T cell survival differentially in different tissues
Although MD treatment with certain glycolipids protects NOD mice from T1D, the iNK T cell frequency in the spleen of these mice can be reduced significantly, due possibly to activation-induced cell death (AICD) [18,31]. To test the therapeutic value of a glycolipid drug in humans at risk for T1D, it is essential that the treatment regimen does not compromise the immune responses of an individual. Because several immune responses are triggered by glycolipid-primed iNK T cells [3], long-term depletion of these cells may have adverse effects and lead to increased risk of infection. As anergic iNK T cells are more susceptible to AICD than non-anergic iNK T cells [27], we investigated whether the faster recovery from iNK T cell anergy observed after MD administration with C16:0 compared to C8:0 and α-GalCer results in less AICD and greater survival of iNK T cells. At 24 h, 72 h and 1 week post-treatment, the frequencies of iNK T cells in the spleen and PLN activated by C16:0, C8:0 or α-GalCer were determined by flow cytometry and compared to those observed after treatment with vehicle. At all time-points, the frequency of iNK T cells in the spleen of C16:0-treated mice was significantly greater than that detected in the spleens from C8:0- and α-GalCer-treated mice (Fig. 7b, upper panels). Similarly, the absolute numbers of iNK T cells in the spleen of C16:0-treated mice were significantly greater at the three time-points analysed than that detected in the spleens from vehicle-treated mice (Table 1). Technical problems precluded a similar analysis of the numbers of iNK T cells in the PLN. Conversely, treatment with C8:0 but not C16:0 or α-GalCer reduced (P ≤ 0·05) the iNK T cell frequency in the PLN (Fig. 7b, lower panels). Thus, the C16:0-induced increase in iNK T cell survival in the spleen and C8:0-induced decrease in iNK T cell survival in the PLN correlate well with disease outcome, as C16:0 but not C8:0 protects from T1D.
Table 1.
Kinetic analysis of number of iNK T cells in the spleen at various times after multi-dose treatment with glycolipid
| Day post-MLD (glycolipid) | Total cells (×10−6) | iNK T cells (×10−6) |
|---|---|---|
| α-GalCer | ||
| 1 | 121·2 ± 20·3 | 0·6 ± 0·2 |
| 3 | 96·8 ± 24·2 | 0·3 ± 0·1* |
| 7 | 78·2 ± 11·9 | 0·2 ± 0·0* |
| C16:0 | ||
| 1 | 122·5 ± 10·8 | 0·5 ± 0·1 |
| 3 | 94·8 ± 26·0 | 0·6 ± 0·0 |
| 7 | 109 ± 12·2 | 0·5 ± 0·0 |
| C8:0 | ||
| 1 | 109·0 ± 12·2 | 0·2 ± 0·1* |
| 3 | 134·9 ± 22·0 | 0·2 ± 0·1* |
| 7 | 57·0 ± 21·3 | 0·1 ± 0·0* |
| Vehicle | ||
| 1 | 120·9 ± 20·5 | 0·4 ± 0·1 |
| 3 | 75·9 ± 12·0 | 0·7 ± 0·1 |
| 7 | 64·1 ± 22·7 | 0·5 ± 0·1 |
Female non-obese diabetic (NOD) mice (n = 5, 3–5-week-old) were treated every other day for 3 weeks with glycolipid (4 µg) or vehicle, and were then rested for 1 day, 3 days or 7 days. Absolute numbers of iNK T cells in the spleen were calculated by multiplying the percentages of iNK T cells obtained by flow cytometry (Fig. 7b) by the total lymphocyte counts per spleen from individual mice. Data are presented as the mean ± standard deviation for each group of five mice analysed. For each mouse, a minimum of 2 × 105 gated lymphocytes and 2 × 104 iNK T cells were enumerated by flow cytometry. An asterisk (*) indicates statistical significance (P ≤ 0·05) less than vehicle control values. α-GalCer: α-galactosylceramide; MLD, multi-low dose.
Discussion
Structure-guided design of novel iNK T cell agonists is an approach with significant clinical potential for the treatment of several diseases, including infectious diseases, cancer and autoimmune diseases [33]. Based on recent structure–function analyses of novel synthetic glycolipid agonists with modifications in their polar head and/or lipid tails, several properties of such agonists have been defined. While the desirable properties include enhanced adjuvant activity for antigen-specific responses, differential (Th1- or Th2-bias) cytokine production by iNK T cells and preferential presentation by selected APCs (DCs and B cells), the undesirable properties include cytokine-induced inflammation, target cell lysis by iNK T cells and long-term iNK T cell anergy upon repeated agonist administration [42–48]. Here, we adopted this structure-guided approach to the systematic design of an optimal iNK T cell agonist(s) for protection against T1D. Our results obtained in the NOD mouse model of T1D demonstrate that, of the panel of five (C8:0–C16:) acyl chain derivatives of α-GalCer analysed, C16:0 elicited the highest protection from insulitis and T1D, increased iNK T cell survival and frequency in the PLN stimulated the reduced production of IL-12 by proinflammatory CD8α+ mDCs in the PLN, and more rapid kinetics of entry into and recovery of iNK T cells from anergy. These findings demonstrate further that structural modification of the length of the acyl chain of α-GalCer is critical for the design of an iNK T agonist that optimally protects from T1D, and that C16:0 may fulfil the properties of such an agonist.
Shortening the length of either lipid chain in α-GalCer increases the rate of lipid dissociation from CD1d and the stability of glycolipid binding to CD1d [28]. In this study, we show that the length of the acyl chain also influences the strength of initial activation and subsequent downstream responses of iNK T cells. Both C16:0 and C8:0 stimulate splenic iNK T cell proliferation and cytokine secretion in vitro, with C16:0 being a less potent activator of iNK T cells than C8:0. Moreover, these two derivatives differ in their ability to expand the iNK T cell population and transactivate bystander cells in vivo.
The difference between the ability of C8:0 to stimulate the proliferation and expansion of iNK T cells in vitro (Fig. 2), but not in vivo (Fig. 3), may be attributable to the differential binding of a C8:0/CD1d complex to lyso-phosphatidyl choline (LPC) in vitro and in vivo. Recent reports suggest that LPC, an 18-carbon-long glycolipid (structure is C18:1), is a major spacer lipid in the A′ pocket of CD1d [49,50]. Due to size and conformation constraints, LPC would be expected to bind in the A′ pocket of a C8:0/CD1d complex but not C16:0/CD1d complex (see below). A complex such as C8:0/LPC/CD1d may have a short half-life, inhibit antigen presentation and block certain iNK T cell responses, as proposed recently [50]. Although LPC is present at high concentration in blood and other fluids during chronic inflammation in vivo, this is unlikely to be the case in the serum (10% inactivated FBS) of iNK T/APC cell cultures maintained for a short term (72 h) in vitro. Thus, the presence of C8:0/LPC/CD1d complexes on APCs in vivo and the relatively low concentration (or absence) of LPC from C8:0/LPC/CD1d complexes on APCs in short-term in-vitro cultures may explain why C8:0 stimulates significantly more proliferation of iNK T cells in vitro than in vivo.
Our results regarding C16:0- and C8:0-induced iNK T cell activation in vitro support those of Goff et al. [34], who identified an inverse correlation between the length of an acyl chain and the IL-4 : IFN-γ ratio of cytokines secreted by iNK T cells in vitro. We confirmed their findings and extended them to include activated NOD iNK T cells. Interestingly, even though C8:0 yielded the most Th2-biased cytokine response of all the glycolipids tested, it did not protect NOD mice from T1D using our MD protocol. This result may arise from the presence of C8:0/LPC/CD1d complexes on APCs, as discussed above. Thus, it is critical that the selection of a synthetic glycolipid(s) for the in vivo treatment of T1D in humans be based on functional analyses of human iNK T cells stimulated by this glycolipid(s) in vivo. Recent technology involving adoptive cell transfer coupled with multi-photon microscopy was developed to identify the target site of iNK T cell activation and function in vivo in mice [45]. The application of such technology to humans may prove valuable in future studies.
Anergy induction in iNK T cells may be important for protection against T1D because anergic iNK T cells can stimulate DCs to become tolerogenic, and thereby avoid the chronic production of inflammatory cytokines and host target cell damage. However, anergic iNK T cells are also more susceptible to apoptosis [18,27]. Thus, an optimal glycolipid for the treatment of T1D should elicit short-term iNK T cell anergy, i.e. for a time long enough to induce tolerogenic DCs but not too long as to result in iNK T cell depletion. We found that NOD splenic iNK T cells from C16:0-treated mice enter into and recover from anergy more rapidly than α-GalCer. This faster recovery may enable these iNK T cells to mediate subsequent immune responses to pathogens – an important consideration for human clinical trials with iNK T agonists. Our finding that C8:0-treated iNK T cells do not become anergic supports our conclusion reached using the C20:2 N-acyl variant of α-GalCer that iNK T cell anergy is an important contributor to glycolipid-mediated protection against T1D [51].
Interaction between the inhibitory signalling molecule programmed death-1 (PD-1) and its programmed death ligand-1 (PD-L1) is crucial for the induction and maintenance of iNK T cell anergy [52–54]. While α-GalCer up-regulates PD-1 and PD-L1 expression on iNK T cells in the spleen and PLN [51], we do not know whether C16:0 and C8:0 yield similar or different iNK T responses. If C16:0, but not C8:0, up-regulates PD-1 and PD-L1 expression on NOD iNK T cells, this might explain why C16:0 and α-GalCer, but not C8:0, induces iNK T cell anergy. Note that while anergic iNK T cells induce tolerogenic DCs that mediate protection from T1D, chronic administration of α-GalCer also results in long-lasting anergy accompanied by significantly reduced iNK T cell frequencies, which has raised concerns about its long-term therapeutic use. Importantly, we showed in this report that C16:0-stimulated iNK T cells recover from anergy more rapidly than α-GalCer-stimulated iNK T cells. Thus, C16:0 may be a more promising candidate than α-GalCer for the therapy of T1D in humans.
Tolerogenic DCs induced by anergic iNK T cells are characterized phenotypically by their decreased IL-12 and increased IL-10 secretion responses [15]. We reasoned, therefore, that our finding that mDCs from C16:0-treated mice possess this tolerogenic DC phenotype (Fig. 6a) may be informative about the mechanism of action of C16:0 in protection from T1D. Based on our data and those in the literature reviewed in Lehuen et al. [55], C16:0 may induce tolerogenic mDCs in the PLN that can suppress autoreactive T cells responsible for islet beta cell death and also generate Th2 and Treg cell responses that aid in protection against T1D. Indeed, we discovered that the rapid recovery of C16:0-treated iNK T cells from anergy correlated with the stimulation of immunosuppressive CD8α+ mDCs to produce a significant amount of intracellular IL-10 but not IL-12 (Fig. 7). Furthermore, MD treatment with α-GalCer and C16:0, but not C8:0, reduced the frequency of these CD8α+ mDCs in the PLN. The ability of C16:0-stimulated iNK T cells to modulate CD8α+ mDC function and frequency agrees with reports that activated iNK T cells can enhance antigen-specific immune responses by impairing the suppressive activity of mDCs [48,51]. Thus, our data strengthen the notion that iNK T cells can regulate self-tolerance by influencing the state of activation of DCs. If influenced by an appropriate iNK T agonist, such as C16:0, this would lead to efficient protection from T1D. Further experimentation is required to test this possibility in humans with T1D and/or at high risk for developing T1D.
It is also noteworthy that iNK T cells can inhibit NK cell cytotoxic activity and tumour metastasis in the liver by the production of IL-10 [56]. Thus, IL-10 production by iNK T cells may protect from T1D and tumour metastasis by blocking NK cell cytolysis of target islet beta cells in the pancreas and tumour cells in the liver, respectively. This result further emphasizes the important role that IL-10 may play in protection from T1D.
Structural analyses of glycolipid/CD1d/TCR complexes demonstrate that a spacer lipid buried deep in the A′ pocket of CD1d can stabilize this pocket when the acyl chain is not long enough to occupy the entire channel [44,57,58]. As this spacer lipid may be displaced from the pocket by a glycolipid, its identification may be important for the design of an optimal iNK T cell agonist that contains a shortened acyl chain and protects from disease, as in the case of C16:0 for T1D. A candidate spacer lipid is LPC (structure is C18:1), a relatively abundant glycolipid in mammalian cells [49] that accumulates at high concentration in blood and other fluids during chronic inflammation [59] and activates human iNK T cells [50]. Hence, the presence of LPC (18 carbons) in the A′ pocket of CD1d, and the ability of this pocket to accommodate an acyl chain with ≤26 carbons, may explain why C14:0 and C16:0 are more protective from T1D than C8:0. Whereas C8:0 may accommodate LPC in the A′ pocket, α-GalCer (C26:0), C16:0, C14:0, C12:0 and C10:0 are expected to displace LPC from pocket. Given that high concentrations of CD1d-bound LPC have a short half-life, do not activate iNK T cells and inhibit antigen presentation, it has been suggested that increased binding of LPC by CD1d may block certain iNK T cell responses [50]. Thus, C8:0/CD1d/LPC complexes may diminish the relative capacity of iNK T cells to protect from T1D. Further structural analyses of C16:0/CD1d/TCR and C8:0/CD1d/TCR complexes derived from mammalian cells and the characterization of the spacer lipid in these complexes are required to explain how the fatty acyl chain length of a glycosphingolipid can regulate protection against T1D.
In summary, our data suggest that the relative strength of activation of iNK T cells by an agonist may not be the critical factor that determines protection from T1D. Rather, the length, structure and conformational constraints of the acyl chain of a glycolipid agonist, and its ability to accommodate, or not, an endogenous spacer lipid (e.g. lyso-PC) in the A′ pocket of CD1d, are more important for the regulation of protection against T1D. Thus, the distinguishing parameters of an optimal agonist for protection from T1D, as elicited by C16:0 but not C8:0, include the increased iNK T cell survival and frequency in the PLN, reduced production of IL-12 and increased production of IL-10 by CD8α+ mDCs in the PLN, and more rapid kinetics of entry into and recovery of iNK T cells from anergy.
Acknowledgments
We kindly thank Delfina Mazzuca Siroen for her expertise in operating the flow cytometer as well as the staff of the Robarts Barrier Animal Facility for their assistance with the breeding and maintenance of our mice. We also thank Dalam Ly and Lakshmimathy Subramanian in our laboratory at the Robarts Research Institute for their continued support and advice during the preparation of this manuscript. This work was supported by Juvenile Diabetes Research Foundation International grant 24–2007-388 and Canadian Institutes of Health Research grant MOP 64386 to T. L. Delovitch, Canadian Institutes of Health Research grant MOP 86601 to S. M. M. Haeryfar and the National Institutes of Allergy and Infectious Disease grant to P. B. Savage. S. M. M. Haeryfar is the Canada Research Chair in Viral Immunity and Pathogenesis. H. Blumenfeld was the recipient of a Schulich Graduate Enhancement Scholarship and Department of Microbiology and Immunology Graduate Entrance Fellowship. R. Tohn was the recipient of a Schulich Graduate Enhancement Scholarship, a Department of Microbiology and Immunology Graduate Entrance Fellowship and an Ontario Graduate Scholarship.
Disclosure
All the authors declare there are no conflicts of interest.
References
- 1.Taniguchi M, Seino K, Nakayama T. The NKT cell system: bridging innate and acquired immunity. Nat Immunol. 2003;4:1164–65. doi: 10.1038/ni1203-1164. [DOI] [PubMed] [Google Scholar]
- 2.Wu L, Gabriel CL, Parekh VV, Van Kaer L. Invariant natural killer T cells: innate-like T cells with potent immunomodulatory activities. Tissue Antigens. 2009;73:535–45. doi: 10.1111/j.1399-0039.2009.01256.x. [DOI] [PubMed] [Google Scholar]
- 3.Wilson SB, Delovitch TL. Janus-like role of regulatory iNK T cells in autoimmune disease and tumour immunity. Nat Rev Immunol. 2003;3:211–22. doi: 10.1038/nri1028. [DOI] [PubMed] [Google Scholar]
- 4.Hong S, Wilson MT, Serizawa I, et al. The natural killer T-cell ligand alpha-galactosylceramide prevents autoimmune diabetes in non-obese diabetic mice. Nat Med. 2001;7:1052–56. doi: 10.1038/nm0901-1052. [DOI] [PubMed] [Google Scholar]
- 5.Sharif S, Arreaza GA, Zucker P, et al. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune Type 1 diabetes. Nat Med. 2001;7:1057–62. doi: 10.1038/nm0901-1057. [DOI] [PubMed] [Google Scholar]
- 6.Wang B, Geng YB, Wang CR. CD1-restricted NK T cells protect nonobese diabetic mice from developing diabetes. J Exp Med. 2001;194:313–20. doi: 10.1084/jem.194.3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naumov YN, Bahjat KS, Gausling R, et al. Activation of CD1d-restricted T cells protects NOD mice from developing diabetes by regulating dendritic cell subsets. Proc Natl Acad Sci USA. 2001;98:13838–43. doi: 10.1073/pnas.251531798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mieza MA, Itoh T, Cui JQ, et al. Selective reduction of V alpha 14+ NK T cells associated with disease development in autoimmune-prone mice. J Immunol. 1996;156:4035–40. [PubMed] [Google Scholar]
- 9.van der Vliet HJ, von Blomberg BM, Nishi N, et al. Circulating V(alpha24+) Vbeta11+ NKT cell numbers are decreased in a wide variety of diseases that are characterized by autoreactive tissue damage. Clin Immunol. 2001;100:144–8. doi: 10.1006/clim.2001.5060. [DOI] [PubMed] [Google Scholar]
- 10.Lee PT, Putnam A, Benlagha K, Teyton L, Gottlieb PA, Bendelac A. Testing the NKT cell hypothesis of human IDDM pathogenesis. J Clin Invest. 2002;110:793–800. doi: 10.1172/JCI15832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson SB, Kent SC, Patton KT, et al. Extreme Th1 bias of invariant Valpha24JalphaQ T cells in type 1 diabetes. Nature. 1998;391:177–81. doi: 10.1038/34419. [DOI] [PubMed] [Google Scholar]
- 12.Mi QS, Ly D, Zucker P, McGarry M, Delovitch TL. Interleukin-4 but not interleukin-10 protects against spontaneous and recurrent type 1 diabetes by activated CD1d-restricted invariant natural killer T-cells. Diabetes. 2004;53:1303–10. doi: 10.2337/diabetes.53.5.1303. [DOI] [PubMed] [Google Scholar]
- 13.Saxena V, Ondr JK, Magnusen AF, Munn DH, Katz JD. The countervailing actions of myeloid and plasmacytoid dendritic cells control autoimmune diabetes in the nonobese diabetic mouse. J Immunol. 2007;179:5041–53. doi: 10.4049/jimmunol.179.8.5041. [DOI] [PubMed] [Google Scholar]
- 14.Chen YG, Choisy-Rossi CM, Holl TM, et al. Activated NKT cells inhibit autoimmune diabetes through tolerogenic recruitment of dendritic cells to pancreatic lymph nodes. J Immunol. 2005;174:1196–204. doi: 10.4049/jimmunol.174.3.1196. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Cho S, Ueno A, et al. Ligand-dependent induction of noninflammatory dendritic cells by anergic invariant NKT cells minimizes autoimmune inflammation. J Immunol. 2008;181:2438–45. doi: 10.4049/jimmunol.181.4.2438. [DOI] [PubMed] [Google Scholar]
- 16.Joyee AG, Uzonna J, Yang X. Invariant NKT cells preferentially modulate the function of CD8 alpha+ dendritic cell subset in inducing type 1 immunity against infection. J Immunol. 2010;184:2095–106. doi: 10.4049/jimmunol.0901348. [DOI] [PubMed] [Google Scholar]
- 17.Diana J, Griseri T, Lagaye S, et al. NKT cell–plasmacytoid dendritic cell cooperation via OX40 controls viral infection in a tissue-specific manner. Immunity. 2009;30:289–99. doi: 10.1016/j.immuni.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 18.Van Kaer L. alpha-Galactosylceramide therapy for autoimmune diseases: prospects and obstacles. Nat Rev Immunol. 2005;5:31–42. doi: 10.1038/nri1531. [DOI] [PubMed] [Google Scholar]
- 19.Okai M, Nieda M, Tazbirkova A, et al. Human peripheral blood Valpha24+ Vbeta11+ NKT cells expand following administration of alpha-galactosylceramide-pulsed dendritic cells. Vox Sang. 2002;83:250–3. doi: 10.1046/j.1423-0410.2002.00217.x. [DOI] [PubMed] [Google Scholar]
- 20.Nieda M, Okai M, Tazbirkova A, et al. Therapeutic activation of Valpha24+Vbeta11+ NKT cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood. 2004;103:383–9. doi: 10.1182/blood-2003-04-1155. [DOI] [PubMed] [Google Scholar]
- 21.Chang DH, Osman K, Connolly J, et al. Sustained expansion of NKT cells and antigen-specific T cells after injection of alpha-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J Exp Med. 2005;201:1503–17. doi: 10.1084/jem.20042592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Motohashi S, Nagato K, Kunii N, et al. A phase I–II study of alpha-galactosylceramide-pulsed IL-2/GM-CSF-cultured peripheral blood mononuclear cells in patients with advanced and recurrent non-small cell lung cancer. J Immunol. 2009;182:2492–501. doi: 10.4049/jimmunol.0800126. [DOI] [PubMed] [Google Scholar]
- 23.Osman Y, Kawamura T, Naito T, et al. Activation of hepatic NKT cells and subsequent liver injury following administration of alpha-galactosylceramide. Eur J Immunol. 2000;30:1919–28. doi: 10.1002/1521-4141(200007)30:7<1919::AID-IMMU1919>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 24.Tupin E, Nicoletti A, Elhage R, et al. CD1d-dependent activation of NKT cells aggravates atherosclerosis. J Exp Med. 2004;199:417–22. doi: 10.1084/jem.20030997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ito K, Karasawa M, Kawano T, et al. Involvement of decidual Valpha14 NKT cells in abortion. Proc Natl Acad Sci USA. 2000;97:740–4. doi: 10.1073/pnas.97.2.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bilenki L, Yang J, Fan Y, Wang S, Yang X. Natural killer T cells contribute to airway eosinophilic inflammation induced by ragweed through enhanced IL-4 and eotaxin production. Eur J Immunol. 2004;34:345–54. doi: 10.1002/eji.200324303. [DOI] [PubMed] [Google Scholar]
- 27.Parekh VV, Wilson MT, Olivares-Villagomez D, et al. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J Clin Invest. 2005;115:2572–83. doi: 10.1172/JCI24762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCarthy C, Shepherd D, Fleire S, et al. The length of lipids bound to human CD1d molecules modulates the affinity of NKT cell TCR and the threshold of NKT cell activation. J Exp Med. 2007;204:1131–44. doi: 10.1084/jem.20062342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizuno M, Masumura M, Tomi C, et al. Synthetic glycolipid OCH prevents insulitis and diabetes in NOD mice. J Autoimmun. 2004;23:293–300. doi: 10.1016/j.jaut.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 30.Yu KO, Im JS, Molano A, et al. Modulation of CD1d-restricted NKT cell responses by using N-acyl variants of alpha-galactosylceramides. Proc Natl Acad Sci USA. 2005;102:3383–8. doi: 10.1073/pnas.0407488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forestier C, Takaki T, Molano A, et al. Improved outcomes in NOD mice treated with a novel Th2 cytokine-biasing NKT cell activator. J Immunol. 2007;178:1415–25. doi: 10.4049/jimmunol.178.3.1415. [DOI] [PubMed] [Google Scholar]
- 32.Ly D, Tohn R, Rubin B, et al. An alpha-galactosylceramide C20:2 N-acyl variant enhances anti-inflammatory and regulatory T cell-independent responses that prevent type 1 diabetes. Clin Exp Immunol. 2010;160:185–98. doi: 10.1111/j.1365-2249.2009.04074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cerundolo V, Barral P, Batista FD. Synthetic iNK T cell-agonists as vaccine adjuvants – finding the balance. Curr Opin Immunol. 2010;22:417–24. doi: 10.1016/j.coi.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 34.Goff RD, Gao Y, Mattner J, et al. Effects of lipid chain lengths in alpha-galactosylceramides on cytokine release by natural killer T cells. J Am Chem Soc. 2004;126:13602–3. doi: 10.1021/ja045385q. [DOI] [PubMed] [Google Scholar]
- 35.Gombert JM, Herbelin A, Tancrède-Bohin E, Dy M, Carnaud C, Bach JF. Early quantitative and functional deficiency of NK1+-like thymocytes in the NOD mouse. Eur J Immunol. 1996;26:2989–98. doi: 10.1002/eji.1830261226. [DOI] [PubMed] [Google Scholar]
- 36.Fousteri G, Dave A, Bot A, Juntti T, Omid S, von Herrath M. Subcutaneous insulin B:9-23/IFA immunisation induces Tregs that control late-stage prediabetes in NOD mice through IL-10 and IFN-γ. Diabetologia. 2010;53:1958–70. doi: 10.1007/s00125-010-1777-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rapoport MJ, Jaramillo A, Zipris D, et al. Interleukin 4 reverses T cell proliferative unresponsiveness and prevents the onset of diabetes in nonobese diabetic mice. J Exp Med. 1993;178:87–99. doi: 10.1084/jem.178.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–36. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 39.Qin H, Lee IF, Panagiotopoulos C, et al. Natural killer cells from children with type 1 diabetes have defects in NKG2D-dependent function and signaling. Diabetes. 2011;60:857–66. doi: 10.2337/db09-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsuda JL, Gapin L, Baron JL, et al. Mouse V alpha 14i natural killer T cells are resistant to cytokine polarization in vivo. Proc Natl Acad Sci USA. 2003;100:8395–400. doi: 10.1073/pnas.1332805100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujii S, Liu K, Smith C, Bonito AJ, Steinman RM. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med. 2004;199:1607–18. doi: 10.1084/jem.20040317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xia C, Zhang W, Zhang Y, et al. The roles of 3′ and 4′ hydroxy groups in alpha-galactosylceramide stimulation of invariant natural killer T cells. Chem Med Chem. 2009;4:1810–15. doi: 10.1002/cmdc.200900350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmieg J, Yang G, Franck RW, Tsuji M. Superior protection against malaria and melanoma metastases by a C-glycoside analogue of the natural killer T cell ligand alpha-galactosylceramide. J Exp Med. 2003;198:1631–41. doi: 10.1084/jem.20031192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X, Fujio M, Imamura M, et al. Design of a potent CD1d-binding NKT cell ligand as a vaccine adjuvant. Proc Natl Acad Sci USA. 2010;107:13010–15. doi: 10.1073/pnas.1006662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barral P, Polzella P, Bruckbauer A, et al. CD169(+) macrophages present lipid antigens to mediate early activation of iNK T cells in lymph nodes. Nat Immunol. 2010;11:303–12. doi: 10.1038/ni.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emamaullee JA, Davis J, Merani S, et al. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes. 2009;58:1302–11. doi: 10.2337/db08-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kinjo Y, Wu D, Kim G, et al. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. 2005;434:520–5. doi: 10.1038/nature03407. [DOI] [PubMed] [Google Scholar]
- 48.De Santo C, Salio M, Masri SH, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest. 2008;118:4036–48. doi: 10.1172/JCI36264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan W, Kang SJ, Evans JE, Cresswell P. Natural lipid ligands associated with human CD1d targeted to different subcellular compartments. J Immunol. 2009;182:4784–91. doi: 10.4049/jimmunol.0803981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fox LM, Cox DG, Lockridge JL, et al. Recognition of lyso-phospholipids by human natural killer T lymphocytes. PLoS Biol. 2009;7:e1000228. doi: 10.1371/journal.pbio.1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tohn R, Blumenfeld H, Haeryfar SMM, et al. Stimulation of a shorter duration in the state of anergy by an iNK T cell agonist enhances its efficiency of protection from type 1 diabetes. Clin Exp Immunol. 2011;164:26–41. doi: 10.1111/j.1365-2249.2011.04323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keir ME, Butte MJ, Freeman GJ, Sharpe AH, et al. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol. 2007;19:813–24. doi: 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- 54.Parekh VV, Lalani S, Kim S, et al. PD-1/PD-L blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant NKT cells. J Immunol. 2009;182:2816–26. doi: 10.4049/jimmunol.0803648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol. 2010;10:501–13. doi: 10.1038/nri2787. [DOI] [PubMed] [Google Scholar]
- 56.Yang W, Li H, Mayhew E, Mellon J, Chen PW, Niederkorn JY. NKT cell exacerbation of liver metastases arising from melanomas transplanted into either the eyes or spleens of mice. Invest Ophthalmol Vis Sci. 2011;52:3094–102. doi: 10.1167/iovs.10-7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu D, Zajonc DM, Fujio M, et al. Design of natural killer T cell activators: structure and function of a microbial glycosphingolipid bound to mouse CD1d. Proc Natl Acad Sci USA. 2006;103:3972–7. doi: 10.1073/pnas.0600285103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koch M, Stronge VS, Shepherd D, et al. The crystal structure of human CD1d with and without alpha-galactosylceramide. Nat Immunol. 2005;6:819–26. doi: 10.1038/ni1225. [DOI] [PubMed] [Google Scholar]
- 59.Salio M, Cerundolo V. Linking inflammation to natural killer T cell activation. PLoS Biol. 2009;7:e1000226. doi: 10.1371/journal.pbio.1000226. [DOI] [PMC free article] [PubMed] [Google Scholar]