

Abstract
The capsid protein (CA) plays crucial roles in HIV-infection and replication, essential to viral maturation. The absence of high-resolution structural data on unassembled CA hinders the development of antivirals effective in inhibiting assembly. Unlike enzymes that have targetable functional substrate binding sites, the CA does not have a known site that affects catalytic or other innate activity, which can be more readily targeted in drug development efforts. We report the crystal structure of the HIV-1 CA, revealing the domain organization in context of the wild-type full-length (FL) unassembled CA. The FL CA adopts an antiparallel dimer (APD) configuration, exhibiting a domain organization sterically incompatible with capsid assembly. A small compound, generated in-situ during crystallization, is bound tightly at a hinge-site (“H-site”), indicating that binding at this interdomain region stabilizes the ADP conformation. Electron microscopy studies on nascent crystals reveal both dimeric and hexameric lattices coexisting within a single condition, in agreement with the interconvertibility of oligomeric forms and supporting the feasibility of promoting assembly-incompetent dimeric states. Solution characterization in the presence of the H-site ligand shows predominantly unassembled dimeric CA, even under conditions that promote assembly. Our structure elucidation of the HIV-1 FL CA and characterization of a potential allosteric binding site provides 3D views of an assembly-defective conformation, a state targeted in and, thus, directly relevant to, inhibitor development. Based on our findings, we propose an unprecedented means of preventing CA assembly, by ‘conformationally-trapping’ CA in assembly-incompetent conformational states, induced by H-site binding.
Keywords: native full-length HIV-1 capsid crystal structure, assembly inhibitor, hinge- binding site, conformational-trapping, alternative dimer states
Introduction
During virus maturation, the capsid of HIV-1 assembles from multimerization of a single polypeptide. After cleavage of the Gag polyprotein into component peptides, the two-domain capsid protein (CA) forms a polymeric shell comprised of a network of hexagonal CA rings with pentameric interactions to close the capsid shell at vertices 1. Despite low sequence homologies, structures of isolated domains and full-length CA proteins from a variety of retroviruses share similar tertiary folds. The carboxy-terminal domain (CTD) contains some of the key determinants of Gag oligomerization, and mutations or deletions in this domain dramatically affect viral assembly. The most conserved sequence segment in retroviral Gag proteins is the major homology region (MHR, residues 152–172); otherwise the sequence of the CTD is highly variable 2; 3. While the CTD is believed to initiate assembly by dimer formation through two-fold interactions, the NTD is thought to play key roles in stabilizing the assembled state, through six-fold interactions. Intermolecular associations between the NTD and CTD of adjacent subunits also appear to stabilize the assembled capsid 1.
Structural studies on the CA have been focused on the assembled, hexameric and unassembled, dimeric, states. CryoEM reconstructions have revealed general domain relationships in assembled hexameric and pentameric rings of various retroviruses 4; 5; 6. High-resolution structural analyses on the wild-type (wt) full length (FL) CA have been hampered by difficulties in producing well-diffracting crystals, with the dynamic, conformationally-heterogeneous behavior of CA similarly precluding NMR analyses on the intact protein. The domain flexibility of CA has been attributed to the hinge region that connects the NTD to the CTD. While primary sequence conservation is low overall across viral genomes except in the MHR region, the domain organization connected by a flexible interdomain hinge linker represents the other highly conserved topological feature in retroviruses. The hinge linker has been proposed to be the key structural element in enabling CA to adopt the multitude of conformations required for initiating assembly and disassembly of the capsid shell during the viral lifecycle and, thus, responsible for the prolific range of polymorphism found in retroviral capsids4.
Beyond dynamic considerations, structural and molecular studies are complicated by CA’s tendency to aggregate, in-vitro, limiting solubilities and preventing growth of suitable crystals of the native CA. Consequently, attempts to obtain high-resolution 3D structural data on the CA have been restricted to truncated proteins containing a single domain, on proteins bearing multiple-site mutations, and on CA additionally engineered with disulfide bonds crosslinking domain interfaces, all in attempts to limit CA’s conformational mobility7; 8; 9; 10; 11; 12.
Another approach to both restricting dynamics and enhancing solubility was through addition of large antibody fragments to generate fusion Fab-CAs. In all Fab-complexed CA crystals, the lattice contacts enabling crystallization were formed through the Fab molecules, which created the scaffold within which the CA dimer was held 7; 12. However, despite these lattice contacts, large regions of the CTD (residues 152–231) were unresolvable, further highlighting the flexibility inherent in the CTD and the role of the Fab fragments in enabling lattice formation12. Consequently, despite the structure determination of the Fab-CA, uncertainties about the biological veracity of the conformational state together with the incomplete CTD in the models have limited adoption of the structural results. Instead, the truncated, isolated CTD dimer has been suggested to recapitulate the CTD-CTD interactions expected in the assembled CA, despite missing the NTD or ~50% of the intact protein 10; 13.
The direct influence of the hinge angle on intra- and inter-molecular associations and, thus, assembly state, can only be experimentally determined by structural elucidation of the FL native CA protein. Although the recent structural results on the hexameric CA8 provided valuable insights on putative intermolecular contacts in an assembled state, it is unknown whether the structures fully reflect the native assembled CA since multiple mutations and disulfide crosslinks were introduced to ‘lock-down’ the conformation, believed to be requisite for reducing solution molecular dynamics to permit crystallization. We report here the crystal structures of the FL CA, resolving the NTD-CTD domain orientation in context of the wild type, unassembled CA, crystallized without alteration of the native sequence and under lower salt conditions than previously achieved. Our results support the central role of the inter-domain hinge region on the overall monomer conformation in the FL CA, suggesting that the domain (ie NTD-CTD) orientation prior to capsid formation strongly influences the assembly process. Our studies reveal interactions between a novel ligand and residues at a hinge region-binding site (“H-site”), define contacts that strongly influence affinities of associations. The H-site is adjacent to the putative assembly inhibitor, CAP-1, binding site, although a structure of CA in complex with CAP-1 or another inhibitor bound at the H-site, has never been resolved structurally.
Our structural and solution characterization of an alternative dimeric form of CA can provide insight relevant to development of anti-retroviral drugs that prevents or impedes the transition from the preassembled to assembled states. Illuminating the parameters that affect the kinetics and thermodynamics of capsid formation, together with the high-resolution structures of unassembled CA, will be directly beneficial to drug development efforts. We propose that by “conformationally trapping” the HIV-1 CA into assembly-incompetent conformational state(s), induced by small molecule binding at the H-site, could effectively impede capsid shell formation and present preliminary data supporting this idea.
Results
Structure of full length HIV-1 CA
The full-length (FL; residues 1–231) capsid (CA) protein of HIV-1 was crystallized from conditions initially identified through extensive screening followed by condition optimization to obtain well-diffracting, single crystals, as described under Experimental Methods. The asymmetric unit is comprised of an APD dimer, crystallized in two lattices (Table 1; Figure 1A). Associated with each dimer are numerous ions including irons, iodides, and triiodide anions (Figure 1B), generated in situ 14. In the FL CA structure, single iodide ions are usually found within 4 Å of backbone amide groups, near serine Oγ oxygens and hydrophobic side chains. Several iodides are associated with Fe3+/Fe2+ which are further coordinated with waters, bound to the surface of the protein. Augmenting these are numerous ordered waters that bridge ionic and protein backbone and sidechain atoms, mediating hydrogen-bonding networks (Figure 1B). These extensive ionic and water interactions create a secondary hydration shell, sheathing the surface of the protein, contributing to the enhanced solubility. The pervasiveness of solvent-mediated hydrogen-bonding networks stabilizing molecular and domain interfaces in the native FL CA resembles those found in assembled CAs 4; 15. Although the concentrations of ions present in the structural study differ from those present under physiological conditions, this extent of ionic interactions has only been reported in structures of assembled CA, possibly signifying not only the importance of electrostatic neutralization but also mediates the formation of specific oligomeric states in native retroviral CAs.
Table 1.
Data Collection and Refinement
| Data set | Monoclinic | Orthorhombic |
|---|---|---|
| Space Group | P21 | P212121 |
| Unit Cell | a=47.9Å, b=86.5 Å, c=55.6 Å, β=99° | a=53.1 Å, b=87.7 Å, c=103.4 Å |
| Crystal mosaicity (degrees) | 0.7 | 1.0 |
| Total number of observations | 481550 | 48813 |
| Unique reflections | 32758 | 18971 |
| Resolution (Å) | 1.95 – 50.0 | 2.9 – 53.0 |
| Rsym | 0.062 | 0.14 |
| I/σ | 20 (1.9) | 6 (1.8) |
| Completeness (%) | 94.8 (82.4) | 97.0 (80.0) |
| Redundancy | 3.7 (2.9) | 2.6 (2.6) |
| Refinement in PHENIX | ||
| Rwork = 20.1 % | Rwork = 20.5 % (no waters) | |
| Rfree = 24.8 % | Rfree = 25.2 % | |
| RMS Bond lengths (Å) = 0.008 | RMS Bond lengths (Å) = 0.008 | |
| RMS Angles (deg.) = 1.12 | RMS Angles (deg.) = 1.21 |
Figure 1.
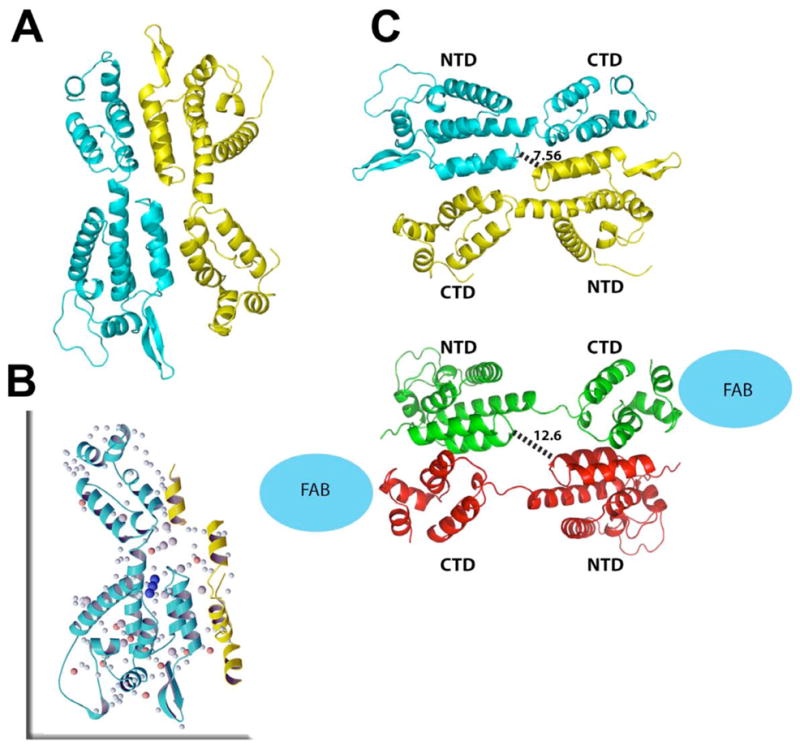
Crystal asymmetric unit (A) with one monomer shown in yellow, one in cyan. Extensive solvent and ion network (B) comprised of irons (orange spheres), iodides (larger gray spheres) and waters (small light cyan spheres) mediate interactions between domains within the FL CA (in blue ribbon) and between protein subunits (neighboring subunit partially shown in yellow ribbon). Binding of triiodide (dark blue spheres) close to the hinge likes stabilizes the conformation. Comparison between the native full-length CA structure to the RH24 complexed to a Fab fragment (C) with native FL CA dimer in cyan and yellow and the RH24 dimer in green and red ribbons. The antiparallel dimer in both structures exhibit similar molecular organization but in RH24, the two halves of the dimer are stretched further apart because of unraveling of 310 helical bridge between the NTD to CTD, possibly due to Fab (blue sphere) binding.
The two FL CA molecules in the asymmetric unit are well resolved, with strong and continuous electron densities for residues 1 – 219, including the cyclophilin (CypA)-binding loop in subunit A (residues 91 – 95). In subunit B, greater flexibility of this loop results in weaker electron densities with average thermal (B-factors) values of 48 A2 compared to 35 A2 for the whole structure. This suggests that in absence of CypA binding, the loop region, centered on Pro90, is flexible unless molecular or solvation interactions are formed, such as those found in the A subunit. Structural differences of the loops are highly localized so that the amino-terminal β-hairpin is visible in both monomers. These include hydrogen bonds between backbone amide of Pro1 to Oδ1 of Asp51 in one subunit and in the other, Pro1 to backbone oxygen of Gln13 and Oδ1 of Asp51.
High affinity intermolecular interactions in the FL CA APD
The APD arrangement found in the FL CA structures are maintained in both monoclinic and orthorhombic lattice systems and, despite changes in monomer orientation between the parallel and antiparallel dimer configurations, the extent of cross-monomer contacts formed in both forms are largely equivalent (Figure 2). Both dimers bury extensive regions upon oligomerization, forming considerable hydrophobic associations in both cases, even slightly greater in the APD configuration. Dimeric conformations similar to the APD configuration have been found in other retroviral CA structures, including a Fab-complexed CA (“RH24”, pdb ID code 1E6J; Berthet-Colominas et al, 1999), where the dimer is comprised of reciprocal NTD-CTD1 interfaces. The conservation of this dimeric organization in multiple lattices, using different protein constructs, decreases the possibility that the APD configuration is a crystallization artifact. Furthermore, the crystallization conditions for the current FL CA and the RH24 are different yet yielding siimilar dimer configuration. The capsid sequence in RH24, the Fab-CA fusion protein, differs from this FL CA in an N-terminal extension containing a His-tag plus 5 amino acids before Pro1 (thereby lacking the amino-terminal β-hairpin). Additionally RH24 C-terminal sequence is PGDLV while the FL sequence in this study is PGHKARVL, otherwise, the sequences are identical. It should be noted that in the FL CA, a cystine bond is formed between residues Cys198 and Cys218. However, the disulfide is unlikely to significantly perturb the structural fold, as native crosslinks have also been found in a number of other CA structures. The recurrence of the APD motif reflects the innate affinities of these surfaces for one another, likely intrinsic to retroviral capsids.
Figure 2.
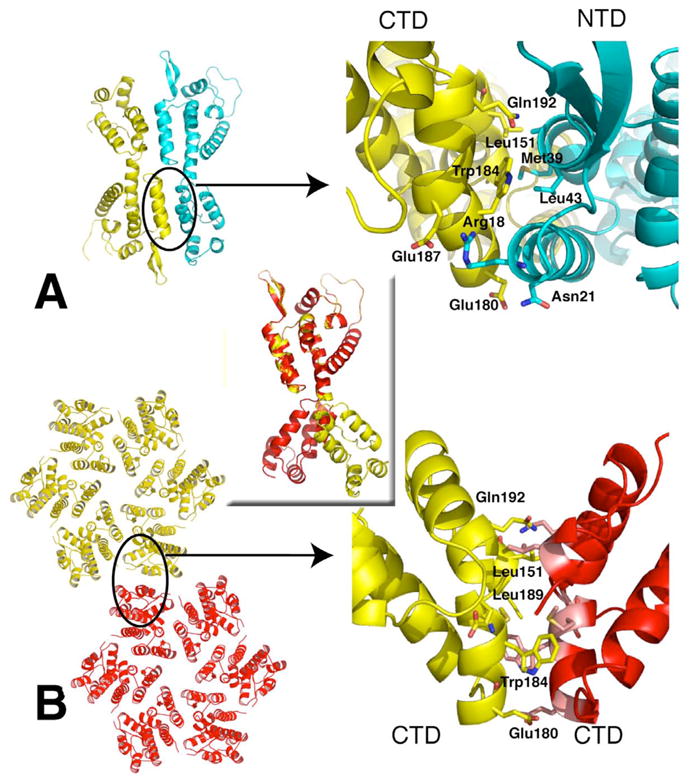
Relationships between CA domains in the FL CA compared to assembled CA.
Crystal asymmetric unit with one monomer shown in yellow the other in cyan (A). Close up view of the CTD-NTD interface with key residues labeled (top right). CryoEM reconstructed pseudo-atomic model from (3DIK) showing two hexamers (B). Proposed hexamer-hexamer interface based on modeling into EM maps isolated CTD dimer structures. Many of the key residues forming the CTD-CTD dimer interface in the EM model are also form NTD-CTD interactions in the FL CA structures. Rotation of 180 degrees is required to superimpose the CTD of a monomer subunit of the FL CA (yellow) onto the hexamer monomer (red; central inset).
Although the overall monomer organization is similar between the native FL CA and the RH24 Fab complex, significant differences are found at the hinge region connecting the NTD to CTD (Figure 1C). In the FL CA structure, the hinge loop motif joining NTD α-helix (H7) to the CTD 310 helix is shorter than the hinge loops of any other CA structures, including RH24 and the hexameric HIV CA15. In our APD conformation, the NTD terminates at Tyr145, joined to the CTD by a short linker loop comprised of Ser146-Pro147, leading into the CTD 310 helix. In the RH24 structure, unwinding of the 310 helix elongates the loop, increasing the separation between the NTD and CTD by an additional ~5 Å (measured between Cα of Tyr145-Ile150). Compared to the domain orientation in a monomer of a hexamerically-packed (accession number 3H47), the distance is increased by ~4 Å in the assembled state. Along with the extended separation between domains, intermolecular interactions between monomer subunits are diminished so that the distance (Cα to Cα) between the Pro34 of one NTD to the same residue in the other NTD is longer by ~4 Å in RH24 (Figure 1C). Among the interactions formed between residues on helix 1 of the NTD to residues on helix 9 of the CTD in the APD configuration of CA, are the pair of H-bonds formed between Arg18-Glu187’ and Arg21-Glu180’ (primes denote second monomer subnit), which stabilizes the domain orientation in the FL CA. These appear to be central to the conformational stability of the hinge angle found in the FL structure. The interactions between the Arg/Glu pair are further augmented by interactions between hydrophobic residues on helix 1 of the NTD and residues on helix 9 of the CTD (Figure 2A). The hinge angle between the NTD and CTD closes by about 30 degrees in the FL CA compared to RH24, decreasing the distance between the NTD-CTD, producing more intimate intra-domain and inter-molecular interactions in the FL APD than in the RH24 or any other dimeric CAs.
The tight intra- and inter-molecular associations in the APD conformation modify the monomer orientation but do not disrupt the overall domain folds. Pairwise differences over all Cα atoms between the individual NTD and CTD domains of the FL CA structures and various published atomic models are less than 3 Å (Table 2). In the NTD, the regions exhibiting greatest differences are at the amino-terminal hairpin, the CypA loop, and the inter-domain hinge region (elaborated further, below). The CTD exhibits greater variability, arising from discrete local changes in structural elements. Most isolated CTD dimers in crystal and NMR structures interact by reciprocal contacts of helix 9, displaying variations of helical crossing angles as the inter-helical contacts are mediated through predominantly hydrophobic interactions, such as between residues Trp184-Trp184’. Conformational variability of the CTD, especially in the helix8 – helix9 loop, was observed in the crystal structure of the hexameric CA 15. Superposition of the CTD in the FL CA onto the CTD dimer of 1A43, 1BAJ, or 1A8O, shows that the loop between helix 8 & 9 and the first part of helix 9 (residues 176 through 189) moves relative to the rest of the domain. Although the same residues, specifically Leu151, Trp184, Glu180, and Gln192, are involved in mediating intermolecular interactions in both FL and isolated CTD dimers (Figure 2A,B), changes in orientation result in dramatically different dimer interactions. Superposition of the NTD of the FL CA to the NTD of a monomeric subunit of a hexamer (3H47) shows a remarkable ~180° rotation of the CTD (Figure 2 inset). In the CTD dimer, this rotation would result in a clash with the corresponding CTD of the second subunit of the dimer so that the two orientations of the CTD domains, represented by the FL CA and the assembled 3H47, precludes the other, defining two distinct conformational states. These results suggest that the conformational flexibility of the CTD may promote formation of various dimeric states, with the most extensive intermolecular interactions found in the APD than in any other dimeric CA structures determined to date.
Table 2.
Pairwise Comparisons of Published CA Domain Structures to the FL CA
| A. NTD# | Structure Method & Residues Analyzed | RMSD Cα (Å) |
|---|---|---|
| 2PXR (Kelly et al., 2007) | NTD: by X-ray with CAP-1, 1 – 145 | 0.7 |
| 1AK4 (Gamble et al., 1996) | NTD: CypA complex, 3 – 145 | 1.0 |
| 1E6J (Berthet-Colominas et al., 1999) | NTD: from FL RH24 - Fab | 0.8 |
| 1GWP (Tang et al., 2002) | NTD: by NMR, 1 – 145, 2 models | 2.8, 2.3 |
| This work | FL CA: NTDA NTDB | 0.7 |
| B. CTD | ||
| 1A43 (Worthylake et al., 1999) | CTD: 146–231 | 1.7 |
| 1A8O (Gamble et al., 1997) | CTD: 151–231 | 2.0 |
| 1BAJ (Worthylake et al., 1999) | CTD: 146–229 plus P2 of GAG | 1.7 |
| 1E6J (Berthet-Colominas et al., 1999) | CTD: from FL RH24 - Fab | 1.5 |
| 2BUO (Ternois et al., 2005) | CTD: plus CAI | 1.7 |
| This work | FL CA: CTD-A CTD-B | 0.8 |
Excluding N-terminal β-hairpin (residues 5–9) and CypA-binding loop (residues 85–93)
EM analysis of nascent crystals
EM analysis of negatively stained samples collected from 3D crystallization drops shows the presence of tubular assemblies, 2D crystalline sheets, and small 3D crystalline nucleates (Figure 3). Two conditions were analyzed, using the nucleated, early-stage crystalline material grown from the same crystallization condition that yielded the single crystals used X-ray data collection (denoted as ‘condition 1’, solution components described in Experimental Methods). Another set of crystals was grown from the same crystallization solution minus iron (‘condition 1B’) and EM analysis was repeated, again on the nucleated, early-stage microcrystals. Tubular shaped assemblies were found at the edges of newly growing 3D rod-shaped crystals in condition 1, while 2D crystalline sheets coexist with tubules and 3D rod-shaped crystals in condition 1B (Figure 3a–d). Consequently, the EM analysis shows that early in the crystallization process, tubular-shaped assemblies coexist with the rod-shaped crystalline form that, with time, grow into the single crystals that are used for X-ray diffraction experiments. In condition 1B, an additional form is found, 2D sheets that coexist with the tubular-shaped assemblies and rod-shaped crystalline forms. These 2D sheets, typically framed by tubules, display hexameric packing of CA with unit cell parameters (axis length a=b=83.0 Å; Figure 3e–g) that are consistent with those reported for the assembled CA1.
Figure 3.

EM Analysis on Crystal Nuclei Showing initial 3D Crystals Coexisting with Multiple-layered Tubes at their Edges.
A projection image (a) of a small negatively stained 3D crystal after 12 hours crystallization. Fourier transforms from the crystal sheet (b, region 1) and from tubular region (c, region 2); respective models of presumed oligomers representing the diffraction images shown at bottom (h). A projection image (d) and its Fourier transform (insert) of a negatively stained multiplelayered tubular crystal. A low magnification image of a 2D crystal (e) edged with tubes and an enlarged view of the 2D crystalline sheet (f). The Fourier transforms of the 2D crystalline sheet (insert of f) and corresponding projection map (g) indicate packing of hexagonal lattice. Scale bars, 100nm in a and d; 50nm in f, 1um in e.
Compared to other published crystallization conditions, these crystals are grown at lower salt concentrations (i.e. 0.2M NaCI) than those generally required for in-vitro tube assembly17. Large single crystals grow from the initial crystalline rod-shaped nucleates and the presence of iron enhances growth whereas without iron, the nuclei do not grow beyond the very small microcrystals (<20 μm). These observations indicate that the tubular form is likely to be less thermodynamically stable than the crystalline form under condition 1, with the equilibrium shifting towards the 3D crystals with time and likely correlated with the stabilization of the hinge region.
Hydrophobic binding site and domain switch
A large tri-lobed area of strong positive (> 8σ) electron density in Fo-Fc maps was found in both the monoclinic and orthorhombic crystal forms, in an area of the NTD that is almost completely buried from solvent. Assessing the crystallization conditions and verified by calculating anomalous difference Fourier maps with delF coefficients from data collected at CuKα wavelength and phases from the protein-only model identified the tri-lobed density belonging to a triiodide (I3−) molecule (Figure 4A). The electron density for the I3− ligand shows three peaks in a linear array, with distances of 2.8–2.9 Å between peak centers, consistent with the geometrical parameters reported for I3− 14; 18. In the FL CA structure, the position of the triiodide anion completely overlaps with the position of Phe32 found in NTD in absence of any ligation (yellow Phe32, Figure 4A,B), which resembled the Phe32 conformation proposed when complexed with the inhibitor, CAP-1 19. The electron density maps generated from data collected from crystals of CAP-1-NTD complex did not show CAP-1 bound at this site due to its low affinity of binding so its binding was inferred from changes of CA residues at the binding site 19. In contrast, in our FL CA structure, there are very strong electron densities for the I3− ligand (Figure 4 panel). In the NTD-CAP-1 complex, the orientation of His62 is altered, which is assumed to propagate the binding event to other neighboring residues. In addition to the position of Phe32, the neighboring Ala31-Phe32 peptide bond exhibits an unfavorable cis conformation. The orientation of the Ala31-Phe32 peptide bond in some CA NTD structures exhibits a strained cis conformation, as seen in the CAP-1 complex 19. Characterization of the hydrophobic binding site indicates that this site may be primed for binding of small hydrophobic molecules such as I3−, with binding facilitated by release of peptide-bond strain induced in the unliganded state by the rotameric conformation of Phe32. Although the I3 and CAP-1 sites are in the same general area, the I3− molecule is deeply buried, forming extensive networks of H-bonds and hydrophobic interactions to residues at the hinge region, resulting in high-affinity binding between I3− to FL CA and limiting hinge angle mobility.
Figure 4.

The Triiodide (I3−) and inhibitor, CAP-1, Binding Sites are Located at the Inter Domain Hydrophobic Pocket Formed Between the NTD-CTD.
(A) A monomer of FL CA is shown in cyan, with dark blue residues labeled to show the hydrophobic side chains (except His64) surrounding the I3−, the difference density map at the site is shown (Fo-Fc, contoured at 6σ). (B) The position of Phe32 in the I3− bound crystal is shown in cyan, in CAP-1 NTD in magenta (2PXR) and the position of Phe32 in unliganded NTD structures is shown in yellow (1AK45). (C) A striking rotation of Tyr145 is found in the CAP-1 complex, where the aromatic ring is swung out of the pocket. (D) In the I3− complexed structure, Tyr-145 remains associated with the binding pocket, forming interactions to Gln176 and Met144, with each side-chains forming additional networks of H-bond interactions that bridge NTD and CTD, stabilizing domain orientation.
Beyond the similarities and subtle shifts of the residues surrounding the hydrophobic pocket, a most striking difference is in the conformation of the Tyr145, which remains associated with the NTD, abutting the site at one end (Figure 4C,D) with the I3− deeply bound. The position of Tyr145 in the CAP-1 structure has been suggested to indicate the mechanism of assembly-inhibition, preventing packing of adjacent subunits in the formation of the hexameric CA20. In the extra-domain conformation, Tyr145 has been proposed to sterically block the formation of intermolecular associations that produce the hexameric lattice. However, other factors likely contribute since Tyr145 is buried in our the FL CA structure and thermodynamic considerations support the shielding of Tyr145 from adopting solvent exposed conformations, an energetically unfavorable state. The position of CAP-1 is >11 Å away from Tyr145 so that its out-of-pocket conformation is unlikely due to direct displacement but through the combination of coordinated side-chain changes in conjunction with the absence of the CTD (Tyr145 is the terminal residue in 2PXR/2JPR). In the FL CA, a network of residues at the domain interface helps to maintain the conformation of Tyr145 (Figure 4D). Presuming that the position of Tyr145 signifies a steric inhibition mechanism, the finding that Tyr145 remains associated with the NTD in the FL CA I3− complex suggests another mechanism. An alternative explanation is that binding of molecules at the hinge region stabilizes an alternative dimeric conformation that is incompetent for, or impedes, assembly. The APD may represent a conformation that is induced when bound to CAP-1, a dimeric configuration that precludes the reciprocal interdomain associations required to form the hexameric lattice.
Induction of Dimeric State of CA by I3− Binding and Inhibition of Assembly In Vitro
To characterize the solution oligomeric states of CA in the presence of I3−, a series of dynamic light scattering analyses were done at various I3− concentrations, verified by a colorimetric assay20. The molecular weight of CA in the absence or presence of I3− was calculated from light scattering measurements and replicated in triplicate. Under the conditions of the analyses (details in Materials and Methods) both monomeric and dimeric forms of CA are found, with predominantly monomeric population in the absence of I3− binding, even at 37μM concentration (Table 3; Figure 5), consistent with the high salt requirement reported for in-vitro CA assembly.24; 28 In contrast, the binding of I3− to CA dramatically increased dimer formation, shifting the population to almost exclusively dimers even at CA concentrations of 17 μM (Table 3; Figure 5). For comparison, the effects of CAP-1 on the oligomeric state of CA were determined by DLS, using similar CA and salt concentrations as those used for characterization of I3− effects. In the presence of CAP-1, at 100 μM concentration to account for its lower CA binding affinity relative to I3−, a dimeric state of CA is found, emulating the effects of I3− and consistent with our proposal that binding of a compound at the interdomain hinge region, the binding site for both I3− and CAP-1, induces a dimeric state of CA.
Table 3.
Light Scattering Analysis for CA and CA-I3− complex.
| [CA] (μM) | [KI/I2] (μM) | Temperature (ºC) | MW (KDa) | Oligomeric state% |
|---|---|---|---|---|
| 17.4 | 0 | 21 | 28.0 | monomer |
| 17.4 | 16 | 21 | 51.0 | dimer |
| 37 | 0 | 21 | 35.0 | monomer-dimer |
| 37 | 40 | 21 | 56.0 | dimer |
| 37 | 80 | 21 | 62.2 | dimer |
The molecular weight of monomeric CA molecule is ~27 KDa.
Figure 5.

Light Scattering Analysis of CA in Solution in Absence and Presence of I3− & CAP-1.
To characterize the solution oligomeric state of CA in the absence and presence of I3−, a series of dynamic light scattering analysis were conducted. All measurements were done in 50 mM sodium phosphate buffer at pH 7.0. (A) CA at 17.4 μM concentration is monomeric; (B) CA at 37 μM is predominantly monomeric at low salt; (C) CA at 17 μM concentration with 16 μM I3−; (D) CA at 37 μM and 40 μM I3- resulted in the enhanced formation of the dimeric state, even at 17 μM concentration; (E) CA at 17 μM concentration with 100 μM CAP-1.
In the absence of other viral constituents, HIV-1 CA can assemble into tubes with structural features that resemble mature core 10; 18. Tube formation leads to increases in sample turbidity that can be monitored spectrophotometrically21; 22; 23 and this assay was used to probe for potential inhibitory effects of the I3−on in-vitro capsid assembly. The effects of I3− on CA assembly rates were determined and compared to two capsid assembly inhibitors, CAP-119 and a Pfizer compound PF-0345007424, referred to as “074”. The rates of CA assembly in-vitro are strongly dependent salt, pH, and protein concentrations, exhibiting higher-order concentration-dependencies that are indicative of nucleated, cooperative, and multi-state polymerization 24. The binding of I3− and CAP-1 decreased the rate of CA assembly, whereas 074 accelerated the rate, consistent with the effects of CAP-1 reported previously (Table 4) 25. The higher turbidity values in the presence of 074 are likely indicative of nonspecific aggregation rather than related to functional assembled CA.
Table 4.
Rates of Assembly in Presence of Inhibitors Determined by Turbidity Assay24
| Compound% | Compound Concentration (μM) | Assembly Rate (mOD/min) |
|---|---|---|
| I3− [KI/I2] | 0 | 100.1 |
| 25 | 49.8 | |
| 50 | 31.8 | |
| 100 | 25.8 | |
| CAP-1 | 0 | 103.7 |
| 100 | 77.2 | |
| 500 | 63.7 | |
| 074 | 0 | 97.3 |
| 25 | 134.7 | |
| 100 | Too fast to catch initial burst |
CA concentration in all assays is 37 μM
To ascertain whether the binding of I3−, CAP-1, and 074 modifies interactions within the oligomeric CA, the endogenous Trp fluorescence was monitored. The binding of I3− and CAP-1 would be predicted to decrease the fluorescence signal due to the known quenching properties of iodide species but not shift the emission peak wavelength if these compounds bound at the binding site identified in the APD conformation of CA. Binding of I3− and CAP-1 reduced the fluorescence emission peak signal in a concentration-dependent manner, as expected from the locations of the two Trp residues at the vicinity of the domain-interface, the hinge-pocket binding site (Figure 6). Even at increased concentrations of I3− and CAP-1, the intermolecular associations are not perturbed, in agreement with structural predictions that I3− and CAP-1 binding augments interactions at the dimer interface, stabilizing its conformation. In contrast to CAP-1 and I3−, binding of 074 causes a bathochromic shift in the fluorescence emission peak, suggesting increased solvation of Trp residues, consistent with the reported mechanism of inhibition of 074, which accelerates dissociation of CA to monomers, inducing aggregation to protect hydrophobic residues. The binding site of 074 has been reported to be between helices 3 and 4 in the NTD of CA25, a distinctly different site from the I3− and CAP-1 binding pocket.
Figure 6.

Fluorescence Analysis of FL CA in Presence of Inhibitors
The effects of I3− binding to assembly were characterized by fluorescence spectroscopy to determine if intermolecular associations were affected by binding and compared to two capsid assembly inhibitors, CAP-119 and a Pfizer compound PF-0345007424, referred to as “074”. These results are consistent with the reported mechanism of inhibition of 074, which accelerates dissociation of CA to monomers, which rapidly aggregates to protect the hydrophobic residues. The similarity in the fluorescence emission profile of CA in the presence of I3− and CAP-1 are also consistent with the structural results, which identified their binding sites to both be at the interdomain hinge region, supporting the idea that their binding promotes a NTD-CTD domain orientation in the FL CA that stabilizes a dimeric conformation that is not competent for assembly, such as the APD configuration.
The data generated by our light scattering, turbidity, and fluorescence analysis on the effects of I3− binding to CA, experiments that included published inhibitors, CAP-1 and 074, for comparative and control purposes, supports the inhibitory activity of I3− on CA assembly in solution. Our findings implicate an inhibitory mechanism whereby binding of I3− and CAP-1 promotes a NTD-CTD domain orientation in the FL CA that stabilizes a dimeric conformation that is incompetent for assembly, such as the APD conformation revealed in our crystal structure.
Discussion
While much attention has been focused on the structure of the assembled retroviral capsid, biochemical and biophysical data indicate that a major fraction of the CA protein within mature HIV-1 particles exist in an unassembled state. Analysis of the mass of immature HIV-1 particles led to an estimate of 5000 molecules of Gag per particle, approximately three times that required for assembling a conical capsid according to current structural models27. Furthermore, isolation of native HIV-1 cores from mature virions results in maximal recovery of only one-third of the viral CA protein, suggesting that two-thirds of the CA molecules are not associated with the assembled viral capsid. The concentration of CA within the virion is approximately 3 mM, yet the CA dimer affinity is approximately 18 μM 28. Thus, the dimer-hexamer equilibrium is likely to play a critical role in controlling assembly and disassembly of CA. Hydrogen-deuterium exchange studies have detected two subpopulations of CA that exhibit different exchange kinetics, suggestive of different protein-protein interactions 26. Kinetic analysis of CA domain interactions indicated the participation of cross NT- and CT-domains in solution during CA protein assembly and led to a prediction that mixed heterodimers inhibited polymerization, suggesting that an NT-domain-CT-domain interaction is involved in the assembly of both Rous sarcoma virus and HIV 28. The feasibility of perturbing the dimer-hexamer equilibrium by stabilizing a dimeric or otherwise unassembled CA state as a means of inhibition is supported not only from kinetic and thermodynamic considerations described above but further justified when temporal aspects are factored in.
The efficacy of perturbing the dimer-hexamer equilibria as a means of assembly inhibition is additionally justified by consideration of the nature and level of intermolecular contacts found in the various oligomeric and conformational forms. Analysis of the contacts formed in the APD relative to those found in the hexamerically- and pentagonally-packed CA indicate that these are largely equivalent. Comparison to those formed in parallel dimers modeled after the assembled structures show that the extents of intra- and inter-molecular contacts are even greater in the APD form. Consequently, the feasibility of stabilizing the CA in a conformation that is less productive or even ‘incompetent’ for assembly is supported by our structural results, consistent with the observed solution behavior of CA, and reasonable with regards to the molecular constraints of the assembly process.
The APD exhibits contacts between subunits utilize surfaces that also participate in intersubunit contacts in the CA hexamer; it is therefore potentially incompatible with HIV-1 capsid assembly. The observation that these crystals grow from a solution containing both crystalline and tubular assembled forms signifies that the CA hinge angle must be somewhat meta-stable under these conditions (Figure 3). Our electron diffraction studies reveal the presence of both dimer and hexameric CA forms in the same crystallization well, suggesting that these multiple oligomeric forms can coexist in solution, interconverting by domain rotation. Consequently, inducing domain organizations that enhances assembly-incompetent forms, by stabilizing intra- and intermolecular contacts such as those formed in the APD shown here, may conformationally-trap the CA so that assembly can be impeded or largely inhibited.
The finding that a triiodide anion is bound at the hinge region substantiates the likelihood of promoting distinct conformational and oligomeric states in CA. In the APD crystal structure, the triiodide anion, a novel ligand not previously reported to interact with CA, occupies a small cavity at an interdomain hinge site. Its binding site overlaps with the putative CAP-1 binding site although notable differences in their interactions (I3− ~>103 higher affinity than CAP-1€) may provide valuable clues to development of small molecules with desired CA binding characteristics. While the CAP-1 molecule is poorly active as an HIV-1 inhibitor, it is still capable of disrupting capsid assembly, in-vitro, despite inabilities to detect the inhibitor in complex with the CA structure reflecting weak affinities. These findings suggest that in-vitro determinants of binding interactions, such as affinity values, may not directly correlate to in-vivo inhibitory activities, as we found here for capsid assembly inhibitors. While triiodide has ~ a thousand fold greater binding affinity for CA than CAP-1, the activities of both compounds in turbidity assays are similar (Table 4). This apparent discrepancy may be due to technical limitations of the turbidity assay (ie narrow dynamic range due to high CA concentration), the complexities of the kinetics involved in the assembly process, and possible differences the uptake or membrane permeability of the compounds in-vivo. While further detailed studies will be required to fully identify and elucidate the complex molecular and dynamic facets governing capsid assembly, our model of conformationally-trapped CA in an assembly-incompetent dimeric state induced by small molecule binding provide an alternative avenue of investigation for inhibitor development.
The commonality of binding sites and in-vitro inhibitory activities indicate that compounds that target the hinge region-binding site may elicit similar antiviral effects. Based on the structural results, binding at the hydrophobic pocket initiates a ‘domain switch’, preventing free rotation of the domains about the hinge to favor the APD configuration. The plausibility of conformationally trapping the CA in an assembly-incompetent APD state is further supported by our spectroscopy results, showing that the binding of triiodide and CAP-1 prevented assembly, by stabilizing the CA in predominantly dimeric state, providing additional solution evidence that supports the crystallographic and electron diffraction findings.
Unlike enzymes that have targetable functional substrate binding sites, the CA protein does not have a known binding site that affects catalytic or other innate activities, which can be more readily targeted in drug development efforts. The structural characterization of a potential allosteric binding site in HIV-1 CA can be highly valuable. We suggest that binding at the H-site promotes transition to conformational states inhibitory to normal CA assembly functions. The APD conformation captured in our FL CA complex may represent one such “assembly-incompetent” conformation, possibly explaining the mechanism of inhibition elicited by CAP-1. Consequently, detailed structural and solution characterization on domain interactions and ligand contacts at the H-site are directly relevant and highly promising for inhibitor development. Design of ligands that promote conformations such as those seen in the APD structure will likely impede, or altogether prevent, the intra- and intermolecular associations required for generating hexameric and/or pentameric lattices. The H-site may be an unexpected allosteric switch in CA, where binding at this hinge region triggers side-chain rotameric transitions that are readily propagated to interdomain residues leading to domain reorganization of the NTD/CTD that ultimately results in assembly-competent (eg parallel) or incompetent (eg APD) form and, thus, represents a key rate-limiting step in capsid formation. We propose that the APD found in the FL CA complex is a ‘conformationally-trapped’ state, induced by triiodide binding at the interdomain H-site, and that these results reveal an unprecedented means of preventing CA assembly. Studies to identify and design novel small molecules targeting the H-site, using the contacts formed by the triiodide ligand as optimizable parameters, may yield additional knowledge valuable for developing effective HIV-1 antivirals with desirable pharmaceutical properties.
Experimental Methods
Protein Expression and Purification
cDNA of CA was cloned from pDAB72 encoding gag polyprotein, pr55gag. The reagent was obtained through NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. Briefly, the cDNA was amplified and subcloned into pET21 (EMD chemicals, Inc. San Diego, CA) using NdeI and XhoI sites. Proteins were expressed in E. coli, Rosetta 2 (DE3), cultured in Luria-Bertani media, with 0.4 mM IPTG at 23 ºC for 16 h. Soluble forms of CA proteins were extracted using sonication and purified over 5 mL Hi-Trap QP (GE Healthcare, Piscataway, NJ) with a buffer containing 25mM sodium phosphate, 1 mM DTT, 0.02% sodium azide. The fractions containing CA were pooled and the pH was adjusted to 5.8 before purification with 5 mL Hi-Trap SP (GE Healthcare, Piscataway, NJ) using 0 – 1 M NaCl gradient. The protein peak was further purified over Hi-Load Superdex200 16/60 (GE Healthcare, Piscataway, NJ) equilibrated with a buffer containing 25 mM sodium phosphate, 100 mM NaCl, 1 mM DTT, and 0.02% sodium azide. CA was concentrated to 10 mg/mL using Amicon cocentrators (Millipore, Billerica, MA) before stored at − 80ºC after flash-frozen with liquid N2. Molecular mass of purified CA was confirmed by LC-TOF mass spectrometry (Bruker Daltonics, Billerica, MA).
Crystallization
FL-CA was concentrated to 11 mg/mL and screening was done in 96-well trays with a Cartesian Honeybee robot, using commercial (Hampton Research, Qiagen, Emerald Biosystems) and in-house crystallization screening kits. Showers of tiny crystals of full-length CA were obtained from conditions containing 25% PEG3350, 0.2 M sodium iodide, and 0.1 M BTP pH 6.5, at 6.4 mg/mL protein. Large crystals were obtained after optimization using additive screens; it was found that addition of 10 mM FeCl3 gave rise to larger rod-shaped crystals which took over two weeks to complete growth. Plate-like crystals were also found, growing from the same clusters that the rods grew out of. The two crystal forms in the crystallization medium are orthorhombic plate form (P212121, a=53.1 Å, b=87.7 Å, c=103.4 Å) and a monoclinic rod form (P21 a=47.9 Å, b=86.5 Å, c=55.6 Å, β=99.73°) (Table 1). The orange rod-shaped crystals grow in multiples; often there are small amounts of satellite crystals.
Cryoprotection and data collection
Crystals were dipped briefly in paraffin oil for cryoprotection, as these crystals are sensitive to manipulation and changes in solution conditions. An interesting observation was that repeated “in-situ annealing” 29; 30 in the cryostream of oil-covered crystals that still had aqueous cloudy areas around them (ice formation) gradually reduced the opaque areas and reduced the ice diffraction – without negatively affecting the protein diffraction.
Molecular replacement and refinement
Molecular replacement using PHASER 31 with various search models for the capsid NTD and CTD separately determined. Rotation and translation search solution of the N-terminal domain was obtained from the cyclophilin-complex structure (1ak4) while the C-terminal domain solution was found with 1A43. Four searches were carried out to position the CA domains in the asymmetric unit: – two NTD and two CTD. The first 14 amino acids (the β hairpin) and the Cyp-binding loop (87 – 97) were removed from the search model. PHASER translation function Z-scores were above 30 for three of the four domains in the asymmetric unit, with a Z-score of 14 for the fourth, and log-likelihood gains were large and positive. The structure was manually rebuilt using COOT 32 and refined with PHENIX 33. Ions were assigned based on peak heights, temperature factors, and the presence of a peak in an anomalous difference Fourier (iodine atom) or no peak (Fe atom).
EM analysis of HIV-CA tubular assemblies and 3D crystals
3D crystallization drops (~1μl) under the respective crystallization conditions were quickly diluted 1:5 with the crystallization buffer and negatively stained with 1% uranyl acetate (pH5) on carbon-coated grids. Low dose (20 – 30 e−/Å2) projection images were recorded on a Gatan 4K x4K CCD camera using a Tecnai F20 electron microscope at a nominal magnification of 50,000 and under focus values ranging from 1 μm to 2 μm. HIV-FL CA proteins (~10 mg/ml) were diluted to 2 mg/ml in high salt buffer (50mM Tris pH8.0, 1M NaCl) and incubated at 37°C for 1hr for tubular assembly. Samples (3–5 μl) were applied to glow discharged perforated Quantifoil grids (Quantifoil Micro Tools, Jena, Germany), blotted from back side with a filter paper and plunge-frozen in liquid ethane using a manual gravity plunger. Low dose (10 e−/Å2) projection data of tubes suspended over a thin layer of vitreous ice were collected using a Tecnai Polara microscope at 200KV cooled at liquid nitrogen temperature, Images were recorded on a Gatan 4Kx4K CCD camera at a nominal magnification of 59,000 and under focus values ranging from 1.5 μm to 2.5 μm.
Light Scattering Analysis
Purified full-length (FL) wild type (wt) capsid (CA) at concentrations of 17.4 and 37 μM in 50 mM sodium phosphate buffer at pH 7.0 were prepared for analysis at 22 ºC. The liability of iodine in solution produces various ionic and molecular states of iodine, with a strong dependency on pH and redox conditions. Triiodide were generated in-situ by mixing KI and I2 in a ratio of 1.5:1, with the [KI/I2] representing the total of [I2] and [I3−] species in solution. The concentration of I3− generated from KI/I2 at various pH and buffer conditions were determined by a colorimetric assay20. Solution conditions maintaining neutral pH, limiting light exposure, and degassing all solutions to purge dissolved oxygen stabilized the generated I3− species, so that dissolution of I3− was negligible during the duration of the analysis. Prior to light scattering measurements, aliquots of I3− at known concentrations were added to CA, mixed, and measurements were taken immediately. The light scattering measurements were carried out by Dynapro Plate Reader of Wyatt and analyzed with the program “Danamics V6”.
In vitro Capsid Assembly
Turbidity assays were performed at 21 °C using a Molecular Devices’ SPECTRAmax PLUS384 UV/VIS microplate spectrophotometer operating at 350 nm wavelength. Concentrated I3− (generated as described for light scattering analysis), CAP-1 (in DMSO), and 074 (in DMSO) were added to an aqueous solution containing 50 mM Na phosphate pH 7.0 and the capsid protein. Particulates were removed by centrifugation and capsid assembly initiated by adding concentrated NaCl solution (3.6 M) in 50 mM Na phosphate pH 7.0 to a final concentration of 2.25 M NaCl. Spectral measurements were made every 20 s, following a short initial delay to allow sample equilibration. Relative assembly rates were estimated from initial slopes of the plots of absorbance versus time.
Kinetic analysis of assembly reactions
For kinetic analysis, assembly reactions were performed at 30 μM CA protein and 2.25 M NaCl. After the addition of buffer containing 50 mM Na phosphate pH 7.0 with 2.25 M NaCl to initiate assembly, the reaction was rapidly mixed and placed into a cuvette. Approximately 20 s elapsed between the time of addition and the first time point measured. The increase in optical density was monitored on a Molecular Devices’ SPECTRAmax PLUS384 UV/VIS microplate spectrophotometer at 350 nm every 20 s for 1 h. To estimate the rate of assembly, the increase in turbidity at initial time points were fit to a straight line by using SoftMax Pro 4.8 software.
Fluorescence Emission Spectroscopy
An excitation scan was done to determine the optimal excitation wavelength of tryptophan in CA. A solution of CA at 40 μM in 50 mM sodium phosphate buffer pH 7.0 was scanned from 250 nm to 450 nm and the excitation peak for tryptophan in CA was found at 291 nm. CA was diluted to 17μM in 1.5 ml of 50 mM sodium phosphate buffer pH 7.0. Aliquots of 074, KI/I2, and CAP-1 were added to the CA solution for each fluorescence measurements and replicated in triplicate at 17 μM and 37 μM concentrations. All intrinsic emission fluorescence spectra were measured using 291 nm as the excitation wavelength on a Photon Technology International EasyLife LS Fluorescence Lifetime System spectrophotometer and scanned from 294 nm to 350 nm.
Accession Numbers
The coordinates and structure factors for the monoclinic data set have been deposited in the Protein Data Bank with the accession number 3NTE.
Acknowledgments
Financial support for this work was provided by NIH grants GM82251 and AI76121 and the Pennsylvania Department of Health. We thank the staff members at the General Medicine and Cancer Institutes Collaborative Access Team (GM/CA-CAT) and the Southeast Regional Collaborative Access Team (SER-CAT), both at the Advanced Photon Source, Argonne National Laboratory for access and technical assistance. GM/CA-CAT has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104). Use of the Advanced Photon Source is supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Footnotes
KD 8.9 μM μM, determined by SPR (data not shown)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ganser-Pornillos BK, Yeager M, Sundquist WI. The structural biology of HIV assembly. Curr Opin Struct Biol. 2008;18:203–17. doi: 10.1016/j.sbi.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang YF, Wang SM, Huang KJ, Wang CT. Mutations in capsid major homology region affect assembly and membrane affinity of HIV-1 Gag. J Mol Biol. 2007;370:585–97. doi: 10.1016/j.jmb.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 3.Chu HH, Chang YF, Wang CT. Mutations in the alpha-helix directly C-terminal to the major homology region of human immunodeficiency virus type 1 capsid protein disrupt Gag multimerization and markedly impair virus particle production. J Biomed Sci. 2006;13:645–56. doi: 10.1007/s11373-006-9094-6. [DOI] [PubMed] [Google Scholar]
- 4.Cardone G, Purdy JG, Cheng N, Craven RC, Steven AC. Visualization of a missing link in retrovirus capsid assembly. Nature. 2009;457:694–8. doi: 10.1038/nature07724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ganser-Pornillos BK, Cheng A, Yeager M. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell. 2007;131:70–9. doi: 10.1016/j.cell.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 6.Wright ER, Schooler JB, Ding HJ, Kieffer C, Fillmore C, Sundquist WI, Jensen GJ. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J. 2007;26:2218–26. doi: 10.1038/sj.emboj.7601664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berthet-Colominas C, Monaco S, Novelli A, Sibai G, Mallet F, Cusack S. Head-to-tail dimers and interdomain flexibility revealed by the crystal structure of HIV-1 capsid protein (p24) complexed with a monoclonal antibody Fab. EMBO J. 1999;18:1124–36. doi: 10.1093/emboj/18.5.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. X-ray structures of the hexameric building block of the HIV capsid. Cell. 2009;137:1282–92. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, Hill CP. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell. 1996;87:1285–94. doi: 10.1016/s0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- 10.Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, Hill CP. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science. 1997;278:849–53. doi: 10.1126/science.278.5339.849. [DOI] [PubMed] [Google Scholar]
- 11.Ivanov D, Tsodikov OV, Kasanov J, Ellenberger T, Wagner G, Collins T. Domain-swapped dimerization of the HIV-1 capsid C-terminal domain. Proc Natl Acad Sci U S A. 2007;104:4353–8. doi: 10.1073/pnas.0609477104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Momany C, Kovari LC, Prongay AJ, Keller W, Gitti RK, Lee BM, Gorbalenya AE, Tong L, McClure J, Ehrlich LS, Summers MF, Carter C, Rossmann MG. Crystal structure of dimeric HIV-1 capsid protein. Nat Struct Biol. 1996;3:763–70. doi: 10.1038/nsb0996-763. [DOI] [PubMed] [Google Scholar]
- 13.Worthylake DK, Wang H, Yoo S, Sundquist WI, Hill CP. Structures of the HIV-1 capsid protein dimerization domain at 2.6 A resolution. Acta Crystallogr D Biol Crystallogr. 1999;55:85–92. doi: 10.1107/S0907444998007689. [DOI] [PubMed] [Google Scholar]
- 14.Evans G, Bricogne G. Triiodide derivatization in protein crystallography. Acta Crystallogr D Biol Crystallogr. 2003;59:1923–9. doi: 10.1107/s0907444903012897. [DOI] [PubMed] [Google Scholar]
- 15.Pornillos OG-PB, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. X-ray structures of the hexameric building block of the HIV capsid. Cell. 2009;137:1282–92. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phillips JM, Murray PS, Murray D, Vogt VM. A molecular switch required for retrovirus assembly participates in the hexagonal immature lattice. EMBO J. 2008;27:1411–20. doi: 10.1038/emboj.2008.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.von Schwedler UK, Stemmler TL, Klishko VY, Li S, Albertine KH, Davis DR, Sundquist WI. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. EMBO J. 1998;17:1555–68. doi: 10.1093/emboj/17.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans G, Bricogne G. Triiodide derivatization and combinatorial counter-ion replacement: two methods for enhancing phasing signal using laboratory Cu Kalpha X-ray equipment. Acta Crystallogr D Biol Crystallogr. 2002;58:976–91. doi: 10.1107/s0907444902005486. [DOI] [PubMed] [Google Scholar]
- 19.Kelly BN, Kyere S, Kinde I, Tang C, Howard BR, Robinson H, Sundquist WI, Summers MF, Hill CP. Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J Mol Biol. 2007;373:355–66. doi: 10.1016/j.jmb.2007.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verhaeghe E, Buisson D, Zekri E, Leblanc C, Potin P, Ambroise Y. A colorimetric assay for steady-state analyses of iodo- and bromoperoxidase activities. Analytical Biochemistry. 2008;379:60–65. doi: 10.1016/j.ab.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 21.Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. Assembly and analysis of conical models for the HIV-1 core. Science. 1999;283:80–3. doi: 10.1126/science.283.5398.80. [DOI] [PubMed] [Google Scholar]
- 22.Li S, Hill CP, Sundquist WI, Finch JT. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature. 2000;407:409–13. doi: 10.1038/35030177. [DOI] [PubMed] [Google Scholar]
- 23.Lanman J, Sexton J, Sakalian M, Prevelige PJ. Kinetic Analysis of the Role of Intersubunit Interactions in Human Immunodeficiency Virus Type I Capsid Protein Assembly In Vitro. Journal of Virology. 2002;76:6900–6908. doi: 10.1128/JVI.76.14.6900-6908.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi J, Morris R, Irving S, Zhou J, Butler S, Pickford C, Whitby K, Aiken C. A small molecule HIV-1 inhibitor that destabilizes the viral capsid. Abstract SADR 2009 Conference 2009 [Google Scholar]
- 25.Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, Barklis E, Sun Y, Huang M, Summers MF. Antiviral inhibition of the HIV-1 capsid protein. J Mol Biol. 2003;327:1013–20. doi: 10.1016/s0022-2836(03)00289-4. [DOI] [PubMed] [Google Scholar]
- 26.Lanman J, Lam T, Emmett M, Marshall A, Sakalian M, Prevelige PJ. Key interactions in HIV-1 maturation identified by hydrogen-deuterium exchange. Nat Struct Mol Biol. 2004;11:676–7. doi: 10.1038/nsmb790. [DOI] [PubMed] [Google Scholar]
- 27.Briggs J, Simon M, Gross I, Kräusslich H, Fuller S, Vogt V, Johnson M. The stoichiometry of Gag protein in HIV-1. Nat Struct Mol Biol. 2004;11:672–675. doi: 10.1038/nsmb785. [DOI] [PubMed] [Google Scholar]
- 28.Yoo S, Myszka DG, Yeh C, McMurray M, Hill CP, Sundquist WI. Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J Mol Biol. 1997;269:780–95. doi: 10.1006/jmbi.1997.1051. [DOI] [PubMed] [Google Scholar]
- 29.Vahedi-Faridi A, Stojanoff V, Yeh JI. The effects of flash-annealing on glycerol kinase crystals. Acta Crystallographica. 2005;D61:982–989. doi: 10.1107/S0907444905012746. [DOI] [PubMed] [Google Scholar]
- 30.Yeh JI, Hol WGJ. A Flash-Annealing Technique to Improve Diffraction Limits and Lower Mosaicity in Crystals of Glycerol Kinase. Acta Crystallographica. 1998;D54:479–480. doi: 10.1107/s0907444998004697. [DOI] [PubMed] [Google Scholar]
- 31.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]