

Abstract
Glioblastomas are cytogenetically heterogeneous tumors that frequently display alterations of chromosomes 7, 9p, and 10q. We used high-density (500K) single-nucleotide polymorphism arrays to investigate genome-wide copy number alterations and loss of heterozygosity in 35 primary glioblastomas. We focused on the identification and detailed characterization of alterations involving the most frequently altered chromosomes (chromosomes 7, 9, and 10), the identification of distinct prognostic subgroups of glioblastomas based on the cytogenetic patterns of alteration for these chromosomes, and validation of their prognostic impact in a larger series of tumors from public databases. Gains of chromosome 7 (97%), with or without epidermal growth factor receptor (EGFR) amplification, and losses of chromosomes 9p (83%) and 10 (91%) were the most frequent alterations. Such alterations defined five different cytogenetic groups with a significant effect on patient survival; notably, EGFR amplification (29%) was associated with a better survival among older patients, as confirmed by multivariate analysis of a larger series of glioblastomas from the literature. In addition, our results provide further evidence about the relevance of other genes (eg, EGFR, CDKN2A/B, MTAP) in the pathogenesis of glioblastomas. Altogether, our results confirm the cytogenetic heterogeneity of glioblastomas and suggest that their stratification based on combined assessment of cytogenetic alterations involving chromosomes 7, 9, and 10 may contribute to the prognostic evaluation of glioblastomas.
Gliomas are a heterogeneous group of malignant tumors that show variable localization, histopathologic features, and genetic profiles, together with a heterogeneous response to therapy but a uniformly fatal outcome.1–11 Although no common genetic signature has been detected in all gliomas, multiple chromosomal changes have been described so far, which frequently include gains of chromosome 7 and deletions of chromosomes 9 and 10 and to a less extent also of chromosomes 1 and 19.12–14 These genetic changes are associated with amplification of oncogenes [eg, epidermal growth factor receptor (EGFR)] together with deletion and/or mutation of tumor suppressor genes [eg, tumor protein p53 (TP53), phosphatase and tensin homolog (PTEN), and cyclin-dependent kinase inhibitor 2A (p16/CDKN2A)].15
Altogether, these results point out the potential involvement of different signaling pathways in gliomas, with alterations of chromosome 7, 9, and 10 participating in the most frequent tumor subtypes (eg, glioblastoma). In line with this hypothesis, we have recently shown the existence of distinct cytogenetic pathways in gliomas, by using interphase fluorescence in situ hybridization (iFISH) analysis of intratumoral patterns of chromosomal alterations, at the single-cell level.16 Notably, specific genomic aberrations and cytogenetic profiles are associated with particular tumor histopathologic features.17–19 Accordingly, amplification (or rearrangement) of EGFR is almost restricted to a fraction of all malignant gliomas, particularly glioblastomas. Among these cases, overexpression of the EGFR variant 3 mutant is most frequently detected.20,21 Although this mutant protein is unable to bind to its ligands, it constitutively signals, conferring proliferation and survival advantages to tumor cells.20–22 In turn, genomic deletions of chromosomes 9 and 10 at regions that harbor tumor suppressor genes are also typically found in glioblastomas, where they have been associated with the development of the tumor, its progression, and a poor prognosis.23–26 Interestingly, monosomy 10 is associated with gain or amplification of the EGFR gene on chromosome 7p11.2, supporting the role of both alterations in gliomagenesis.27,28 Other genetic abnormalities that can be frequently found in low-grade gliomas29,30 [eg, combined del(1p)/del(19q) and TP53 mutation] are less frequently detected in glioblastomas.31–35
In the past, most studies devoted to the identification and characterization of genetic/chromosomal alterations in glioblastomas have used conventional cytogenetic and molecular techniques associated with relatively low-resolution (eg, iFISH and comparative genomic hybridization). Recently, high-density single-nucleotide polymorphism (SNP) arrays have been used to characterize the most frequent genetic alterations of glioblastomas.36–51 Newly available high-density SNP arrays allow the study of copy number (CN) changes and loss of heterozygosity (LOH) at both coding and noncoding DNA regions of the whole tumor cell genome, with high resolution; this provides a more precise map of the genetic alterations associated with CN changes in glioblastomas. Thus, SNP array studies performed in large series of patients with or without gene expression profiling have provided new insights into the potential role of new candidate genes (eg, ERBB2, NF1, and TP53), molecular changes (eg, PIK3R1 and PDGFRA/IDH1 mutations), and signaling pathways into the pathogenesis of glioblastomas.40 In turn, based on gene expression profiles, a molecular classification of glioblastomas has been proposed that reflects the involvement of different neural lineages.42 To the best of our knowledge, however, no classification based on the genetic changes involving the most frequently altered chromosomes (eg, 7, 9, and 10) has been proposed so far for glioblastomas.
We used high-density (500K) SNP array to investigate genome-wide CN alterations and LOH in a group of 35 primary glioblastoma patients; we focused on the identification and detailed characterization of the genetic alterations of those chromosomes more frequently altered in these tumors and the identification of groups of glioblastomas with distinct cytogenetic patterns of alteration for these 3 chromosomes, which are potentially associated with the behavior of the disease. Finally, the prognostic value of the presence of amplification of the EGFR gene was confirmed in a larger number of cases from four different independent series of glioblastoma patients, which have been previously reported in the literature.41,42,45,50
Materials and Methods
Patients and Samples
A total of 70 paired tumor (n = 35) and peripheral blood (PB; n = 35) samples from 35 patients (15 men and 20 women) diagnosed as having glioblastomas (mean ± SD age, 60 ± 14 years; age range, 30 to 84 years) who were admitted to the Neurosurgery Service of the University Hospital of Coimbra (Coimbra, Portugal) were included in this study. Before entering the study, each patient gave written informed consent to participate, and the study was approved by the Hospital's Ethics Committee. Of the 35 patients, 5 underwent complete resection of the tumor; either partial resection or just a diagnostic biopsy was performed in the other 30 cases (Table 1). Distribution according to tumor localization was as follows: 16 tumors were localized in the frontal lobe, 12 were temporal, 3 were parietal, 2 were occipital, 1 was insular, and 1 had a deep localization. Tumors were diagnosed and classified by an experienced neuropathologist according to the World Health Organization criteria.3 At the time of closing the study, all patients had died, with a median overall survival of 11 months (range, 1 week to 67 months).
Table 1.
Clinical Features of the 35 Study Patients Diagnosed as Having Glioblastoma Multiforme
| Case no. | Age, years | Sex | Tumor localization | Number of relapses | Survival after surgery, months⁎ | Karnofsky index, % | Surgical removal |
|---|---|---|---|---|---|---|---|
| G14 | 69 | Female | Frontal | 0 | 0 | 70 | B |
| G35 | 50 | Female | Frontal | 0 | 2 | 50 | ST |
| G54 | 65 | Female | Parietal | 0 | 6 | 60 | ST |
| G31 | 71 | Female | Frontal | 0 | 7 | 80 | ST |
| G43 | 67 | Female | Temporal | 0 | 7 | 70 | ST |
| G30 | 71 | Female | Temporal | 0 | 9 | 60 | B |
| G8 | 67 | Female | Deep | 0 | 9 | 90 | ST |
| G45 | 76 | Female | Temporal | 0 | 10 | 60 | ST |
| G29 | 49 | Female | Parietal | 0 | 12 | 90 | B |
| G63 | 61 | Female | Insular | 0 | 13 | 60 | ST |
| G41 | 44 | Female | Frontal | 0 | 14 | 60 | B |
| G23 | 50 | Female | Frontal | 0 | 14 | 80 | B |
| G40 | 45 | Female | Frontal | 1 | 15 | 80 | ST |
| G10 | 35 | Female | Temporal | 0 | 15 | 80 | ST |
| G55 | 54 | Female | Frontal | 1 | 17 | 80 | ST |
| G62 | 57 | Female | Occipital | 1 | 18 | 90 | T |
| G39 | 70 | Female | Frontal | 1 | 18 | 70 | ST |
| G6 | 70 | Female | Temporal | 1 | 19 | 80 | ST |
| G13 | 39 | Female | Frontal | 1 | 21 | 90 | ST |
| G17 | 30 | Female | Temporal | 3 | 67 | 80 | ST |
| G12 | 74 | Male | Temporal | 0 | 1 | 70 | B |
| G51 | 60 | Male | Temporal | 0 | 2 | 60 | B |
| G42 | 67 | Male | Temporal | 0 | 2 | 80 | ST |
| G46 | 62 | Male | Frontal | 0 | 3 | 60 | ST |
| G34 | 69 | Male | Temporal | 0 | 5 | 80 | B |
| G15 | 79 | Male | Parietal | 0 | 5 | 80 | T |
| G25 | 68 | Male | Frontal | 0 | 7 | 70 | ST |
| G57 | 34 | Male | Frontal | 0 | 8 | 90 | T |
| G64 | 57 | Male | Occipital | 0 | 8 | 60 | ST |
| G50 | 84 | Male | Temporal | 0 | 11 | 70 | ST |
| G56 | 65 | Male | Frontal | 0 | 13 | 80 | ST |
| G52 | 56 | Male | Frontal | 0 | 21 | 90 | B |
| G44 | 48 | Male | Frontal | 0 | 22 | 80 | ST |
| G53 | 74 | Male | Frontal | 0 | 29 | 60 | T |
| G37 | 70 | Male | Temporal | 1 | 32 | 80 | T |
B, biopsy; ST, subtotal; T, total.
At the moment of closing this study, all patients had died.
Representative parts of fresh tumor tissues left after routine diagnostic histopathologic procedures had been performed were immediately snap-frozen in liquid nitrogen and stored at −80°C, until used for iFISH and DNA extraction for SNP array studies. In each case, a section cut from the tissue block used for this purpose was histologically assessed to estimate tumor cell contents. Specimens with 75% or more tumor cells in the absence of contamination by normal brain parenchyma and tumor necrosis were systematically selected for further DNA extraction and SNP array studies.
DNA Extraction and SNP Array Hybridization
DNA from both frozen tumor tissues and their paired PB leukocyte samples was purified using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. DNA yield and purity were determined with a NanoDrop-1000 spectrophotometer (Nano-Drop Technologies Inc., Wilmington, DE). DNA integrity was evaluated by conventional electrophoretic procedures in 1% agarose gel.
DNA samples were processed according to the Mapping 500K Array Set (Affymetrix Inc., Santa Clara, CA) protocol with two arrays, each containing 250,000 SNPs, with a mean intermarker distance of 5.8 kb (250K Nsp and Sty arrays). Briefly, total DNA (250 ng per array) from paired tumor and PB samples was separately digested with the NspI and StyI restriction enzyme and ligated to the corresponding adaptors that recognize overhangs generated by the restriction enzymes. All digested DNA fragments were then used as substrates for adaptor ligation, regardless of their size. A generic primer that recognizes the adaptor sequence was used in triplicate to amplify adaptor-ligated DNA fragments through PCR. The amplified DNA was then fragmented, labeled, and hybridized to the GeneChip Human Mapping 250K Nsp or Sty arrays. After hybridization, the chips were washed and the hybridized sequences were labeled using streptavidin-phycoerythrin and assayed by fluorescence detection. Arrays were washed in an Affymetrix Fluidics Station 450 and scanned using a GeneChip Scanner 3000 (Affymetrix). The allelotype at a locus was then determined based on probe-associated fluorescence intensity data for complementary oligonucleotides to the reference sequences covering the corresponding SNP position.
Identification of CN Alterations and LOH
Identification of CN alterations and LOH was based on the analysis of a total of 500,568 SNPs for paired tumor and normal PB DNA samples. A total of 140 “.CEL” files containing data on the SNP arrays (one for each type of chip) for each type of sample (paired tumor and PB DNA) were obtained for the 35 glioblastomas using the Affymetrix GCOS software (version 1.3). The Copy Number Analysis Tool (CNAT v 4.0; Affymetrix) and the dChip 2007 software52 (Dana Farber Institute, Harvard, MA, http://www.dchip.org, last accessed June 1, 2011) were used to calculate CN values and plot them according to chromosome localization. Genotypes were generated using the BRLMM algorithm included in the Genotyping Console software (version 3.0.2; Affymetrix). Normal PB samples with cutoff values of 1.30 or less and 2.50 or more (arbitrary units) were used to establish CN losses and gains. In addition, the CNAG software (version 3.3.01, The University of Tokyo, Tokyo, Japan)53 was used to explore the state of each of the two alleles corresponding to each chromosome to distinguish between homozygous and heterozygous deletions.
LOH was defined by the presence of homozygous alleles in tumor DNA samples for alleles that were heterozygous in normal PB DNA from the same individual, and it was classified as follows: LOH by true allelic imbalance (loci at which one of the two parental copies of a chromosome is deleted) or copy neutral LOH (cnLOH) (tumor DNA showing two copies of a chromosome region from one allele in the absence of the other allele and a CN value of two).
To confirm further our findings, an additional series of 119 patients with primary glioblastomas, whose tumors had been analyzed by SNP arrays (100K, 250K, and 500K Affymetrix SNP arrays) and reported in the literature, with data on such analyses being available in an individual patient basis, were included in this study. These additional patients corresponded to a total of five different series, with data on four of them being accessed from public databases (access codes: GSE19612,42 E-MEXD-1330,45 and GSE963550), whereas for the other series, it was kindly provided by the authors.41
From these five series of glioblastomas, cases with secondary glioblastomas, tumors with simultaneously normal CN values for chromosomes 7, 9, and 10, and patients lacking survival data and/or showing low SNP call rates in the array file (<90%) were excluded from the analysis.
iFISH Studies
Confirmatory iFISH studies were performed in all cases, according to previously described methods, using dual-color probes directed against different regions of chromosomes 7, 9, and 10. Three genes (EGFR, p16, and PTEN) and three chromosome centromeres (7, 9, and 10) were tested with the following commercially available probes, all obtained from Vysis Inc. (Downers Grove, IL), except the 7p12 (EGFR)/alphasatellite 7 DNA dual-color probe, which was obtained from Q-BIOgene (Carlsbad, CA); for chromosome 9, the LSI 9p21/CEP-9 dual-color probe was used, and for chromosome 10, the LSI PTEN/CEP-10 dual-color probe was used.
Statistical Analyses
To establish the statistical significance of differences observed between groups, the Student's t-test and the Mann-Whitney U-test were used for parametric and nonparametric (continuous) variables, respectively; for qualitative variables, the χ2 test was applied (SPSS software version 15.0, SPSS Inc, Chicago, IL). Survival curves were plotted according to the method of Kaplan and Meier, and the log-rank test was used to assess the statistical significance of differences observed in survival between distinct groups of patients (SPSS software). For the identification of those parameters with an independent prognostic impact on patient overall survival, the Cox regression was used; in the multivariate analysis only those variables that showed a significant impact in the univariate analysis (age and cytogenetic profile) were included. Patient overall survival was measured from the date of diagnosis until the date of death. P < 0.05 were considered to be associated with statistical significance.
Results
CN Changes in Glioblastomas by SNP Arrays
SNP array studies showed genetic alterations for all chromosomes in the 35 cases studied; such alterations involved either entire chromosomes or specific chromosomal regions (Figure 1). Overall, CN changes showed predominance of gains of chromosomes 7 and 20, losses of chromosomes 4, 6, 9p, 10, 15, and 17, and both gains and losses of chromosomes 1, 3, 9, 19, and 22. As could be expected, chromosomes 7, 9p, and 10 were those chromosomes more frequently altered: gains of chromosome 7 were found in all but one case (97%) and losses of chromosomes 9p and 10 were identified in 83% and 91% of all glioblastomas analyzed (Figure 1). A more detailed description of the genetic alterations found for these three chromosomes is shown in Table 2 and detailed below.
Figure 1.
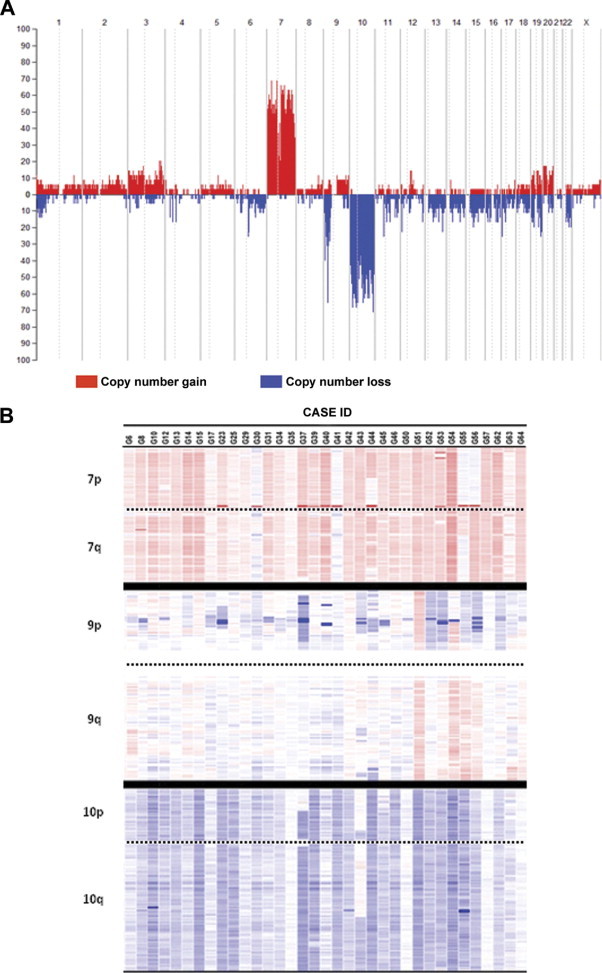
Frequency of CN gains (red areas) and losses (blue areas) along the tumor cell genome of 35 glioblastomas. A: An overview of the frequency of CN changes detected for each individual chromosome is shown. B: A heat map representation of the CN changes detected for chromosomes 7, 9, and 10 is displayed for each case analyzed. The intensity of colors is directly proportional to the frequency of genetic gains (red color) or losses (blue color) identified for each specific chromosomal region. Cutoff values for chromosome gains and losses were defined at CN values of 2.50 or greater and 1.30 or less, respectively.
Table 2.
Frequency of Different Patterns of CN Alterations for Chromosomes 7, 9, and 10 in Glioblastomas as Detected by SNP Arrays (n = 35)
| Chromosome | Genetic alteration | No. of cases/total cases (%) |
|---|---|---|
| 7 | No alterations | 0/35 (0) |
| CN gains | 34/35 (97) | |
| +7 | 23/35 (65) | |
| +7 and EGFR amplification | 9/35 (26) | |
| EGFR amplification | 1/35 (3) | |
| +7 and other amplifications | 1/35 (3) | |
| cnLOH⁎ | 1/35 (3) | |
| 9 | No alterations | 4/35 (11) |
| CN gains† | 2/35 (6) | |
| CN losses | 28/35 (80) | |
| Heterozygous del(9p) | 4/35 (11) | |
| Heterozygous del(9p) and del(9q) | 2/35 (6) | |
| Heterozygous del(9p) and +9q and 9p gains‡ | 2/35 (6) | |
| Heterozygous and homozygous del(9p) | 12/35 (34) | |
| Heterozygous and homozygous del(9p) and cnLOH§ | 2/35 (6) | |
| Heterozygous and homozygous del(9p) and del(9q) | 2/35 (6) | |
| Heterozygous and homozygous del(9p) and +9q¶ | 1/35 (3) | |
| Heterozygous and homozygous del(9p) and +9q and cnLOH | 2/35 (6) | |
| Monosomy 9 | 1/35 (3) | |
| cnLOH⁎ | 1/35 (3) | |
| 10 | No alterations | 3/35 (9) |
| CN gains | 0/35 (0) | |
| CN losses | 28/35 (80) | |
| −10 | 22/35 (63) | |
| −10 and homozygous del(10q) | 4/35 (11) | |
| del(10p) and del(10q) | 2/35 (6) | |
| cnLOH⁎ | 4/35 (11) |
cnLOH involving the whole chromosome.
Chromosome 9 gains without losses of this chromosome.
Gain of 9p24.3 in one tumor and other gains of 9p21.1 in another case.
cnLOH of chromosome 9p was detected in four cases; however, only two are included here (G54 and G55) because the other two cases had +9q21. In one case cnLOH involved the whole chromosome 9.
Two case have cnLOH (G54, G55).
CN Changes of Chromosomes 7, 9, and 10
Gains of chromosome 7 were found in all but one tumor (G41) and consisted of the gain of an entire chromosome (n = 33; 94%) and EGFR amplification (n = 10; 29%). Most of these later cases (n = 9/10) also carried gains of a whole chromosome 7. One tumor showed cnLOH of chromosome 7 (Table 2). Two cases (6%) presented amplification 7p in association with cnLOH, involving a whole chromosome 7 in one case and chromosome 7p in the other tumor. Figure 2A delineates the amplified segments at the 7p11.2 chromosomal region and their extension. As displayed, the amplified regions at chromosome 7p11.2 were variable in size, with a mean ± SD length of 908,412 ± 281,717 bp (range, 554,485 to 1284,332 bp) (Figure 2A). A more detailed analysis of the amplified 7p11.2 regions in each individual tumor showed that they typically involved a region systematically containing the EGFR gene, in association or not with another two genes: LANCL2 (n = 5/10) and ECOP (n = 1/10) genes (Figure 2A).
Figure 2.
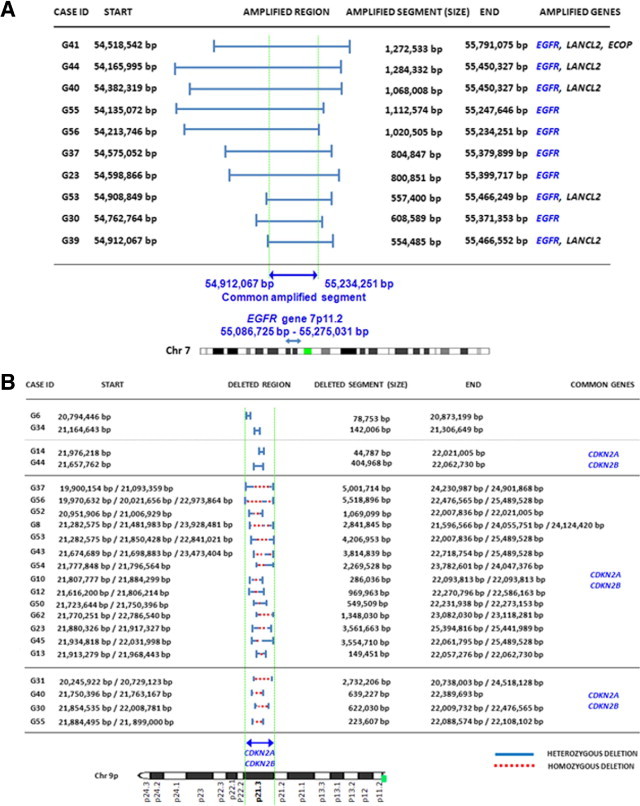
Detailed characterization of the amplified 7p11.2 chromosomal segments (n = 10, A) and the deleted chromosome 9p21.3 sequences (n = 22, B) in glioblastomas. Note that the EGFR gene extends from 55,086,725 bp to 55,275,031 bp from pter positions (Entrez Gene, GeneID1956); in the 500K SNP array, the SNPs assayed in this region of chromosome 7p11.2 extended from the 55,062,691-bp to the 55,236,410-bp positions. Regarding del(9p21) no single gene was systematically deleted; however, both CDKN2A and CDKN2B were lost in all except one case.
Allelic loss of chromosome 9p21 was the most common alteration found for chromosome 9 (27/35 cases; 77%); in addition, monosomy 9 in association with homozygous del(9p21) was detected in one case (case G17; 3%), and cnLOH of an entire chromosome 9 was found in another case (case G57, 3%) (Table 2; see also Supplemental Table S1 at http://jmd.amjpathol.org). Seven tumors showed gains of chromosome 9q, consisting of partial gains (n = 5; cases G6, G14, G54, G55, G56, and G63) or gain of an entire chromosome 9 (n = 1; case G51); some of these cases (n = 5/7) showed additional coexisting losses of chromosome 9p (cases G6, G14, G54, G55, and G56). A more detailed analysis of chromosome 9 sequences in cases with del(9p) revealed a wide spectrum of allelic losses regarding the size of the deleted regions, ranging from 44,787 to 5518,896 bp. Overall, deletions within the short arm occurred much more frequently than in the long arm of chromosome 9 (n = 27 versus 4 cases), with several different patterns: i) heterozygous del(9p) (n = 8) (cases G41, G39, G6, G14, G29, G34, G64, and G35); ii) combined heterozygous and homozygous del(9p) (n = 15) (cases G23, G53, G44, G56, G30, G37, G13, G52, G62, G12, G45, G8, G10, G43, and G50); and iii) cnLOH combined with heterozygous and homozygous del(9p) (n = 4) (cases G55, G40, G31, and G54). From those cases showing cnLOH with or without del(9p) (n = 5), complete loss of chromosome 9p was found in 3 cases (9%); the other two glioblastomas had cnLOH involving the whole chromosome 9, in association with heterozygous and homozygous del(9p21) in one tumor (case G40) (Table 2; see also Supplemental Table S1 at http://jmd.amjpathol.org). Despite all these patterns, cases with del(9p21.3) (n = 22) or monosomy 9 (n = 1) almost systematically displayed in common loss of the CDKN2B and the CDKN2A genes (21 of 22 cases), in association with loss of the MTAP gene in 15 cases (Table 3). Other frequently deleted genes included the MLLT3 (4/35 cases), KIAA1797 (6/35 tumors), PTPLAD2 (5/35 cases), IFNA4 (6/35 patients), IFNA14 (6/35 cases), KLHL9 (7/35 tumors), and ELAVL2 (9/35 cases) genes (Table 3). One additional tumor (G17) presented loss of an entire chromosome 9 in association with homozygous del(9p21) also involving the CDKN2A/CDKN2B genes.
Table 3.
Detailed Characterization of the Localization and Deletion Size of 9p and Commonly Lost Chromosomal Segments at 9p21.3 Detected in Glioblastomas
| Type of deletion | Case ID | Deleted segment |
No. of deleted genes | Deleted genes |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Start (bp) | End (bp) | MLLT3 | KIAA1797 | PTPLAD2 | IFNA4 | IFNA14 | KLHL9 | MTAP | CDKN2A | CDKN2B | ELAVL2 | |||
| Heterozygous (n = 4) | G6 | 20,794,446 | 20,873,199 | 10 | ∘ | |||||||||
| G34 | 21,164,643 | 21,306,649 | 6 | ∘ | ∘ | |||||||||
| G14 | 21,976,218 | 22,021,005 | 3 | ∘ | ∘ | |||||||||
| G44 | 21,657,762 | 22,062,730 | 16 | ∘ | ∘ | |||||||||
| Heterozygous & homozygous (n = 14) | G37 | 19,649,652 | 24,901,868 | 128 | ∘ | ∘ | ∘ | • | • | • | • | • | • | • |
| G56 | 19,970,632 | 26,131,011 | 10 | • | • | • | • | • | • | • | • | • | • | |
| G52 | 20,951,906 | 22,021,005 | 53 | ∘ | • | • | • | • | • | • | • | |||
| G8 | 21,282,575 | 24,124,420 | 7 | ∘ | ∘ | ∘ | ∘ | ∘ | ||||||
| G53 | 21,282,575 | 22,021,005 | 146 | ∘ | • | • | • | |||||||
| G43 | 21,674,689 | 26,027,837 | 32 | • | • | • | • | |||||||
| G54 | 21,777,848 | 24,047,376 | 4 | • | • | • | • | |||||||
| G10 | 21,807,777 | 22,093,813 | 3 | ∘ | • | • | ||||||||
| G12 | 21,616,200 | 22,586,163 | 6 | • | ∘ | ∘ | ||||||||
| G50 | 21,723,644 | 22,273,153 | 3 | • | • | • | ||||||||
| G62 | 21,770,251 | 23,118,281 | 10 | ∘ | ∘ | ∘ | ||||||||
| G23 | 21,880,326 | 25,441,989 | 33 | • | • | • | ||||||||
| G45 | 21,934,818 | 26,741,666 | 5 | ∘ | ∘ | • | ||||||||
| G13 | 21,913,279 | 22,062,040 | 2 | • | • | |||||||||
| cnLOH plus Heterozygous & homozygous (n = 4) | G31 | 20,245,922 | 24,518,128 | 10 | ∘ | • | ∘ | ∘ | ∘ | ∘ | ∘ | ∘ | ∘ | ∘ |
| G40 | 21,750,396 | 22,389,693 | 3 | • | • | • | ||||||||
| G30 | 21,854,535 | 22,476,565 | 30 | ∘ | ∘ | ∘ | ||||||||
| G55 | 21,884,495 | 22,108,102 | 3 | • | • | |||||||||
Deleted genes are noted with a circle; genes for which homozygous deletions were observed are noted with a solid circle. Case G17 presented monosomy 9 and homozygous losses from 9p22.1 to p21.3. (n = 22/35 cases).
Genetic losses of chromosome 10 consisted of monosomy 10 in 26 of 35 glioblastomas (74%) in association with homozygous del(10q) in 4 cases (11%), isolated del(10p) coexisting with del(10q) in two cases (6%), and cnLOH for the entire chromosome 10 in four cases (11%); three tumors (9%) did not show any CN change for chromosome 10, and gains of chromosome 10 were systematically absent (Table 2; see also Supplemental Table S1 at http://jmd.amjpathol.org).
Cytogenetic CN Profiles of Glioblastomas According to the Alterations of Chromosomes 7, 9, and 10
On the basis of the pattern of CN alterations observed for chromosomes 7, 9, and 10, glioblastomas were grouped into five distinct cytogenetic profiles (Table 4; see also Supplemental Table S1 at http://jmd.amjpathol.org): i) tumors exhibiting amplification of the EGFR gene (n = 10; 29%); ii) glioblastomas with gains of chromosome 7, losses along chromosome 10, and del(9p) or cnLOH 9 (n = 17; 48%); iii) tumors displaying gains of chromosome 7 without monosomy 10 (n = 3; 9%); iv) tumors that had gains of chromosome 7 and monosomy 10, in the absence of del(9p21.3) (n = 4; 11%); and v) tumors with gains of an entire chromosome 9 (n = 1; 3%).
Table 4.
Cytogenetic Subgroups of Glioblastomas as Defined by the CN Alterations Detected by SNP Arrays for Chromosomes 7, 9, and 10 and Association With Patient Overall Survival (n = 35)
| Pattern | Chromosomal abnormalities |
No. of cases (%)⁎ | Overall survival (range)† | ||
|---|---|---|---|---|---|
| Chr7 | Chr9 | Chr10 | |||
| I | EGFR AMP | del(9p21) | −10 or del(10p) and del(10q) or cnLOH | 10/35 (29) | 16 (9–32) |
| II | +7 | del(9p) or cnLOH | −10 or del(10p) and del(10q) or cnLOH | 17/35 (48) | 8 (0–21) |
| III | +7 | del(9p21) or +9q | No monosomy 10 | 3/35 (9) | 13 (11–67) |
| IV | +7 | Normal 9p21.3 | −10 | 4/35 (11) | 4 (2–7) |
| V | +7 | +9 | −10 | 1/35 (3) | 2 |
AMP, amplification.
Results are expressed as number of cases/total cases analyzed with percentages.
Median (range) overall survival in months.
Overall, no clear association was found between these cytogenetic profiles and other clinical variables, including tumor localization, except for patient survival. Accordingly, despite the dismal outcome observed in all cases, the cytogenetic profile of the tumor (as defined by the cytogenetic pattern of CN alterations observed for chromosomes 7, 9, and 10 by SNP arrays) had a significant effect on overall survival (P < 0.0001) (Figure 3A): tumors with EGFR gene amplification exhibited the longest (P = 0.005) survival rates versus all other cases (Figure 3B and Table 4), specifically among older (>60-year-old) patients (P = 0.01). This also holds true when patients undergoing complete tumor resection and those not undergoing resection were separately considered (data not shown).
Figure 3.
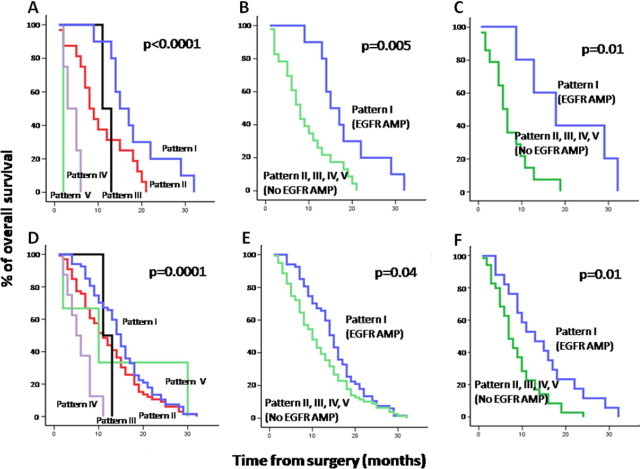
Overall survival curves of glioblastoma patients (n = 35) from our patients (A–C) and our patients plus cases from five other series (154 total cases) from the literature (D–F), according to the different cytogenetic patterns of alteration detected for chromosomes 7, 9, and 10 (A and D) and the presence of EGFR amplification in the whole series (B and E) and among patients older than 60 years (C and F). Cytogenetic patterns by SNP array studies corresponded to the following profiles: pattern 1, EGFR amplification; pattern 2, +7/del(9p) or cnLOH 9/−10 or del(10p) with del(10q); pattern 3, +7/without monosomy 10; pattern 4, +7/−10/absence of del(9p21.3); and pattern 5, +7 and + 9.
The presence and frequency of these five cytogenetic groups were then confirmed among the 119 glioblastoma cases collected from other series in the literature: pattern 1, 61 of 119 cases (51%); pattern 2, 57 of 119 (45%); pattern 3, 0 of 119 (0%); pattern 4, 4 of 119 (3%); and pattern 5, 2 of 119 cases (2%) (see Supplemental Table S2 at http://jmd.amjpathol.org). In addition, the impact of the cytogenetic profiles on overall survival was also confirmed when the whole series of glioblastoma patients (n = 154) was evaluated, both when the five different cytogenetic subgroups were considered (P = 0.0001; Figure 3D) and when cases with EGFR amplification were compared with all other cases in the whole series (P = 0.04; Figure 3E) and among older (>60-year-old) patients (P = 0.01; Figure 1F). Noteworthy, amplification of the EGFR gene (P = 0.01) together with age (P = 0.04) emerged as the best combination of independent variables to predict overall survival in the multivariate analysis.
CN Alterations by SNP Arrays Versus iFISH Analyses
CN changes identified by SNP arrays for chromosomes 7, 9, and 10 were confirmed in most cases by iFISH studies. However, discrepancies were observed by iFISH in two tumors (see Supplemental Table S3 and Supplemental Figure S1 at http://jmd.amjpathol.org). One of these cases showed tetrasomy and trisomy for chromosome 9p (n = 1) but a diploid profile by SNP arrays, whereas the other displayed coexistence of nulisomy 9p plus monosomy 9p and monosomy 10 by iFISH but an SNP array profile with both gain and loss of small regions of chromosome 9 and a diploid 10q23 profile.
Discussion
In recent years, increasingly heterogeneous genotypic profiles have been identified in glioblastomas. Accumulating evidence indicates that such variability reflects progressive acquisition and accumulation of multiple combined genetic events in single cells, accounting for gliomagenesis and potentially also for the behavior of the disease. Detailed characterization of common genetic changes in single chromosomes and shared genetic profiles in individual tumors will contribute to the identification of commonly altered genes and molecular profiles for a better understanding of the molecular mechanisms of the disease and its variable biological, histopathologic, and clinical features. Overall, chromosomes 7, 9, and 10 have been reported as those more frequently altered in glioblastomas.46,50,51,54 Despite this, to the best of our knowledge, no study has attempted to classify glioblastomas on the basis of the distinct patterns of combined alterations of these three chromosomes.
We used high-density SNP arrays for detailed characterization of those CN changes and genotypic profiles involving the three most frequently altered chromosomes in a group of 35 cases of glioblastomas. Overall, our results show that with very few exceptions, SNP arrays allow detection of underlying genetic changes of chromosomes 7, 9, and/or 10 whenever specimens contain 75% or more tumor cells, as confirmed by iFISH studies. Through this approach, we confirmed the existence of previously reported genomic abnormalities for these three chromosomes.13–15 In addition, use of high-resolution SNP arrays allowed accurate and detailed delineation of those sequences affected by CN changes (eg, gains, amplifications, and homozygous or heterozygous deletions) and allelic imbalances (eg, LOH and cnLOH) and identification of the specific genes involved; overall, five different patterns of combined alterations for these three chromosomes were observed.
Noteworthy, gains of chromosome 7 were identified in virtually every case. This highlights the relevance of complete gains of this chromosome in the development of glioblastomas and the potential pathogenic contribution of multiple oncogenes coded in it. In addition, we also confirm and extend on previous observations that have suggested that EGFR is the most frequently amplified oncogene in glioblastomas5,11,55,56 because EGFR was the only oncogene found to be amplified in common in cases with multiple copies of the 7p11.2 chromosomal region. Interestingly, precise localization of the EGFR amplicons revealed amplification of other adjacent genes in many cases, particularly the LANCL2 gene. Noteworthy, from those genes involved in 7p11.2 amplification, only EGFR showed increased expression in gene expression profiling,7,57 further supporting the unique and relevant role of this oncogene in glioblastomas versus the other genes (eg, LANCL2).
Regarding chromosome 9, more heterogeneous patterns of CN changes were observed, from which heterozygous and/or homozygous del(9p) was the most common alteration. Interestingly, although homozygous deletions were restricted to relatively small sequences of chromosome 9p21, heterozygous del(9p) extended to larger chromosomal regions. Notably, common deleted segments at 9p21 almost systematically involved the CDKN2B/p15 tumor suppressor gene in association with CDKN2A/p16 and the MTAP housekeeping genes. Del(9p21) is known to play an important role in the development and progression of many different types of cancer through deregulation of cell cycle and/or apoptosis.58 The CDKN2A locus has been claimed to play a crucial role in this regard. CDKN2A codes for two gene products, p16 and p14, that control both the Rb and the p53 pathways; p16 binds to CDK4 and CDK6 and inhibits the catalytic activity of CDK/cyclin D complexes to activate cell cycle through RB phosphorylation. In turn, p14 blocks MDM2 inhibition of p53 activity, thereby leading to stabilization of p53.11 Because deletion of the CDKN2A/B locus causes deregulation of two crucial pathways involved in many types of cancer, loss of the MTAP gene activity could be viewed as potentially irrelevant. However, deficiency of the MTAP protein (an enzyme involved in the metabolism of methionine and purines) has also been detected in multiple types of malignant neoplasms in association with deletion of the CDKN2A and CDKN2B loci,59 as also found in our glioblastoma cases. Most interestingly, it has been shown that MTAP can be lost independently of CDKN2A/p16, which suggests that loss of MTAP may indeed play a role in tumor biology.60–62
Taken together, these results raise the question about which of these three genes is/are critical target genes in glioblastomas. On the basis of our results, CDKN2A/p16 and CDKN2B/p15 are the most frequently altered in cases with heterozygous and homozygous deletions in line with previous large-scale multidimensional analyses performed by The Cancer Genome Atlas Research Network.40 The CDKN2B gene (p15; INK4b) is located adjacent to p16 (INK4a) on 9p21 and is co-deleted in a high proportion of human cancers. p15 (INK4b) is a member of the family of cyclin-dependent protein kinases that inhibits CDK4B. Because expression of CDKN2B is induced by transforming growth factor β, p15 may act as an effector of the transforming growth factor β–mediated cell cycle arrest pathway. In line with our results, data from both mutational and functional studies indicate that CDKN2B/p15 deletion could likely be the target of del(9p21).63
Regarding the specific mechanism by which these genes are inhibited, LOH at 9p21 was a relatively rare event, whereas combined homozygous and heterozygous deletions (associated or not with cnLOH events) were relatively common in our and other studies64; this finding suggests that all three genes (CDKN2A/p16, CDKN2B/p15, and MTAP) may be inactivated in glioblastomas by a large deletion event. In line with this hypothesis, a large mapping study of 545 primary tumors65 showed that tumors containing homozygous del(9p21) minimally have a 170-kb region deleted that includes both the MTAP and p16 loci, as also found here. However, homozygous deletion does not seem to be the only mechanism leading to inactivation of these tumor suppressor genes in glioblastomas because cases with heterozygous deletions were also found at higher frequency in our study. In another study on 85 brain tumor samples of different histologic features and grade, CDKN2B/p15 and CDKN2A/p16 genes were found to be methylated in only 4% and 7% of the cases, respectively; interestingly, CDKN2A was methylated only in glioblastoma samples (6% of the cases), and none of the samples showed simultaneous methylation of both the p15 and p16 genes66; this finding suggests that methylation of these genes does not play a major role in the development of glioblastomas. Interestingly, however, gene expression profiling of glioblastomas shows a significant impact on the expression of CDKN2A in cases with not only homozygous but also heterozygous del(9p21), whereas this does not affect the expression of the other two genes (data not shown). In any case, point mutations of these genes should be investigated in parallel in these cases. Because emerging CN analyses of glioblastoma samples confirmed the CDKN2A/CDKN2B locus to be the most common homozygous deletion at 9p21, detailed characterization of the deletion at chromosome 9p21 and the lost genes becomes particularly relevant. In this study, detailed mapping of the 9p21.3 region shows distinct patterns and extents of del(9p21) among the tumors analyzed. In addition, our results also show that the deleted locus encompassed not only genes with well-established tumor suppressor functions in glioblastomas but also multiple other less known genes (eg, the ELAVL2, MLLT3, KIAA1797, PTPLAD2, and KLHL9 genes). These findings strengthen the hypothesis that suggests the presence of additional candidate tumor suppressor genes mapped to this region.67
Overall, approximately three-quarters of all glioblastomas analyzed showed chromosome 10 losses, which most frequently consisted of monosomy 10 and cnLOH of an entire chromosome 10. These findings point to the loss of more than one tumor suppressor gene, localized both in the short and the long arms of this chromosome. In this regard, extensive losses of chromosome 10 sequences have been associated with progression of astrocytoma,68 and several regions along this chromosome (eg, 10q23, 10q24, 10q25-26, 10p13, and 10p14-p15) have been consistently proposed to harbor tumor suppressor genes (eg, the PTEN/MMAC1, DMBT1, and LGI1 genes).69 Although it has been previously suggested that the PTEN gene could be a preferential target of del(10q),70 in our series, losses of chromosome 10 mainly involved the entire chromosome. Despite this, our results highlight the fact that other regions at 10q11.21, 10q21.3, and 10q.23.33 (with loss of the HNRPF, PAKDB, and CUL2 genes, the CXXCC, CCPRL1, STOX1, and DDX50 genes; and the IRE gene, respectively) were more frequently lost and could act as potential preferential targets of deletion in glioblastomas. Likewise, those genes encompassed within these deleted loci could also represent novel candidate tumor suppressor genes involved in glioblastoma tumorigenesis, in addition to PTEN.
In this study, as in other larger series of glioblastomas,14,18,19,27,71 gains of chromosome 7 and losses of chromosomes 9 and 10 frequently coexisted in the same tumor, but different patterns were observed for these abnormalities. Accordingly, glioblastomas that exhibited EGFR gene amplification also showed extensive losses of chromosome 10, del(9p21), and trisomy 7 in all but one case. Conversely, in more than half of the cases, monosomy 10 coexisted with trisomy 7 in the absence of EGFR gene amplification with or without del(9p21). Altogether, these findings suggest that these alterations may occur independently from each other, with EGFR amplification appearing to be a later event in the development of glioblastomas versus trisomy 7 and monosomy 10. Nevertheless, their combination could be crucial in the malignant transformation process for which the underlying mechanism is still poorly understood. In this regard, several candidate genes in chromosome 10 with putative reciprocal relationship to EGFR have been identified, with great emphasis on the PTEN gene. Complementary deregulation of the EGFR and PTEN pathways often results in constitutional signaling through PI3-kinase and Akt, leading to altered cell proliferation and survival.72 A recent study by Yadav et al28 also suggests a tumorigenic synergism between loss of the annexin A7 (ANXA7) gene at 10q21.1-q21.2 and EGFR amplification, with ANXA7 haploinsufficiency acting as a positive regulator of EGFR signaling in glioblastomas. This study also demonstrates a cross-talk among the ANXA7, PTEN, and EGFR genes, which leads to constitutive activation of the PI3K-AKT signaling pathway and, ultimately, to malignant transformation. Taken together, these findings suggest that cytogenetic profiles, more than isolated chromosomal alterations, should be considered in evaluating the impact of CN alterations in disease behavior.
On the basis of CN alterations of chromosomes 7, 9, and 10, five different genetic profiles were identified in our series and confirmed to be present in other series from the literature41,42,45,50 from which cases with amplification of the EGFR gene, in association with monosomy 10 and del(9p21), clearly showed a better outcome in our 35 cases and when data on 119 additional glioblastoma patients from four previously reported series41,42,45,50 were considered. Controversial results have been reported about the prognostic value of EGFR amplification/overexpression in glioblastomas. Although some authors claim there is no association with survival,73,74 others state that this aberration is a negative prognostic factor.75,76 In turn, an association between EGFR overexpression and a better prognosis in older glioblastoma patients has also been reported,25,26,33,77 in line with our observations. Noteworthy, we did not find an association between tumor cytogenetics and other disease characteristics, such as patient age76 and tumor localization, among other features.55,78
Simmons et al78 and Batchelor et al5 have previously found that EGFR overexpression is associated with a trend toward a worse prognosis in young patients and a better outcome in older cases; likewise, in a series of 220 primary glioblastomas Houillier et al55 also documented an association between EGFR amplification and increased survival in older patients, which could be associated with the existence of additional as-yet-unidentified specific molecular alterations in older patients. In the present study, we confirm the prognostic value of EGFR amplification in patients older than 60 years in our small patient series and in a larger series of patients from four independent studies previously reported in the literature.5,55,78,79
In summary, our high-density analysis of the CN alterations of chromosomes 7, 9, and 10 disclosed five subgroups of patients defined by unique cytogenetic profiles, which are associated with patient outcome, with tumors with EGFR amplification showing a longer overall survival among older patients. In addition, our results provide further evidence about the relevance of the EGFR, CDKN2A/B, and MTAP genes, together with other genes coded in chromosome 10, in the malignant transformation of glioblastomas. Further studies in larger series of glioblastoma patients are necessary to investigate the functional interaction between these genes and more precisely delineate their pathogenetic role and clinical impact in glioblastomas.
Acknowledgments
We thank Dr. Pim J. French (Josephine Nefkens Institute, Department of Neurology, Erasmus Medical Center, Rotterdam, The Netherlands) for his valuable collaboration with 21.CEL files and patient survival data.41
Footnotes
Supported by the Portuguese Foundation for Science and Technology (FCT) grant PIC/IC/83108/2007, FCT PhD fellowships SFRH/BD/23086/2005 and SFRH/BD/11820/2003, and the Spanish Network of Cancer Research Centers (Red Temática de Investigación Cooperativa en Cáncer) grant RD06/0020/0035 from the Instituto de Salud Carlos III, Ministry of Science and Innovation, Madrid, Spain.
Supplemental material for this article can be found at http://jmd.amjpathol.org or at doi: 10.1016/j.jmoldx.2011.06.003.
The authors did not disclose any relevant financial relationships.
Supplementary data
Complete SNP array profiles for the discrepant karyotypic findings between SNP arrays and iFISH, detected in chromosome 9 (cases G15 and G63; A) and chromosome 10 (case G63; B).
References
- 1.DeAngelis L.M. Brain tumors. N Engl J Med. 2001;344:114–123. doi: 10.1056/NEJM200101113440207. [DOI] [PubMed] [Google Scholar]
- 2.Malmer B., Iselius L., Holmberg E., Collins A., Henriksson R., Gronberg H. Genetic epidemiology of glioma. Br J Cancer. 2001;84:429–434. doi: 10.1054/bjoc.2000.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Louis D.N., Ohgaki H., Wiestler O.D., Cavenee W.K., Burger P.C., Jouvet A., Scheithauer B.W., Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamborn K.R., Chang S.M., Prados M.D. Prognostic factors for survival of patients with glioblastoma: recursive partitioning analysis. Neuro Oncol. 2004;6:227–235. doi: 10.1215/S1152851703000620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Batchelor T.T., Betensky R.A., Esposito J.M., Pham L.D., Dorfman M.V., Piscatelli N., Jhung S., Rhee D., Louis D.N. Age-dependent prognostic effects of genetic alterations in glioblastoma. Clin Cancer Res. 2004;10:228–233. doi: 10.1158/1078-0432.ccr-0841-3. [DOI] [PubMed] [Google Scholar]
- 6.Bussiere M., Hopman W., Day A., Pombo A.P., Neves T., Espinosa F. Indicators of functional status for primary malignant brain tumour patients. Can J Neurol Sci. 2005;32:50–56. doi: 10.1017/s0317167100016875. [DOI] [PubMed] [Google Scholar]
- 7.Mischel P.S., Nelson S.F., Cloughesy T.F. Molecular analysis of glioblastoma: pathway profiling and its implications for patient therapy. Cancer Biol Ther. 2003;2:242–247. doi: 10.4161/cbt.2.3.369. [DOI] [PubMed] [Google Scholar]
- 8.Hassler M., Seidl S., Fazeny-Doerner B., Preusser M., Hainfellner J., Rossler K., Prayer D., Marosi C. Diversity of cytogenetic and pathohistologic profiles in glioblastoma. Cancer Genet Cytogenet. 2006;166:46–55. doi: 10.1016/j.cancergencyto.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 9.Nakamura M., Shimada K., Ishida E., Nakase H., Konishi N. Genetic analysis to complement histopathological diagnosis of brain tumors. Histol Histopathol. 2007;22:327–335. doi: 10.14670/HH-22.327. [DOI] [PubMed] [Google Scholar]
- 10.Louis D.N. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117. doi: 10.1146/annurev.pathol.1.110304.100043. [DOI] [PubMed] [Google Scholar]
- 11.Kanu O.O., Hughes B., Di C., Lin N., Fu J., Bigner D.D., Yan H., Adamson C. Glioblastoma multiforme oncogenomics and signaling pathways. Clin Med Oncol. 2009;3:39–52. doi: 10.4137/cmo.s1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohgaki H., Kleihues P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009;100:2235–2241. doi: 10.1111/j.1349-7006.2009.01308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vranova V., Necesalova E., Kuglik P., Cejpek P., Pesakova M., Budinska E., Relichova J., Veselska R. Screening of genomic imbalances in glioblastoma multiforme using high-resolution comparative genomic hybridization. Oncol Rep. 2007;17:457–464. [PubMed] [Google Scholar]
- 14.Dahlback H.S., Brandal P., Meling T.R., Gorunova L., Scheie D., Heim S. Genomic aberrations in 80 cases of primary glioblastoma multiforme: pathogenetic heterogeneity and putative cytogenetic pathways. Genes Chromosomes Cancer. 2009;48:908–924. doi: 10.1002/gcc.20690. [DOI] [PubMed] [Google Scholar]
- 15.Parsons D.W., Jones S., Zhang X., Lin J.C., Leary R.J., Angenendt P., Mankoo P., Carter H., Siu I.M., Gallia G.L., Olivi A., McLendon R., Rasheed B.A., Keir S., Nikolskaya T., Nikolsky Y., Busam D.A., Tekleab H., Diaz L.A., Jr., Hartigan J., Smith D.R., Strausberg R.L., Marie S.K., Shinjo S.M., Yan H., Riggins G.J., Bigner D.D., Karchin R., Papadopoulos N., Parmigiani G., Vogelstein B., Velculescu V.E., Kinzler K.W. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vital A.L., Tabernero M.D., Crespo I., Rebelo O., Tao H., Gomes F., Lopes M.C., Orfao A. Intratumoral patterns of clonal evolution in gliomas. Neurogenetics. 2010;11:227–239. doi: 10.1007/s10048-009-0217-x. [DOI] [PubMed] [Google Scholar]
- 17.Misra A., Pellarin M., Nigro J., Smirnov I., Moore D., Lamborn K.R., Pinkel D., Albertson D.G., Feuerstein B.G. Array comparative genomic hybridization identifies genetic subgroups in grade 4 human astrocytoma. Clin Cancer Res. 2005;11:2907–2918. doi: 10.1158/1078-0432.CCR-04-0708. [DOI] [PubMed] [Google Scholar]
- 18.Korshunov A., Sycheva R., Golanov A. Genetically distinct and clinically relevant subtypes of glioblastoma defined by array-based comparative genomic hybridization (array-CGH) Acta Neuropathol. 2006;111:465–474. doi: 10.1007/s00401-006-0057-9. [DOI] [PubMed] [Google Scholar]
- 19.Mohapatra G., Bollen A.W., Kim D.H., Lamborn K., Moore D.H., Prados M.D., Feuerstein B.G. Genetic analysis of glioblastoma multiforme provides evidence for subgroups within the grade. Genes Chromosomes Cancer. 1998;21:195–206. [PubMed] [Google Scholar]
- 20.Viana-Pereira M., Lopes J.M., Little S., Milanezi F., Basto D., Pardal F., Jones C., Reis R.M. Analysis of EGFR overexpression: EGFR gene amplification and the EGFRvIII mutation in Portuguese high-grade gliomas. Anticancer Res. 2008;28:913–920. [PubMed] [Google Scholar]
- 21.Liu L., Backlund L.M., Nilsson B.R., Grander D., Ichimura K., Goike H.M., Collins V.P. Clinical significance of EGFR amplification and the aberrant EGFRvIII transcript in conventionally treated astrocytic gliomas. J Mol Med. 2005;83:917–926. doi: 10.1007/s00109-005-0700-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicholas M.K., Lukas R.V., Jafri N.F., Faoro L., Salgia R. Epidermal growth factor receptor– mediated signal transduction in the development and therapy of gliomas. Clin Cancer Res. 2006;12:7261–7270. doi: 10.1158/1078-0432.CCR-06-0874. [DOI] [PubMed] [Google Scholar]
- 23.Wemmert S., Ketter R., Rahnenfuhrer J., Beerenwinkel N., Strowitzki M., Feiden W., Hartmann C., Lengauer T., Stockhammer F., Zang K.D., Meese E., Steudel W.I., von Deimling A., Urbschat S. Patients with high-grade gliomas harboring deletions of chromosomes 9p and 10q benefit from temozolomide treatment. Neoplasia. 2005;7:883–893. doi: 10.1593/neo.05307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tada K., Shiraishi S., Kamiryo T., Nakamura H., Hirano H., Kuratsu J., Kochi M., Saya H., Ushio Y. Analysis of loss of heterozygosity on chromosome 10 in patients with malignant astrocytic tumors: correlation with patient age and survival. J Neurosurg. 2001;95:651–659. doi: 10.3171/jns.2001.95.4.0651. [DOI] [PubMed] [Google Scholar]
- 25.Daido S., Takao S., Tamiya T., Ono Y., Terada K., Ito S., Ouchida M., Date I., Ohmoto T., Shimizu K. Loss of heterozygosity on chromosome 10q associated with malignancy and prognosis in astrocytic tumors, and discovery of novel loss regions. Oncol Rep. 2004;12:789–795. [PubMed] [Google Scholar]
- 26.Brat D.J., Seiferheld W.F., Perry A., Hammond E.H., Murray K.J., Schulsinger A.R., Mehta M.P., Curran W.J. Analysis of 1p, 19q, 9p, and 10q as prognostic markers for high-grade astrocytomas using fluorescence in situ hybridization on tissue microarrays from Radiation Therapy Oncology Group trials. Neuro Oncol. 2004;6:96–103. doi: 10.1215/S1152851703000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez-Gines C., Cerda-Nicolas M., Gil-Benso R., Pellin A., Lopez-Guerrero J.A., Callaghan R., Benito R., Roldan P., Piquer J., Llacer J., Barbera J. Association of chromosome 7, chromosome 10 and EGFR gene amplification in glioblastoma multiforme. Clin Neuropathol. 2005;24:209–218. [PubMed] [Google Scholar]
- 28.Yadav A.K., Renfrow J.J., Scholtens D.M., Xie H., Duran G.E., Bredel C., Vogel H., Chandler J.P., Chakravarti A., Robe P.A., Das S., Scheck A.C., Kessler J.A., Soares M.B., Sikic B.I., Harsh G.R., Bredel M. Monosomy of chromosome 10 associated with dysregulation of epidermal growth factor signaling in glioblastomas. JAMA. 2009;302:276–289. doi: 10.1001/jama.2009.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gil-Perotin S., Marin-Husstege M., Li J., Soriano-Navarro M., Zindy F., Roussel M.F., Garcia-Verdugo J.M., Casaccia-Bonnefil P. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci. 2006;26:1107–1116. doi: 10.1523/JNEUROSCI.3970-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Deimling A., Eibl R.H., Ohgaki H., Louis D.N., von Ammon K., Petersen I., Kleihues P., Chung R.Y., Wiestler O.D., Seizinger B.R. p53 mutations are associated with 17p allelic loss in grade II and grade III astrocytoma. Cancer Res. 1992;52:2987–2990. [PubMed] [Google Scholar]
- 31.Vordermark D., Ruprecht K., Rieckmann P., Roggendorf W., Vince G.H., Warmuth-Metz M., Kolbl O., Flentje M. Glioblastoma multiforme with oligodendroglial component (GBMO): favorable outcome after post-operative radiotherapy and chemotherapy with nimustine (ACNU) and teniposide (VM26) BMC Cancer. 2006;6:247. doi: 10.1186/1471-2407-6-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salvati M., Formichella A.I., D'Elia A., Brogna C., Frati A., Giangaspero F., Delfini R., Santoro A. Cerebral glioblastoma with oligodendrogliomal component: analysis of 36 cases. J Neurooncol. 2009;94:129–134. doi: 10.1007/s11060-009-9815-6. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt M.C., Antweiler S., Urban N., Mueller W., Kuklik A., Meyer-Puttlitz B., Wiestler O.D., Louis D.N., Fimmers R., von Deimling A. Impact of genotype and morphology on the prognosis of glioblastoma. J Neuropathol Exp Neurol. 2002;61:321–328. doi: 10.1093/jnen/61.4.321. [DOI] [PubMed] [Google Scholar]
- 34.Shiraishi S., Tada K., Nakamura H., Makino K., Kochi M., Saya H., Kuratsu J., Ushio Y. Influence of p53 mutations on prognosis of patients with glioblastoma. Cancer. 2002;95:249–257. doi: 10.1002/cncr.10677. [DOI] [PubMed] [Google Scholar]
- 35.Kato H., Kato S., Kumabe T., Sonoda Y., Yoshimoto T., Han S.Y., Suzuki T., Shibata H., Kanamaru R., Ishioka C. Functional evaluation of p53 and PTEN gene mutations in gliomas. Clin Cancer Res. 2000;6:3937–3943. [PubMed] [Google Scholar]
- 36.Bredel M., Bredel C., Juric D., Harsh G.R., Vogel H., Recht L.D., Sikic B.I. High-resolution genome-wide mapping of genetic alterations in human glial brain tumors. Cancer Res. 2005;65:4088–4096. doi: 10.1158/0008-5472.CAN-04-4229. [DOI] [PubMed] [Google Scholar]
- 37.Arslantas A., Artan S., Oner U., Muslumanoglu H., Durmaz R., Cosan E., Atasoy M.A., Basaran N., Tel E. The importance of genomic copy number changes in the prognosis of glioblastoma multiforme. Neurosurg Rev. 2004;27:58–64. doi: 10.1007/s10143-003-0279-4. [DOI] [PubMed] [Google Scholar]
- 38.Verhaak R.G., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., Miller C.R., Ding L., Golub T., Mesirov J.P., Alexe G., Lawrence M., O'Kelly M., Tamayo P., Weir B.A., Gabriel S., Winckler W., Gupta S., Jakkula L., Feiler H.S., Hodgson J.G., James C.D., Sarkaria J.N., Brennan C., Kahn A., Spellman P.T., Wilson R.K., Speed T.P., Gray J.W., Meyerson M., Getz G., Perou C.M., Hayes D.N. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA. IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beroukhim R., Mermel C.H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J.S., Dobson J., Urashima M., Mc Henry K.T., Pinchback R.M., Ligon A.H., Cho Y.J., Haery L., Greulich H., Reich M., Winckler W., Lawrence M.S., Weir B.A., Tanaka K.E., Chiang D.Y., Bass A.J., Loo A., Hoffman C., Prensner J., Liefeld T., Gao Q., Yecies D., Signoretti S., Maher E., Kaye F.J., Sasaki H., Tepper J.E., Fletcher J.A., Tabernero J., Baselga J., Tsao M.S., Demichelis F., Rubin M.A., Janne P.A., Daly M.J., Nucera C., Levine R.L., Ebert B.L., Gabriel S., Rustgi A.K., Antonescu C.R., Ladanyi M., Letai A., Garraway L.A., Loda M., Beer D.G., True L.D., Okamoto A., Pomeroy S.L., Singer S., Golub T.R., Lander E.S., Getz G., Sellers W.R., Meyerson M. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bralten L.B., Kloosterhof N.K., Gravendeel L.A., Sacchetti A., Duijm E.J., Kros J.M., van den Bent M.J., Hoogenraad C.C., Sillevis Smitt P.A., French P.J. Integrated genomic profiling identifies candidate genes implicated in glioma-genesis and a novel LEO1-SLC12A1 fusion gene. Genes Chromosomes Cancer. 2010;49:509–517. doi: 10.1002/gcc.20760. [DOI] [PubMed] [Google Scholar]
- 42.Chen R., Nishimura M.C., Bumbaca S.M., Kharbanda S., Forrest W.F., Kasman I.M., Greve J.M., Soriano R.H., Gilmour L.L., Rivers C.S., Modrusan Z., Nacu S., Guerrero S., Edgar K.A., Wallin J.J., Lamszus K., Westphal M., Heim S., James C.D., VandenBerg S.R., Costello J.F., Moorefield S., Cowdrey C.J., Prados M., Phillips H.S. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010;17:362–375. doi: 10.1016/j.ccr.2009.12.049. [DOI] [PubMed] [Google Scholar]
- 43.Gardina P.J., Lo K.C., Lee W., Cowell J.K., Turpaz Y. Ploidy status and copy number aberrations in primary glioblastomas defined by integrated analysis of allelic ratios, signal ratios and loss of heterozygosity using 500K SNP Mapping Arrays. BMC Genomics. 2008;9:489. doi: 10.1186/1471-2164-9-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solomon D.A., Kim J.S., Cronin J.C., Sibenaller Z., Ryken T., Rosenberg S.A., Ressom H., Jean W., Bigner D., Yan H., Samuels Y., Waldman T. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 2008;68:10300–10306. doi: 10.1158/0008-5472.CAN-08-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yin D., Ogawa S., Kawamata N., Tunici P., Finocchiaro G., Eoli M., Ruckert C., Huynh T., Liu G., Kato M., Sanada M., Jauch A., Dugas M., Black K.L., Koeffler H.P. High-resolution genomic copy number profiling of glioblastoma multiforme by single nucleotide polymorphism DNA microarray. Mol Cancer Res. 2009;7:665–677. doi: 10.1158/1541-7786.MCR-08-0270. [DOI] [PubMed] [Google Scholar]
- 46.Cowell J.K., Lo K.C., Luce J., Hawthorn L. Interpreting aCGH-defined karyotypic changes in gliomas using copy number status, loss of heterozygosity and allelic ratios. Exp Mol Pathol. 2010;88:82–89. doi: 10.1016/j.yexmp.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paugh B.S., Qu C., Jones C., Liu Z., Adamowicz-Brice M., Zhang J., Bax D.A., Coyle B., Barrow J., Hargrave D., Lowe J., Gajjar A., Zhao W., Broniscer A., Ellison D.W., Grundy R.G., Baker S.J. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol. 2010;28:3061–3068. doi: 10.1200/JCO.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qu H.Q., Jacob K., Fatet S., Ge B., Barnett D., Delattre O., Faury D., Montpetit A., Solomon L., Hauser P., Garami M., Bognar L., Hansely Z., Mio R., Farmer J.P., Albrecht S., Polychronakos C., Hawkins C., Jabado N. Genome-wide profiling using single-nucleotide polymorphism arrays identifies novel chromosomal imbalances in pediatric glioblastomas. Neuro Oncol. 2010;12:153–163. doi: 10.1093/neuonc/nop001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zarghooni M., Bartels U., Lee E., Buczkowicz P., Morrison A., Huang A., Bouffet E., Hawkins C. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol. 2010;28:1337–1344. doi: 10.1200/JCO.2009.25.5463. [DOI] [PubMed] [Google Scholar]
- 50.Beroukhim R., Getz G., Nghiemphu L., Barretina J., Hsueh T., Linhart D., Vivanco I., Lee J.C., Huang J.H., Alexander S., Du J., Kau T., Thomas R.K., Shah K., Soto H., Perner S., Prensner J., Debiasi R.M., Demichelis F., Hatton C., Rubin M.A., Garraway L.A., Nelson S.F., Liau L., Mischel P.S., Cloughesy T.F., Meyerson M., Golub T.A., Lander E.S., Mellinghoff I.K., Sellers W.R. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kotliarov Y., Steed M.E., Christopher N., Walling J., Su Q., Center A., Heiss J., Rosenblum M., Mikkelsen T., Zenklusen J.C., Fine H.A. High-resolution global genomic survey of 178 gliomas reveals novel regions of copy number alteration and allelic imbalances. Cancer Res. 2006;66:9428–9436. doi: 10.1158/0008-5472.CAN-06-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin M., Wei L.J., Sellers W.R., Lieberfarb M., Wong W.H., Li C. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics. 2004;20:1233–1240. doi: 10.1093/bioinformatics/bth069. [DOI] [PubMed] [Google Scholar]
- 53.Nannya Y., Sanada M., Nakazaki K., Hosoya N., Wang L., Hangaishi A., Kurokawa M., Chiba S., Bailey D.K., Kennedy G.C., Ogawa S. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–6079. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- 54.Lo K.C., Bailey D., Burkhardt T., Gardina P., Turpaz Y., Cowell J.K. Comprehensive analysis of loss of heterozygosity events in glioblastoma using the 100K SNP mapping arrays and comparison with copy number abnormalities defined by BAC array comparative genomic hybridization. Genes Chromosomes Cancer. 2008;47:221–237. doi: 10.1002/gcc.20524. [DOI] [PubMed] [Google Scholar]
- 55.Houillier C., Lejeune J., Benouaich-Amiel A., Laigle-Donadey F., Criniere E., Mokhtari K., Thillet J., Delattre J.Y., Hoang-Xuan K., Sanson M. Prognostic impact of molecular markers in a series of 220 primary glioblastomas. Cancer. 2006;106:2218–2223. doi: 10.1002/cncr.21819. [DOI] [PubMed] [Google Scholar]
- 56.Lopez-Gines C., Gil-Benso R., Ferrer-Luna R., Benito R., Serna E., Gonzalez-Darder J., Quilis V., Monleon D., Celda B., Cerda-Nicolas M. New pattern of EGFR amplification in glioblastoma and the relationship of gene copy number with gene expression profile. Mod Pathol. 2010;23:856–865. doi: 10.1038/modpathol.2010.62. [DOI] [PubMed] [Google Scholar]
- 57.Rossi M.R., La Duca J., Matsui S., Nowak N.J., Hawthorn L., Cowell J.K. Novel amplicons on the short arm of chromosome 7 identified using high resolution array CGH contain over expressed genes in addition to EGFR in glioblastoma multiforme. Genes Chromosomes Cancer. 2005;44:392–404. doi: 10.1002/gcc.20256. [DOI] [PubMed] [Google Scholar]
- 58.Serrano M., Lee H., Chin L., Cordon-Cardo C., Beach D., DePinho R.A. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 59.Nobori T., Takabayashi K., Tran P., Orvis L., Batova A., Yu A.L., Carson D.A. Genomic cloning of methylthioadenosine phosphorylase: a purine metabolic enzyme deficient in multiple different cancers. Proc Natl Acad Sci U S A. 1996;93:6203–6208. doi: 10.1073/pnas.93.12.6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hori Y., Hori H., Yamada Y., Carrera C.J., Tomonaga M., Kamihira S., Carson D.A., Nobori T. The methylthioadenosine phosphorylase gene is frequently co-deleted with the p16INK4a gene in acute type adult T-cell leukemia. Int J Cancer. 1998;75:51–56. doi: 10.1002/(sici)1097-0215(19980105)75:1<51::aid-ijc9>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 61.Schmid M., Malicki D., Nobori T., Rosenbach M.D., Campbell K., Carson D.A., Carrera C.J. Homozygous deletions of methylthioadenosine phosphorylase (MTAP) are more frequent than p16INK4A (CDKN2) homozygous deletions in primary non-small cell lung cancers (NSCLC) Oncogene. 1998;17:2669–2675. doi: 10.1038/sj.onc.1202205. [DOI] [PubMed] [Google Scholar]
- 62.Christopher S.A., Diegelman P., Porter C.W., Kruger W.D. Methylthioadenosine phosphorylase, a gene frequently codeleted with p16(cdkN2a/ARF), acts as a tumor suppressor in a breast cancer cell line. Cancer Res. 2002;62:6639–6644. [PubMed] [Google Scholar]
- 63.Jen J., Harper J.W., Bigner S.H., Bigner D.D., Papadopoulos N., Markowitz S., Willson J.K., Kinzler K.W., Vogelstein B. Deletion of p16 and p15 genes in brain tumors. Cancer Res. 1994;54:6353–6358. [PubMed] [Google Scholar]
- 64.Kawamata N., Ogawa S., Zimmermann M., Kato M., Sanada M., Hemminki K., Yamatomo G., Nannya Y., Koehler R., Flohr T., Miller C.W., Harbott J., Ludwig W.D., Stanulla M., Schrappe M., Bartram C.R., Koeffler H.P. Molecular allelokaryotyping of pediatric acute lymphoblastic leukemias by high-resolution single nucleotide polymorphism oligonucleotide genomic microarray. Blood. 2008;111:776–784. doi: 10.1182/blood-2007-05-088310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cairns P., Polascik T.J., Eby Y., Tokino K., Califano J., Merlo A., Mao L., Herath J., Jenkins R., Westra W. Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Genet. 1995;11:210–212. doi: 10.1038/ng1095-210. [DOI] [PubMed] [Google Scholar]
- 66.Yin D., Xie D., Hofmann W.K., Miller C.W., Black K.L., Koeffler H.P. Methylation, expression, and mutation analysis of the cell cycle control genes in human brain tumors. Oncogene. 2002;21:8372–8378. doi: 10.1038/sj.onc.1206031. [DOI] [PubMed] [Google Scholar]
- 67.Nord H., Hartmann C., Andersson R., Menzel U., Pfeifer S., Piotrowski A., Bogdan A., Kloc W., Sandgren J., Olofsson T., Hesselager G., Blomquist E., Komorowski J., von Deimling A., Bruder C.E., Dumanski J.P., Diaz de Stahl T. Characterization of novel and complex genomic aberrations in glioblastoma using a 32K BAC array. Neuro Oncol. 2009;11:803–818. doi: 10.1215/15228517-2009-013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Voesten A.M., Bijleveld E.H., Westerveld A., Hulsebos T.J. Fine mapping of a region of common deletion on chromosome arm 10p in human glioma. Genes Chromosomes Cancer. 1997;20:167–172. [PubMed] [Google Scholar]
- 69.Ichimura K., Schmidt E.E., Miyakawa A., Goike H.M., Collins V.P. Distinct patterns of deletion on 10p and 10q suggest involvement of multiple tumor suppressor genes in the development of astrocytic gliomas of different malignancy grades. Genes Chromosomes Cancer. 1998;22:9–15. doi: 10.1002/(sici)1098-2264(199805)22:1<9::aid-gcc2>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 70.Li J., Yen C., Liaw D., Podsypanina K., Bose S., Wang S.I., Puc J., Miliaresis C., Rodgers L., McCombie R., Bigner S.H., Giovanella B.C., Ittmann M., Tycko B., Hibshoosh H., Wigler M.H., Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 71.Inda M.M., Fan X., Munoz J., Perot C., Fauvet D., Danglot G., Palacio A., Madero P., Zazpe I., Portillo E., Tunon T., Martinez-Penuela J.M., Alfaro J., Eiras J., Bernheim A., Castresana J.S. Chromosomal abnormalities in human glioblastomas: gain in chromosome 7p correlating with loss in chromosome 10q. Mol Carcinog. 2003;36:6–14. doi: 10.1002/mc.10085. [DOI] [PubMed] [Google Scholar]
- 72.Knobbe C.B., Reifenberger G. Genetic alterations and aberrant expression of genes related to the phosphatidyl-inositol-3′-kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol. 2003;13:507–518. doi: 10.1111/j.1750-3639.2003.tb00481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Quan A.L., Barnett G.H., Lee S.Y., Vogelbaum M.A., Toms S.A., Staugaitis S.M., Prayson R.A., Peereboom D.M., Stevens G.H., Cohen B.H., Suh J.H. Epidermal growth factor receptor amplification does not have prognostic significance in patients with glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 2005;63:695–703. doi: 10.1016/j.ijrobp.2005.03.051. [DOI] [PubMed] [Google Scholar]
- 74.Newcomb E.W., Cohen H., Lee S.R., Bhalla S.K., Bloom J., Hayes R.L., Miller D.C. Survival of patients with glioblastoma multiforme is not influenced by altered expression of p16, p53. EGFR, MDM2 or Bcl-2 genes. Brain Pathol. 1998;8:655–667. doi: 10.1111/j.1750-3639.1998.tb00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhu A., Shaeffer J., Leslie S., Kolm P., El-Mahdi A.M. Epidermal growth factor receptor: an independent predictor of survival in astrocytic tumors given definitive irradiation. Int J Radiat Oncol Biol Phys. 1996;34:809–815. doi: 10.1016/0360-3016(95)02184-1. [DOI] [PubMed] [Google Scholar]
- 76.Etienne M.C., Formento J.L., Lebrun-Frenay C., Gioanni J., Chatel M., Paquis P., Bernard C., Courdi A., Bensadoun R.J., Pignol J.P., Francoual M., Grellier P., Frenay M., Milano G. Epidermal growth factor receptor and labeling index are independent prognostic factors in glial tumor outcome. Clin Cancer Res. 1998;4:2383–2390. [PubMed] [Google Scholar]
- 77.Korshunov A., Sycheva R., Golanov A. The prognostic relevance of molecular alterations in glioblastomas for patients age < 50 years. Cancer. 2005;104:825–832. doi: 10.1002/cncr.21221. [DOI] [PubMed] [Google Scholar]
- 78.Simmons M.L., Lamborn K.R., Takahashi M., Chen P., Israel M.A., Berger M.S., Godfrey T., Nigro J., Prados M., Chang S., Barker F.G., 2nd, Aldape K. Analysis of complex relationships between age, p53, epidermal growth factor receptor, and survival in glioblastoma patients. Cancer Res. 2001;61:1122–1128. [PubMed] [Google Scholar]
- 79.Srividya M.R., Thota B., Arivazhagan A., Thennarasu K., Balasubramaniam A., Chandramouli B.A., Hegde A.S., Santosh V. Age-dependent prognostic effects of EGFR/p53 alterations in glioblastoma: study on a prospective cohort of 140 uniformly treated adult patients. J Clin Pathol. 2010;63:687–691. doi: 10.1136/jcp.2009.074898. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete SNP array profiles for the discrepant karyotypic findings between SNP arrays and iFISH, detected in chromosome 9 (cases G15 and G63; A) and chromosome 10 (case G63; B).