

Abstract
In previous studies (Delker et al. (2010), J. Am. Chem. Soc. 132, 5437-5442) we determined the crystal structures of neuronal nitric oxide synthase (nNOS) complexed with nNOS-selective chiral pyrrolidine inhibitors, designed to have an aminopyridine group bound over the heme where it can electrostatically interact with the conserved active site Glu residue. However, in addition to the expected binding mode with the (S, S)-cis inhibitors an unexpected “flipped” orientation was observed for the (R, R)-cis enanthiomers. In the flipped mode the aminopyridine extends out of the active site where it interacts with one heme propionate. This prompted us to design and synthesize symmetric “double headed” inhibitors with an aminopyridine at each end of a bridging ring structure (Xue, F., Delker, S. L., Li, H., Fang, J., Jamal, J., Martásek, P., Roman, L. J., L., P. T., and Silverman, R. B. Symmetric double-headed aminopyridines, a novel strategy for potent and membrane-permeable inhibitors of neuronal nitric oxide synthase, J. Med. Chem. submitted). One aminopyridine should interact with the active site Glu and the other with the heme propionate. Crystal structures of these “double headed” aminopyridine inhibitors complexed to nNOS show unexpected and significant protein and heme conformational changes induced by inhibitor binding that result in removal of the tetrahydrobiopterin (H4B) cofactor and creation of a new Zn2+ site. These changes are due to binding of a second inhibitor molecule that results in displacement of H4B and the placement of the inhibitor pyridine group in position to serve as a Zn2+ ligand together with Asp, His, and a chloride ion. Binding of the second inhibitor molecule and generation of the Zn2+ site does not occur in eNOS. Structural requirements for creation of the new Zn2+ site in nNOS were analyzed in detail. These observations open the way for the potential design of novel inhibitors selective for nNOS.
Nitric oxide synthases (NOSs) catalyze the oxidation of L-arginine to nitric oxide (NO) and L-citrulline (1). Mammals contain three NOS isoforms: neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS) (2). NO produced from these different NOS isoforms is involved in a wide range of physiologic functions in the nervous, immune, and cardiovascular systems (3). Unregulated NO production can lead to pathologic conditions such as stroke (4), inflammation (5), and hypertension (6). Therefore, the control of NOS activity by isoform selective NOS inhibitors has great potential for therapeutic treatments of NO-related diseases (7).
The three NOS isoforms share a similar domain architecture with an N-terminal catalytic domain containing the heme active site and a tetrahydrobiopterin (H4B) nearby as a redox active cofactor and a C-terminal reductase domain consisting of FMN, FAD, and NADPH binding sites (8). The active sites for the various NOS isoforms, however, are nearly identical, which has presented a serious challenge in the development of isoform selective inhibitors. Nevertheless, we found (9) that a single amino acid difference between nNOS and eNOS, Asp597 in nNOS vs. Asn368 in eNOS, is responsible for why a series of dipeptide inhibitors (10-12) bind much more tightly to nNOS than eNOS. In this earlier work we observed that a series of dipeptide inhibitors bind quite differently in eNOS and nNOS. In nNOS the inhibitors adopt a “curled” conformation which enables the didpeptide α-amino group to be in position to interact with Asp597. However, in eNOS the dipeptide inhibitors adopt an extended conformation since Asp597 is replaced by Asn368 in eNOS and thus there is no additional electrostatic incentive in eNOS for the inhibitors to “curl”. The N368D mutant in eNOS and D697N mutant in nNOS confirmed that the Asn/Asp difference is the primary structural basis for why the dipepetide inhibitors bind much better to nNOS (9).This led to the design of a series of chiral pyrrolidine inhibitors (Fig. 1) that exhibit Ki values in the low nanomolar range with some exhibiting up to 4,000-fold selectivity for nNOS over eNOS (13-16). The most potent of these inhibitors have dramatic in vivo effects and can protect newborn rabbit kits from experimentally induced ischemic brain damage (14).
Figure 1.

Two different binding orientations of cis-pyrrolidine compounds. (A) The aminopyridine of (3′S, 4′S)-2 interacts (dashed lines) with Glu592 while (B) the aminopyridine of (3′R, 4′R)-2 is flipped and interacts with heme propionate D. All of the structural figures were prepared with PyMOL (www.pymol.org).
We had anticipated that these pyrrolidine inhibitors would bind so that the aminopyridine group would mimic the substrate L-Arg guanidinium and be situated over the heme and hydrogen bond/ion pair with the active site Glu592. Indeed, this is the case for the (3′S, 4′R) and (3′R, 4′S)-trans inhibitors and the cis-(3′S, 4′S) inhibitors (e.g., (3′S, 4′S)-2, Fig. 1A) whose structures were determined in a previous study (17). However, the crystal structures (17) showed that cis-(3′R, 4′R) compounds (e.g., (3′R, 4′R)-2) bind with the inhibitor flipped 180° so that the “tail” fluorophenyl end is situated over the heme, leaving the aminopyridine in position to hydrogen bond/ion pair with heme propionate D (Fig. 1B). Binding in the flipped orientation requires the movement of Tyr706. Because the aminopyridine portion of the inhibitor can bind in two flipped orientations, we reasoned that a symmetric, “double headed” inhibitor that has an aminopyridine at each end should bind especially well, displacing Tyr706 for direct interactions between one aminopyridine and the heme propionate and the other aminopyridine with Glu592. A series of double headed inhibitors now have been designed, synthesized, and Ki values measured (18). Here we report the crystal structures of these inhibitors bound to both nNOS and eNOS.
Materials and Methods
Inhibitor design, syntheses, and in vitro inhibitory assay
The design, syntheses, and the structure-activity relationship studies of the double headed aminopyridine inhibitors are reported elsewhere (18). Ki values and selectivity for the best of the inhibitors used in this study are summarized in Table 1.
Table 1.
Ki values (18) and selectivity of inhibitors used in this study.
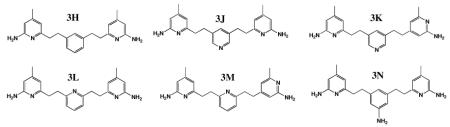
| compound | nNOS (nM) | eNOS (nM) | iNOS (nM) | n/ea | n/ia |
|---|---|---|---|---|---|
| 3h | 38 | 4200 | 2730 | 111 | 71.8 |
| 3j | 25 | 2680 | 1450 | 96 | 52 |
| 3k | 103 | 16100 | 1910 | 156 | 19 |
| 3l | 99 | 9400 | 4750 | 95 | 48 |
| 3m | 195 | 20000 | 12600 | 102 | 64 |
| 3n | 85 | 4950 | 3400 | 58 | 40 |
The ratio of Ki (eNOS or iNOS) to Ki (nNOS).
Protein preparation and crystallization
The nNOS S602H and eNOS H373S mutants were prepared using the QuikChange mutagenesis kit from Stratagene. The single point mutation was introduced into the wild type full length nNOS or eNOS coding sequence in the pCWori plasmid according to the manufacturer’s instructions. The mutation sites were confirmed by DNA sequencing. The nNOS or eNOS heme domain proteins, wild type or mutants, used for crystallographic studies were produced by limited trypsin digestion from the corresponding full length enzymes and further purified through a Superdex 200 gel filtration column (GE Healthcare), as described previously (9, 19). The enzyme-inhibitor complex crystals were obtained by soaking rather than co-crystallization as reported in the earlier structural work (9). The nNOS heme domain at 7-9 mg/mL containing 20mM histidine or the eNOS heme domain at 20 mg/mL with 2 mM imidazole were used for the sitting drop vapor diffusion crystallization setup under the conditions reported before (19, 20). Fresh crystals (1-2 day old) were first passed stepwise through cryo-protectant solutions described (19, 20) and then soaked with 10 mM inhibitor (1.5 mM Zn2+ acetate was included in soaks for eNOS) for 4-6 h at 4 °C before being flash cooled by plunging into liquid nitrogen. Crystals were stored in liquid nitrogen until data collection.
Identification of metal site with anomalous difference Fourier
The new metal site discovered in the nNOS-3j or nNOS-3n complex structure is surrounded by Asp600, His692 (subunit B), the pyridine nitrogen of 3j (a chlorine anion in the case of nNOS-3n complex), and a solvent molecule in a tetrahedral geometry. The strong difference density at 15 σ and the coordination geometry indicate a site for the first row transition metal ions. We identified the unknown metal using anomalous difference Fourier techniques similar to how we identified the native Zn2+ site while working out the initial crystal structure of eNOS (20). Fluorescence scans were performed with nNOS-3n crystals at the SSRL beamline 9-2 near the x-ray absorption edges of Fe, Co, Ni, Cu, and Zn. The absorption signals were detected at the edges of Fe, Cu, and Zn. The anomalous diffraction data were collected at two wavelengths for each of the three metals, one at the peak position of the absorption edge (e.g. E1 = 9667 eV for Zn) and the other at 60-80 eV away from the edge toward the low energy side (E2 = 9600 eV for Zn). To get the anomalous signals accurately measured an inverse beam data collection protocol was used with 10 degree wedge size and 2 s exposure time per frame (1 degree in scanning Φ angle). For each metal, two anomalous data sets at E1 and E2 were processed separately with HKL2000 (21). The anomalous data collection statistics collected at the Zn, Fe, and Cu edges are summarized in Table 2. To carry out the anomalous difference Fourier map calculation using FFT in CCP4 (22), the model phases were obtained by running a rigid body refinement with REFMAC (23) of the known nNOS heme domain model against each of the new anomalous data set (E1 or E2) and the Fourier coefficient was the anomalous difference, ΔF = F(+) − F(−). There is one homodimer in the asymmetric unit of the nNOS structure. The resulting maps (Fig. 2) for Zn edge data showed 5 peaks above 10 σ (the noise peaks were all below 5 σ) at E1, consisting of the native Zn, two new metal sites, and two heme sites; while only 2 peaks (two heme sites) at E2. The disappearance of the anomalous signals from the native Zn and the two new metal sites at the wavelength E2 (where the f ” of Zn is less than 0.5 electron) indicated that the unknown metal at the new sites is Zn. The anomalous data collected at the Fe edge confirmed that the Fe located at the two heme sites only, while the data for Cu detected no specific Cu binding site in structure (data not shown). The Cu fluorescence signals shown during scan may represent the random binding of the metal in solvent channels of crystal lattice.
Table 2.
Anomalous data collection statistics with nNOS-3n complex crystals
| Data set | nNOS-3n Zn E1 | nNOS-3n Zn E2 | nNOS-3n Fe E1 | nNOS-3n Fe E2 | nNOS-3n Cu E1 | nNOS-3n Cu E2 |
|---|---|---|---|---|---|---|
| X-ray wavelength (Å) | 1.28249 | 1.29146 | 1.73875 | 1.75361 | 1.37786 | 1.38835 |
| X-ray energy (eV) | 9667 | 9600 | 7130 | 7070 | 8998 | 8930 |
| Resolution (Å) | 2.80 (2.87-2.80) | 2.80 (2.87-2.80) | 2.95 (3.02-2.95) | 2.95 (3.02-2.95) | 2.85 (2.92-2.85) | 2.85 (2.92-2.85) |
| R merge | 0.075 (0.51) | 0.076 (0.49) | 0.089 (0.60) | 0.090 (0.64) | 0.082 (0.53) | 0.082 (0.52) |
| I / σI | 18.9 (3.0) | 18.8 (2.9) | 15.0 (2.0) | 15.7 (2.0) | 17.0 (2.4) | 16.8 (2.4) |
| No. unique reflections |
44,318 | 44,290 | 38,273 | 38,583 | 42,589 | 42,007 |
| Completeness (%) | 98.7 (99.7) | 98.6 (98.2) | 99.0 (96.4) | 98.9 (95.4) | 98.3 (93.4) | 98.4 (94.3) |
| Redundancy | 3.9 (3.8) | 3.9 (3.6) | 3.8 (3.4) | 3.8 (3.2) | 3.9 (3.4) | 3.9 (3.3) |
Figure 2.

Display of the anomalous difference density of a nNOS crystal bound with 3n at 5 σ contour level calculated at (A) the Zn absorption edge (9667 eV, 1.28 Å) and (B) 67 eV away from the edge on the low energy side (9600 eV, 1.29 Å). The Zn peaks are disappeared in panel B because of the weak anomalous signal of Zn at the x-ray wavelength of 1.29 Å, but the anomalous signals from the two heme iron atoms are retained.
X-ray diffraction data collection, processing, and structure refinement
The cryogenic (100K) X-ray diffraction data were collected remotely at various beamlines at SSRL through data collection control software Blu-Ice (24) and the crystal mounting robot. Raw data frames were indexed, integrated, and scaled using HKL2000 (21). Typically, each data set consisted of 90 to 100 degrees of data with 0.5 degree frame width for both nNOS and eNOS crystals because of their identical orthorhombic P212121 space group symmetry.
The binding of inhibitors was detected by the initial difference Fourier maps calculated with CNS (25). The inhibitor molecules were then modeled in O (26) or COOT (27) and refined using CNS. Refinements were continued with REFMAC (23) in order to implement the TLS (28) protocol with each subunit as one TLS group. Water molecules were added and checked by COOT. The sigmaA weighted omit Fo – Fc electron density maps were calculated with REFMAC by running TLS + Restrained refinement with the interested ligand(s) omitted from the coordinate file. The refined structures were validated through COOT as well as the RCSB validation server with no outlier in the Ramachondron plot. The crystallographic data collection and structure refinement statistics are summarized in Table 3 and Table S1 (Supporting Information). Coordinates of all the crystal structures reported in this work have been deposited in the RCSB protein data bank. Their accession codes are listed in both Tables and as a footnote.
Table 3.
Crystallographic data collection and refinement statistics (part 1)
| Data set1 | nNOS-3j | nNOS-3l | nNOS-3n | nNOS D597N-3j | nNOS D597N/ M336V-3j |
eNOS-3j | eNOS-3n |
|---|---|---|---|---|---|---|---|
| Data collection | |||||||
| PDB code | 3N5W | 3N5Y | 3N60 | 3N62 | 3N61 | 3N5T | 3N5Q |
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 |
Cell dimensions 
|
|||||||
| a, b, c (Å) | 52.3,111.2,164.8 | 51.7,110.6,164.1 | 51.9,111.2,164.0 | 51.8,110.4,164.1 | 51.8,110.5,164.3 | 58.1,106.7,157.4 | 57.9,107.1,157.3 |
| Resolution (Å) | 1.73 (1.76-1.73) | 2.05 (2.09-2.05) | 1.98 (2.01-1.98) | 1.95 (1.98-1.95) | 1.95 (1.98-1.95) | 2.52 (2.56-2.52) | 3.00 (3.05-3.00) |
| Rsym or Rmerge | 0.051 (0.59) | 0.066 (0.56) | 0.054 (0.64) | 0.069 (0.58) | 0.066 (0.50) | 0.083 (0.559) | 0.135 (0.638) |
| I / σI | 29.1 (2.1) | 21.2 (1.9) | 28.7 (2.1) | 21.5 (2.4) | 21.1 (1.8) | 15.0 (1.9) | 9.7 (2.1) |
| No. unique reflections |
101,279 | 60,097 | 67,020 | 69,795 | 69,511 | 33,841 | 19,856 |
| Completeness (%) | 99.5 (99.8) | 99.4 (94.6) | 99.4 (89.8) | 99.8 (100.0) | 99.7 (98.4) | 99.8 (99.8) | 97.9 (99.1) |
| Redundancy | 4.0 (4.0) | 4.0 (3.7) | 4.5 (4.2) | 4.0 (4.1) | 4.0 (3.8) | 3.6 (3.6) | 4.0 (4.0) |
| Refinement | |||||||
| Resolution (Å) | 1.73 | 2.05 | 1.98 | 1.95 | 1.95 | 2.52 | 3.00 |
| No. reflections used |
95,347 | 56,712 | 63,638 | 66,262 | 65,998 | 32,111 | 18,829 |
| Rwork / Rfree | 0.182/0.205 | 0.184/0.228 | 0.191/0.232 | 0.173/0.208 | 0.179/0.213 | 0.186/0.248 | 0.181/0.269 |
| No. atoms | |||||||
| Protein | 6,678 | 6,658 | 6,659 | 6,700 | 6,699 | 6,429 | 6,413 |
| Ligand/ion | 205 | 182 | 145 | 275 | 192 | 199 | 181 |
| Water | 594 | 358 | 347 | 398 | 403 | 138 | 43 |
| R.m.s. deviations | |||||||
| Bond lengths (Å) | 0.012 | 0.012 | 0.015 | 0.013 | 0.013 | 0.014 | 0.016 |
Bond angles ( ) ) |
1.241 | 1.316 | 1.476 | 1.393 | 1.352 | 1.531 | 1.620 |
See Table 1 for nomenclature and chemical formula of inhibitors.
5% of reflections has been set aside during the entire course of refinement for the free R calculation only. For each NOS isoform the choice of free R flags has been kept the same as those used in the starting model
Results and Discussion
Discovery of a new Zn2+ site upon binding of 3j (3k) to nNOS
The basic idea behind the double-headed inhibitors is for one aminopyridine to bind in the active site and interact with Glu592 while the second to extend out of the active site, displace Tyr706, and interact with heme propionate D. The inhibitory constants (Ki) of these compounds have been determined (18) and the results relevant to the present study are summarized in Table 1. Some of these compounds also exhibited IC50 values in the 5μM range in cell-based assays, thus indicating the potential for in vivo applications (18).
To determine if our expectation of how these inhibitors should bind was correct, we determined crystal structures nNOS and eNOS in complex with compounds that showed good inhibitory potency. Compound 3j (Table 1) binds as expected with both aminopyridine rings involved in hydrogen bonding interactions with Glu592 and the heme (Fig. 3). Quite unexpectedly, however, a second molecule of 3j (3jB) binds with one aminopyridine group situated in the H4B binding pocket. Moreover, there is strong difference density (15 σ) near the bridging pyridine nitrogen atom of 3jB. The electron density also is near Asp600 and His692 of subunit B (His692B) in the nNOS dimer. These two residues, the 3j pyridine, and a large solvent ion (probably chloride) are tetrahedrally arranged around the large lobe of density highly reminiscent of a metal binding site. To determine the identity of the metal ion, a series of data sets were collected at different wavelengths near the absorption edge of the most likely metal candidates (Zn2+, Cu2+, Fe3+/Fe2+,, Ni2+, and Co2+) as well at 50-80 eV lower energies from each metal absorption edge. Using this method the metal bound was unambiguously identified as Zn2+ (Fig. 2 and Table 2).
Figure 3.

The nNOS active site with one molecule of 3j bound above the heme and the other in the pterin binding pocket. The sigmaA-weighted Fo-Fc omit density map for 3j is shown at a 3.0 σ contour level. The ligation bonds around the new Zn2+ site and hydrogen bonds are depicted with dashed lines. Two alternate side chain conformations are shown for residue Tyr706. NOS dimerizes through the heme domains with the pterin binding in a pocket at the dimer interface. Residues in subunit A are depicted with green bonds and those of subunit B with cyan bonds. Four pyrrole rings of heme are labeled.
Zinc was not included during purification or crystallization so the source of zinc remains unclear. NOS dimerizes through the heme domain with a Zn+2 coordinated to four Cys residues at the dimer interface. If we assume this dimer interface Zn2+ is at full occupancy, then the new Zn2+ site has an occupancy ≈ 0.7. In order for Zn2+ to bind, substantial conformational rearrangements must occur in addition to displacement of the H4B by 3jB. The Arg596 side chain, which H-bonds with the H4B, must swing out of the way and adopts a new conformation where it now forms hydrogen bonds to both Glu592 and Asp597 (Fig. 3). The imidazole ring of His692B rotates 180° to allow the NE2 atom to provide one of the Zn2+ ligands. This also requires a slight movement of His692B toward the new Zn2+ site, resulting in a tightening of the dimer interface. This new ring orientation of His692B is only possible when Arg596 swings out of the way. Another inhibitor analogous to 3j, namely 3k, which has its aminopyridine ring nitrogen located at a different position (Table 1), shows a nearly identical two inhibitor bound structure to that of 3j (Fig. S1A).
Structure requirements for Zn2+ binding
We next explored the structural requirements for the novel Zn2+ site. Since the bridging pyridine N atom of 3jB provides a Zn2+ ligand, then its removal should prevent Zn2+ binding. Compound 3h, with the bridging pyridine replaced by a benzene ring, binds with one molecule at the substrate binding site without a second molecule that replaces the H4B and there is no new Zn2+ site found with this inhibitor (Fig. S1B). We next asked if how the bridging pyridine is attached to the two aminopyridines is important. The nNOS-3j structure indicates that attachment of the aminopyridines to the bridging pyridine at the meta positions is the only way to properly position the pyridine nitrogen for Zn2+ coordination. To test this idea, an analogue of 3j, 3l (Table 1), was synthesized that has its nitrogen atom in the bridging pyridine adjacent (ortho) to the two substituents. As expected, there is no second molecule of 3l bound to nNOS (Fig. 4). The H4B remains bound and, therefore, no new Zn2+ site is found. However, the first molecule of 3l is not bound to nNOS the same as 3j. The interaction between the new pyridine nitrogen of 3l and heme propionate A moves the second aminopyridine out of position for interaction with heme propionate D (Fig. 4). A structural analog of 3l, namely 3m (Table 1), gives a similar one inhibitor bound structure (supplementary Fig. S2).
Figure 4.

The nNOS active site with one molecule of 3l bound above the heme. The sigmaA-weighted Fo-Fc omit density map for 3l is shown at a 3.0 σ contour level. Major hydrogen bonds are depicted with dashed lines. The second aminopyridine is partially disordered.
While testing the variations at the bridging ring of these double-headed aminopyridine compounds, one of the inhibitors, 3n (Table 1), with an aniline group as the bridging ring, leads to a structure with even more surprising results. This time, only one inhibitor is bound, with its first aminopyridine interacting with Glu592 as usual, but with its second aminopyridine directly replacing the H4B and stacking against the Trp678 side chain (Fig. 5). Since the H4B is no longer in place and the Arg596 side chain swings out, the same new Zn2+ site is created. The only difference with the Zn2+ site seen in the nNOS-3j structure is that the inhibitor 3n is not part of the Zn2+ coordination sphere. Rather, a chloride ion and a water molecule are the metal ligands in addition to the side chains of Asp600 and His692 (subunit B). The binding of 3n causes the largest distortion to the heme propionate (pyrrole A) so far observed in any available nNOS inhibitor complex structures. This propionate is now hydrogen bonded to the side chain of Arg596 in its new rotamer position (Fig. 5). The same heme propionate is important to H4B binding because of hydrogen bonding interactions. Any distortion to this propionate position would cause the weakening of cofactor binding, which is the common feature we have found in the nNOS complex structures with 3j, 3k, and 3n. To briefly summarize, our results thus far indicate that the key to forming the new Zn2+ site is the movement of Arg596, which provides room for a Zn2+ ion to bind and coordinate with Asp600 and His692 from subunit B.
Figure 5.

The nNOS active site with compound 3n bound. The Fo-Fc omit electron density for the inhibitor is contoured at 3.0 σ. The ligation bonds of the new Zn2+ site and hydrogen bonds are depicted with dashed lines. The heme and H4B in the substrate bound structure (PDB code 1OM4) are overlaid to illustrate the large conformational change of heme propionate A and the displacement of H4B by the second aminopyridine of inhibitor.
No Zn2+ site found in eNOS
Although these double aminopyridine compounds bind to eNOS with only micromolar affinity, it was important to determine if inhibitors such as 3j, 3k, and 3n, which are capable of creating the new Zn2+ binding site in nNOS, can do the same to eNOS. Even though the Zn2+ coordinating residues in nNOS, Asp600 and His692B, are conserved in eNOS, Asp371 and His463B, we did not detect any trace of Zn2+ binding in eNOS structures, even after inclusion of 1.5 mM Zn2+ in the cryo-soaks (Fig. 6A). Only one molecule of 3j binds, similar to the nNOS-3jA complex, but there is no disturbance of the heme propionate or Arg367 as when the second 3jB molecule binds to nNOS. As with the nNOS-3n complex, only one 3n molecule binds to eNOS but the second aminopyridine of 3n adopts a substantially different conformation than in nNOS (Fig. 6B). The first aminopyridine of 3n interacts with Glu363, the same as in nNOS, but the bridging aniline ring does not press against heme propionate A resulting in a repositioning of the heme propionate as in nNOS. Instead, it is situated between the two heme propionate groups and does not result in any changes in the heme propionate. In addition, Arg367 does not move, H4B remains bound, and thus Zn2+ cannot bind.
Figure 6.

The eNOS active site with compound 3j (A) and 3n (B) bound. The Fo-Fc omit electron density for the inhibitor is contoured at 2.5 σ. Hydrogen bonds are depicted with dashed lines. In panel A, Tyr477 moves to allow interaction of 3j with the heme propionate. In panel B, the second aminopyridine of 3n is partially disordered. The H4B is not disturbed by ligand binding illustrated by the omit electron density for the pterin at 2.5 σ.
The most obvious difference between nNOS and eNOS that might contribute to Zn2+ binding in nNOS is Asp597 in nNOS, which is Asn368 in eNOS. In nNOS Arg596 must swing out of the way to H-bond with Asp597, and because eNOS has an Asn367 instead of Asp, there may not be the electrostatic energetic incentive for Arg367 to move in eNOS. Another difference between nNOS and eNOS that we have found contributes to selectivity in previous studies is Val106 in eNOS which is Met336 in nNOS (9). However, a series of crystal structures of the eNOS N368D/V106M double mutant complexed with double headed inhibitors shows that Arg367 remains in place, and thus a second inhibitor molecule and Zn2+ cannot bind (Supplementary Figs. S3-S6). We next examined the inverse mutation in nNOS and converted Asp597 to Asn. The D597N mutant binds 3n the same as wild type nNOS, which includes Zn2+ (Supplementary Fig. S7). In the presence of 3n the Arg596 side chain still swings out in a position of hydrogen bonding to N597 even though the residue no longer bears a negative charge as D597 in the wild type nNOS. Clearly the ability of Arg596 to move does not depend on ion pairing with Asp597.
Is removal of H4B necessary in creation of Zn2+ site?
The next question addressed is whether or not displacement of H4B is required for Zn2+ binding. The answer is no. In the D597N nNOS-3j complex the first 3j molecule binds to nNOS the same as in wild type. However, the electron density for the second molecule is not well defined and could best be accounted for by assuming partial occupancy for both the inhibitor and H4B (Fig. 7A). The new Zn2+ is still present but one of the four ligands is either the partially occupied inhibitor or a partially occupied water molecule (Fig. 7A). The heme propionate that must move when two inhibitors bind also exhibits partial occupancy in two different conformations. The picture becomes a little more complicated in the D597N/M336V nNOS double mutant (Met336 is Val in eNOS). In this mutant only one 3j molecule is bound (Fig. 7B). In the wild type protein the Met336 SD atom is 3.8Å from the inhibitor A and 4.4Å from inhibitor B, the one that displaces H4B. These interactions are lost when Met366 is changed to Val thus decreasing the affinity for the second inhibitor molecule.
Figure 7.

The active site of (A) D597N nNOS or (B) D597N/M336V nNOS mutant with compound 3j bound. The Fo-Fc omit density map for the inhibitor is shown at a 2.5 σ (A) or 3.0 σ (B) contour level. The new Zn site ligation bonds and hydrogen bonds are drawn with dashed lines. Heme propionate A in each case has alternate conformations. In panel A, the second molecule of 3j only partially occupies the pterin site, while H4B together with two water molecules, Wat1 and Wat2, account for the rest. In panel B, there is no second 3j molecule; hence, H4B binds with full occupancy indicated by the Fo-Fc omit density map at 3.0 σ.
What prevents eNOS from binding a new Zn2+ ?
What remains a puzzle is why eNOS and the eNOS mutants designed to mimic nNOS fail to bind a second inhibitor molecule and thus do not bind Zn2+. In examining the structures more closely one additional difference may be contributing. His373 in eNOS would be about 4.0Å from inhibitor B (if it would have bound) and thus might interfere with binding. This residue is Ser602 in nNOS. However, the nNOS S602H mutant still binds two molecules of 3j with Zn2+ bound although inhibitor B and H4B are at partial occupancy (Fig. S8A). The nNOS S602H-3n structure shows one inhibitor bound that displaces the H4B and also promotes the formation of the new Zn2+ site just like wild type nNOS (Fig. S8B). However, the inverse mutant in eNOS, H373S, binds 3j and 3n just like wild type eNOS with only one inhibitor bound (Figs. S9A and S9B). Therefore, the involvement of this His373 of eNOS in prevention of binding a new Zn2+ in eNOS has been ruled out.
While the various mutants do perturb the binding of the double headed inhibitors, other more subtle factors that are not due to obvious amino acid differences near the inhibitors or Zn2+ control why nNOS can bind Zn2+ but not eNOS. Although the results we have obtained are somewhat complicated by the partial occupancy of inhibitor B in the nNOS mutants designed to mimic eNOS, there are three criteria that must be met in forming the new Zn2+ site. First, Arg596 must move from its H-bonding position with H4B in order to make room for the Zn2+. Second, the heme propionate A normally H-bonding with H4B must move in order to make room for the second inhibitor that coordinates the Zn2+. Third, one of the Zn+2 ligands, His692B, is from subunit B at the dimer interface. In order for His692B to coordinate Zn2+, the His692B side chain must adopt a new rotamer conformation and move about 0.5Å closer to subunit A, which requires some flexibility at the dimer interface. eNOS cannot undergo any of these changes required for binding of the second inhibitor and/or Zn2+ binding. It is well known that the eNOS dimer is more stable than that of either nNOS or iNOS, indicating a more rigid and less flexible dimer interface in eNOS (29). Such rigidity could prevent the movement of His463B required to bind Zn2+. In addition, a tighter interface may make it more difficult to disrupt the heme propionate-H4B H-bonding interactions, which are required for binding the second inhibitor molecule. Although not an easily testable hypothesis, the best explanation for the primary causal differences between eNOS and nNOS in binding these double headed inhibitors is due to the more rigid dimer interface in eNOS and the greater flexibility in nNOS, which allows the required motions needed for binding Zn2+.
Significance for NOS inhibitor design
The significant conformational changes observed upon inhibitor binding and formation of the new Zn+2 site has identified three key flexible regions in nNOS, but not in eNOS, which may prove useful in the design of a new generation of NOS inhibitors. First, heme propionate A, which interacts with the H4B, is quite flexible and adopts various conformations in response to the bulkiness and chemical nature of the ligand bound at the substrate binding pocket. The conformation of this heme propionate will, in turn, affect the binding of the H4B cofactor. Second, Arg596 must move to provide room for the new Zn2+ ion. Since Arg596 interacts with the H4B, movement of Arg596 and the heme propionate weakens the H4B binding, enabling easier displacement by the second inhibitor molecule. Third, we hypothesize that the relatively weaker dimer interface interactions in nNOS relative to eNOS allows one of the Zn2+ ligands, His692B, to move in order to optimize the Zn2+ coordination sphere. Since none of this occurs in eNOS, owing to a more rigid dimer interface, it should be possible to exploit these nNOS-specific movements for novel nNOS selective inhibitor design. With respect to selectivity, it also is worth noting that the new Zn2+ site is probably not “natural”, since we only observe the Zn2+ binding in the presence of inhibitors. In no case was a Zn2+ ion found in either nNOS or eNOS crystals pre-soaked in the presence of either 10 mM arginine or 5 mM S-ethylisothiourea and 5 mM Zn2+ acetate for 4 hours (unpublished observations).
Supplementary Material
Acknowledgments
We thank the beamline staff at Stanford Synchrotron Radiation Lightsource for their assistance during remote x-ray diffraction data collections.
Footnotes
PDB accession codes: nNOS-3h (3N5V), nNOS-3j ( 3N5W), nNOS-3k (3N5X), nNOS-3l (3N5Y), nNOS-3m (3N5Z), nNOS-3n (3N60), nNOS D597N-3j (3N62), nNOS D597N-3n (3N64), nNOS D597N/M336V-3j (3N61), nNOS D597N/M336V-3n (3N63), nNOS S602H-3j (3N65), nNOS S602H-3n (3N66), eNOS-3h (3N5R), eNOS-3j (3N5T), eNOS-3k (3N5S), eNOS-3m (3N5P), eNOS-3n (3N5Q), eNOS N368D-3j (3N6F), eNOS N368D-3k (3N6G), eNOS N368D-3n (3N6E), eNOS N368D/V106M-3h (3N68), eNOS N368D/V106M-3j (3N69), eNOS N368D/V106M-3k (3N6A), eNOS N368D/V106M-3n (3N67), eNOS H373S-3j (3N6B), eNOS H373S-3m (3N6C), eNOS H373S-3n (3N6D)
Supporting Information Available Crystallographic statistics tables and supplementary figures. This material is available free of charge via the Internet at http://pub.acs.org.
Reference
- 1.Stuehr DJ, Griffith OW. Mammalian nitric oxide synthases. Adv Enzymol Relat Areas Mol Biol. 1992;65:287–346. doi: 10.1002/9780470123119.ch8. [DOI] [PubMed] [Google Scholar]
- 2.Knowles RG, Moncada S. Nitric oxide synthases in mammals. Biochem J. 1994;298:249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moncada S, Higgs EA. Endogenous nitric oxide: physiology, pathology and clinical relevance. Eur J Clin Invest. 1991;21:361–374. doi: 10.1111/j.1365-2362.1991.tb01383.x. [DOI] [PubMed] [Google Scholar]
- 4.Sims NR, Anderson MF. Mitochondrial contributions to tissue damage in stroke. Neurochem. Int. 2002;40:511–526. doi: 10.1016/s0197-0186(01)00122-x. [DOI] [PubMed] [Google Scholar]
- 5.Nathan C. Inducible nitric oxide synthase: what difference does it make? J Clin Invest. 1997;100:2417–2423. doi: 10.1172/JCI119782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taddei S, Virdis A, Ghiadoni L, Sudano I, Salvetti A. Endothelial dysfunction in hypertension. J. Cardiovascular Pharmacol. 2001;38(Suppl. 2):S11–14. doi: 10.1097/00005344-200111002-00004. [DOI] [PubMed] [Google Scholar]
- 7.Hobbs AJ, Higgs A, Moncada S. Inhibition of nitric oxide synthase as a potential therapeutic target. Annu Rev Pharmacol Toxicol. 1999;39:191–220. doi: 10.1146/annurev.pharmtox.39.1.191. [DOI] [PubMed] [Google Scholar]
- 8.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flinspach ML, Li H, Jamal J, Yang W, Huang H, Hah JM, Gomez-Vidal JA, Litzinger EA, Silverman RB, Poulos TL. Structural basis for dipeptide amide isoform-selective inhibition of neuronal nitric oxide synthase. Nat Struct Mol Biol. 2004;11:54–59. doi: 10.1038/nsmb704. [DOI] [PubMed] [Google Scholar]
- 10.Huang H, Martasek P, Roman LJ, Masters BS, Silverman RB. N(omega)-Nitroarginine-containing dipeptide amides. Potent and highly selective inhibitors of neuronal nitric oxide synthase. J Med Chem. 1999;42:3147–3153. doi: 10.1021/jm990111c. [DOI] [PubMed] [Google Scholar]
- 11.Huang H, Martasek P, Roman LJ, Silverman RB. Synthesis and evaluation of peptidomimetics as selective inhibitors and active site probes of nitric oxide synthases. J Med Chem. 2000;43:2938–2945. doi: 10.1021/jm000127z. [DOI] [PubMed] [Google Scholar]
- 12.Hah JM, Roman LJ, Martasek P, Silverman RB. Reduced amide bond peptidomimetics. (4S)-N-(4-amino-5-[aminoakyl]aminopentyl)-N’-nitroguanidines, potent and highly selective inhibitors of neuronal nitric oxide synthase. J Med Chem. 2001;44:2667–2670. doi: 10.1021/jm0101491. [DOI] [PubMed] [Google Scholar]
- 13.Ji H, Stanton BZ, Igarashi J, Li H, Martasek P, Roman LJ, Poulos TL, Silverman RB. Minimal pharmacophoric elements and fragment hopping, an approach directed at molecular diversity and isozyme selectivity. Design of selective neuronal nitric oxide synthase inhibitors. J Am Chem Soc. 2008;130:3900–3914. doi: 10.1021/ja0772041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji H, Tan S, Igarashi J, Li H, Derrick M, Martasek P, Roman LJ, Vasquez-Vivar J, Poulos TL, Silverman RB. Selective neuronal nitric oxide synthase inhibitors and the prevention of cerebral palsy. Ann Neurol. 2009;65:209–217. doi: 10.1002/ana.21555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Igarashi J, Li H, Jamal J, Ji H, Fang J, Lawton GR, Silverman RB, Poulos TL. Crystal structures of constitutive nitric oxide synthases in complex with de novo designed inhibitors. J Med Chem. 2009;52:2060–2066. doi: 10.1021/jm900007a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lawton GR, Ranaivo H. Ralay, Chico LK, Ji H, Xue F, Martasek P, Roman LJ, Watterson DM, Silverman RB. Analogues of 2-aminopyridine-based selective inhibitors of neuronal nitric oxide synthase with increased bioavailability. Bioorg Med Chem. 2009;17:2371–2380. doi: 10.1016/j.bmc.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delker SL, Ji H, Li H, Jamal J, Fang J, Xue F, Silverman RB, Poulos TL. Unexpected binding modes of nitric oxide synthase inhibitors effective in the prevention of a cerebral palsy phenotype in an animal model. J Am Chem Soc. 2010;132:5437–5442. doi: 10.1021/ja910228a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue F, Delker SL, Li H, Fang J, Jamal J, Martásek P, Roman LJ, Poulos TL, Silverman RB. Symmetric double-headed aminopyridines, a novel strategy for potent and membrane-permeable inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 2010 doi: 10.1021/jm101071n. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Wen EZ, Jia Q, Wang PG, Poulos TL. The novel binding mode of N-alkyl-N’-hydroxyguanidine to neuronal nitric oxide synthase provides mechanistic insights into NO biosynthesis. Biochemistry. 2002;41:13868–13875. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 20.Raman CS, Li H, Martasek P, Kral V, Masters BS, Poulos TL. Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 21.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 22.Collaborative, Computational, Project, and Number4 The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 23.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 24.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 25.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 26.Jones TA, Zou J-Y, Cowan SW, Kjeldgaarrd M. Improved methods for building models in electron density and the location of errors in these models. Acta cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Lohkamp B, Scott W, Cowtan K. Features and development of Coot. Acta Cryst. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Cryst. 2001;D57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 29.Panda K, Rosenfeld RJ, Ghosh S, Meade AL, Getzoff ED, Stuehr DJ. Distinct dimer interaction and regulation in nitric-oxide synthase types I, II, and III. J Biol Chem. 2002;277:31020–31030. doi: 10.1074/jbc.M203749200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.