

Abstract
Recently, genome-wide association studies have identified the major histocompatibility complex class I protein HLA-C as an important molecule that affects HIV disease progression. The association between HLA-C and HIV disease outcome was originally determined through a single nucleotide polymorphism (SNP) 35 kb upstream of the HLA-C locus. More recent work has focused on elucidating the functional significance of the -35 SNP, and several groups now have demonstrated HLA-C surface expression to be a key element in control of HIV viral load, with higher surface expression associating with slower disease progression. Most recently, control of HLA-C surface expression has been correlated with the presence of microRNA binding sites that affect HLA-C expression and control of HIV disease. This review highlights these results and explores the ways in which HLA-C surface expression could affect immune system function in the setting of HIV disease.
Keywords: HLA-C, HIV, MHC-I, -35SNP, KIR
Introduction
Infection with HIV is a global health problem, with over 2 million AIDS-related deaths per year (http://www.una-ids.org/en/KnowledgeCentre/HIVData/EpiUpdate/EpiUpdArchive/2009/default.asp). Currently the only effective treatment for the disease is Highly Active Anti-Retroviral Therapy (HAART). Although HAART controls the disease, complete eradication of the virus has yet to be achieved. Addressing this significant global health problem calls for the development of preventative and therapeutic vaccines. A greater understanding of the immune response to HIV and the mechanisms of viral immune evasion will facilitate this goal as well as further the development of therapeutics to eliminate HIV infection.
Major histocompatibility complex class I molecules (MHC-I) are necessary for an effective host immune response to HIV infection. MHC-I present viral peptides to CD8+ T cells and serve as ligands for killer cell immunoglobulin-like receptors (KIRs). A subset of MHC-I allotypes are associated with delayed disease progression and effective control of viral replication, most notably HLA-B*5701. Recently, genome-wide association studies have implicated a role for high HLA-C surface expression in the control of HIV.1–3 This review will summarize these findings, supporting a role for HLA-C in HIV disease progression. Insight gained from these studies will shed light on the role of these complex molecules in the innate and adaptive immune responses.
HLA-C
The MHC-I locus on chromosome 6 in the human genome comprises two groups, the highly polymorphic classical class I molecules (HLA-A, -B and -C)and the conserved non-classical molecules (HLA-E, -F and –G) (reviewed in ref. 4). There is evidence that evolution of the HLA-A, -B and -C loci is driven by pathogen-mediated selection. The HLA-A and -B molecules in particular are highly polymorphic with the greatest variability found within the peptide-binding region, which supports their function in presentation of diverse antigens to cytotoxic T lymphocytes (CTLs).5 HLA-C is the most recently evolved of the classical MHC-I alleles and is restricted to humans and great apes;4,6 and fewer alleles of HLA-C have been identified. There are also functional differences among these molecules that may restrict diversity; KIRs recognize all HLA-C allotypes, but only some HLA-A and -B allotypes.7 HLA-C biology is still under active investigation to define its role in the immune response and the selective pressures that define its diversity (for recent review see ref. 8).
The HLA-C protein is a heterodimer composed of a membrane-bound mature heavy chain and a light chain, β2-microglobulin (β2M). HLA-C plays a dual role in that it can present antigens to CTLs and it can inhibit natural killer (NK) cell (and possibly also CTL) lysis via its interaction with inhibitory receptors. For reasons that are poorly understood, HLA-C is normally expressed on the cell surface at levels approximately 10-fold less than most HLA-A and HLA-B allotypes.9–11
Regulation of HLA-C surface expression
Maintenance of a low HLA-C surface expression phenotype is achieved at the transcriptional, translational and post-translational levels (Fig. 1). It has been known since 1995 that HLA-C heavy chain mRNA turns over at a rapid rate.10 However, a new study has found an explanation for this observation. MicroRNA (miRNA) target sequences have now been identified in the 3′ untranslated region (UTR) of the HLA-C gene.12 One target site in particular was found to have functional significance for HLA-C surface expression; a single base pair insertion or deletion at position 263 downstream of the HLA-C stop codon affects the binding of miRNA-148a and correspondingly affects HLA-C surface expression (Fig. 1).12
Figure 1.
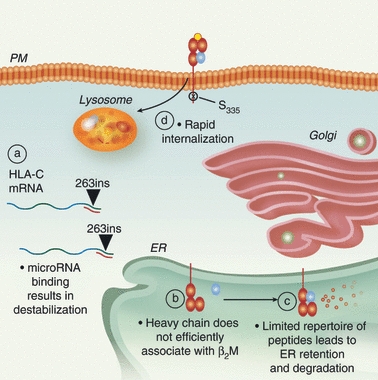
HLA-C surface expression is regulated through multiple mechanisms. (a) The presence of a single base pair insertion polymorphism in the 3′-untranslated region (263ins) of HLA-C mRNA (wavy line) results in the creation of a microRNA binding site causing post-transcriptional cleavage or destabilization. (b) Once translated, the HLA-C heavy chain does not efficiently associate with β2-microglobulin (β2M; blue circle) in the endoplasmic reticulum (ER). (c) HLA-C is able to bind a limited repertoire of peptides, and molecules accumulating in the ER waiting for peptide loading are eventually targeted for degradation. (d) Once on the plasma membrane (PM), the rate of HLA-C internalization and lysosomal degradation is regulated by phosphorylation of serine residue 335.
In addition, HLA-C heavy chain proteins do not efficiently associate with β2M.10,13–15 The HLA-C molecules present a more restricted repertoire of peptides than HLA-A or -B, which leads to accumulation of HLA-C heavy chains in the endoplasmic reticulum and ultimately to degradation of the retained immature HLA-C molecules waiting for peptide loading (Fig. 1).11,13,16
Recent studies have demonstrated that a di-hydrophobic signal in the HLA-C cytoplasmic tail (Fig. 2) also limits cell surface expression by promoting internalization and degradation of HLA-C16 (Fig. 1). Interestingly, Schaefer et al.16 found that antigen-presenting cells (APCs) express higher levels of HLA-C on the cell surface than other cell types. The up-regulation of HLA-C occurs upon macrophage differentiation and is regulated by phosphorylation of a serine residue adjacent to the di-hydrophobic signal (S335)16 (Fig. 2). The tightly regulated cell surface expression of HLA-C is one indication that HLA-C plays an important role in an effective immune response.
Figure 2.
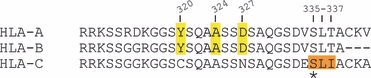
HLA-A, HLA-B and HLA-C cytoplasmic tail sequences. Y320, A324 and D327 are required for Nef-mediated AP-1 recruitment and MHC-I down-modulation of HLA-A and HLA-B cytoplasmic tails. Y320 and D327 are absent in HLA-C cytoplasmic tail sequences, which are not down-modulated by Nef. HLA-C is internalized more rapidly than HLA-A and HLA-B because of a di-hydrophobic motif (L336I337) present in the cytoplasmic tail that is regulated by phosphorylation of an adjacent residue (S335).
High HLA-C expression on APCs suggests that HLA-C may serve a specific role in MHC-I pathways unique to these cell types. For example, APCs can cross-present exogenous antigens in association with co-stimulatory molecules, a process essential for the efficient priming of naive CD8+ T cells. However, uninfected APCs cross-presenting foreign antigens in association with MHC-I could also be a target of CTLs once they have developed effector functions. Lysis of cross-presenting APCs would limit the duration of effective naive T-cell priming. Because HLA-C binds to inhibitory KIR that are up-regulated upon acquisition of CTL effector functions,17 it is possible that HLA-C plays a role in limiting CTL lysis of cross-presenting APCs. Prolonged survival of cross-presenting APCs could facilitate persistent priming of naive CD8+ T cells, which might be beneficial in chronic infection with evolving epitopes (Fig. 3).
Figure 3.
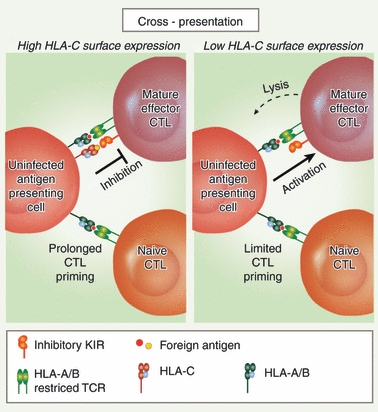
High HLA-C expression may enhance cytotoxic T lymphocyte (CTL) priming. During cross-presentation of foreign peptides by antigen-presenting cells (APCs), high expression of HLA-C molecules may allow more effective stimulation of the CD8+ T-cell response through several mechanisms. Left panel: MHC class I molecules cross present foreign peptides to naive CD8+ T cells to induce maturation into epitope-specific effector CTLs. Because of its function as an inhibitory killer cell immunoglobulin-like receptor (KIR) ligand, high HLA-C surface expression may inhibit lysis of uninfected, cross-presenting cells by fully mature effector CTLs that up-regulate KIRs. This would promote persistent priming of naive T cells in chronic infection. Right panel: Lower HLA-C surface expression may result in the loss of KIR inhibitory ligand protection for the uninfected APCs and in loss of cross-presenting cells once mature effector CTLs have developed. Loss of cross-presenting APCs following CTL maturation may be beneficial to turn off cross-presentation and down-regulate the immune response but may be detrimental in chronic infection of a host by a virus that can vary its epitopes.
HLA-C and NK cell recognition
As mentioned above, almost all HLA-C allotypes are recognized by KIRs on NK cells and mature effector CTLs. On NK cells, inhibitory KIRs monitor for ‘missing self’ resulting from MHC class I down-modulation on virally infected or malignant cells which otherwise would escape CTL recognition and lysis.18,19 HLA-C allotypes dominate over HLA-A and HLA-B allotypes as a source of ligands for KIRs6 and inhibitory receptors for HLA-C ligands are dominant until activating signals overcome inhibition.16,20,21 Activating KIRs are thought to have a weaker binding affinity for the same ligand and are probably involved in recognizing ‘changed self’, such as presentation of stress peptides associated with infection or malignancy by MHC class I molecules.18 Inhibitory receptors are characterized by a long cytoplasmic tail with an immunoreceptor tyrosine inhibitory motif whereas activating KIR have a short tail lacking the tyrosine residue.21
The KIR locus, which is located in the leucocyte receptor complex, is highly polymorphic and is comprised of a family of 14 genes, eight inhibitory, six activating and two pseudo genes.22–24 Most of the ligands for KIRs are still undefined but the dominant ligands that have been identified so far are the C1 and C2 epitopes of HLA-C.25–28 C1 allotypes (HLA-Cw1, -Cw3, -Cw7, -Cw8, -Cw13 and -Cw14) are defined as having an asparagine at residue 80 and are the ligands for the inhibitory receptors KIR2DL2 and KIR2DL3.20,28,29 C2 allotypes (HLA-Cw2, -Cw4, -Cw5, -Cw6, -Cw17 and -Cw18) have a lysine at this residue and can act as ligand for inhibitory receptor KIR2DL1 and for activating receptor KIR2DS1.20
The MHC and KIR loci are physically unlinked and located on separate chromosomes in the human genome. Both loci are highly polymorphic, and for any functional KIR-HLA allelic pairing, it is possible that both, only one, or neither are present in an individual.19,20 The presence of only one allele of the KIR-HLA pair would be functionally null.19 In addition, the frequency and intensity with which a particular KIR allele is expressed is determined during maturation and is subsequently fixed.19 The interactions between the inhibitory receptors KIR2DL1, KIR2DL2/3 and KIR3DL1 and their HLA-C inhibitory ligands have an active role in NK cell education during their maturation.6 These interactions are highly variable and result in a diverse response to infection and malignancy. This may be because HLA-C is the most recently evolved of the MHC class I genes and is subject to evolution and pathogen-mediated selection.6,20
The HLA-C allele and HIV disease
To help evade the anti-HIV CTL response, HIV encodes the accessory protein Nef, which disrupts antigen presentation in association with MHC-I by re-directing HLA-A and HLA-B molecules from the cell surface to lysosomal compartments for degradation. The HIV Nef protein limits CTL recognition by reducing the MHC class I molecules presenting HIV-derived peptides on the cell surface.30 Down-modulation by Nef is efficient but not complete, as evidenced by the presence of anti-HIV CTLs and the emergence of HIV escape mutants capable of generating higher viral loads after evolving resistance to CTLs.31
To disrupt the cell surface expression of HLA-A and HLA-B molecules, Nef binds to the cytoplasmic tail domain and promotes the association of the conserved MHC-I cytoplasmic tail tyrosine residue with the clathrin adaptor protein 1 (AP-1)32,33 The AP-1 complex contains a tyrosine binding pocket in the mu subunit that is required for this interaction. Significantly, HLA-C cytoplasmic tails lack two amino acids necessary for this interaction (Fig. 2). As a result, the HIV-1 Nef protein does not down-modulate HLA-C molecules from the cell surface34 and so HLA-C may have a unique role in presenting antigens to CTLs in HIV disease (Fig. 4).
Figure 4.
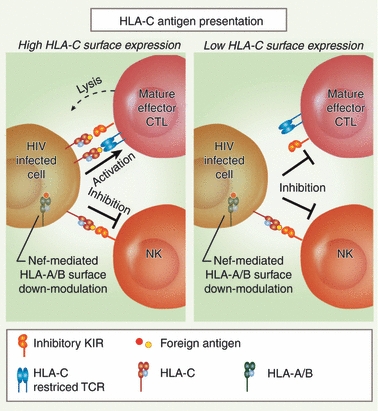
HLA-C surface expression and HLA-C-restricted anti-HIV cytotoxic T lymphocytes (CTLs). Left panel: In HIV-infected individuals, HLA-A and -B molecules are preferentially down-modulated by the accessory protein Nef. Relatively high levels of HLA-C molecules on the cell surface allow for presentation of HIV-derived peptides to antigen-specific CTLs. The engagement of the HLA-C-specific T-cell receptor may overcome the inhibitory signal of HLA-C interaction with killer cell immunoglobulin-like receptors (KIRs) on mature effector CTLs, allowing for efficient recognition and lysis of the infected cell. However, the remaining HLA-C also results in a potent inhibition of natural killer cell-mediated recognition and lysis. Right panel: Lower HLA-C surface expression coupled with HLA-A and -B down-modulation in the context of HIV infection may result in a decrease in efficient CTL recognition and lysis of the infected cell. The lower steady-state surface expression of HLA-C may still allow for inhibition of NK cells expressing KIRs.
HLA-C and host genetic factors that influence HIV disease progression
During primary HIV infection, the host immune response plays a critical role in controlling HIV replication and these responses typically result in a significant decrease in plasma viral load.18,35 At 18–24 months after seroconversion the amount of virus present has reached an equilibrium, termed the viral load set point.36–38 Although there is great variability in the viral set point between infected individuals, many patients have stable levels exceeding 10 000 RNA copies/ml of blood.1 In a typical course of infection, most individuals will progress to AIDS 7–11 years after infection without antiviral therapy; however, a subset of patients will have a more rapid (< 2 years) disease course or alternatively virtually no disease progression at all.39–42
To identify host genetic factors that influence HIV disease progression, a number of investigators have performed genome-wide studies to identify single nucleotide polymorphisms (SNPs) that correlate with disease outcome. One SNP upstream of HLA-C (-35C/T) has been shown to be a major determinant accounting for the viral load set point.1–3 The -35C allele is protective, and correlates with lower viral load set points and higher CD4+ T-cell counts 2 years after seroconversion.2,3,37 These data support a role for HLA-C in early or establishing stages of infection.37
In their second genome-wide association study, Fellay et al.3 reported that analysis of significant SNPs in the MHC class I locus is confounded by the pattern of long range linkage disequilibrium in the region. The two strongest SNPs, the HCP5 and HLA-C variants, were found to be in partial linkage disequilibrium. They report that in 95% of the samples with the rs2395029 minor allele, a polymorphism within the HCP5 gene, which is the proxy for the highly protective HLA-B*5701 allele, the HLA-C -35C SNP is also present.3 Interestingly, they conclude that the effect of viral load set point and slower disease progression is a result of the presence of both alleles, and that analyses that are not adjusted for the presence of the HLA-C variant may in fact be over-estimating HLA-B*5701 as the sole predictor of a protective outcome.3 It should be noted, however, that although the -35 polymorphism is present in African American populations, it does not significantly correlate with control of viral load.43,44 This suggests the possibility of another allelic combination present predominantly in the Caucasian populations used in these analyses that may be contributing to the observed elite controller phenotype.
Higher HLA-C surface expression is protective
Several studies have tried to determine the molecular basis for the protective phenotype of the -35C SNP. It was first reported that the -35C variant associates with higher HLA-C mRNA levels in Epstein–Barr virus-transformed B-cell lines.45 Further studies have now demonstrated that high levels of HLA-C surface expression independently correlate with a significant protection phenotype.44,46 Analysis of multiple -35 SNP genotypes and HLA-C alleles demonstrate variability in surface expression of HLA-C, and those alleles that are in linkage disequilibrium with -35C specifically correlate to a higher overall surface expression, with one study reporting approximately 1·7-fold higher on average for -35CC subjects compared with -35TT subjects.44,46 The -35T SNP does not correlate perfectly with low HLA-C expression levels, however, as there is overlap in the distribution of HLA-C surface expression between CC, CT and TT genotypes.46,47 There are individuals with a -35TT genotype that exhibit low viral-load set points that also express HLA-C mRNA levels as high as those with the -35CC genotype.47 These data suggest that the HLA-C allele and its specific level of surface expression is the controlling factor in viral load.
One of the most significant alleles identified with higher viral loads and more rapid disease progression is HLA-Cw7, which is in strong linkage disequilibrium with -35T.1HLA-Cw7 is a common haplotype, and has been shown to have very low surface expression.44 The HLA-C surface expression in -35TT individuals who were homozygous for HLA-Cw7 was five-fold lower than -35CC individuals who were HLA-Cw7 null.44 In fact, results from Corrah et al.44 suggest that the most significant contribution to the -35 SNP association with HIV disease progression comes from the HLA-Cw7 allele. HLA-Cw7 has been shown previously to associate with more rapid disease progression;1 however, exclusion of individuals who were homozygous for HLA-Cw7 did not lose significance for the association of the -35CC genotype with control of HIV infection.44 These data suggest that although low HLA-C surface expression from HLA-Cw7 is significant for rapid disease progression, the protective effect of the -35CC genotype deserves further characterization.
Significantly, the recent study by Kulkarni et al.12 found the polymorphic miRNA-148a binding site in the HLA-C 3′UTR to be in strong linkage disequilibrium with the -35 SNP. Their analysis found an association between the elite control of HIV infection and a deletion at position 263 in the HLA-C 3′ UTR, which disrupts the miRNA-148a binding site. Correspondingly, there was an association between poor control of HIV infection and an insertion polymorphism that created an miRNA-148a recognition sequence.12 The linkage disequilibrium of the -35SNP with the 3′UTR miRNA-148a binding site polymorphisms offers a functional explanation for the observed differences in HLA-C surface expression among individuals and the associated control of HIV disease.
HLA-C and CTLs in HIV disease
One of the possible explanations for the observed protective effect of higher HLA-C surface expression is improved presentation of antigens by HLA-C to CTLs. Several lines of evidence support the significance of CTLs in the control of HIV infection. The appearance of HIV-specific CD8+ T cells following acute infection correlates with a reduction in viral replication and the establishment of a viral-load set point.48 There is also evidence for the outgrowth of escape mutants within HIV epitopes targeted by CTLs, which demonstrates that CTLs exert pressure on the virus.49–56 Furthermore, a subset of MHC class I alleles confer protection from progression to AIDS.50,57,58
Recent evidence indicates that the HLA-C mediated CTL response to HIV infection may be important. Adnan et al.59 demonstrated inhibition of HIV replication in vitro using HLA-C*03, -07, -15 -restricted HIV-1-specific CTL clones. By examining HLA-driven virus evolution in an African cohort infected with HIV-1 clade C, Rousseau et al.60 also showed that HLA-C*04-restricted CTLs behave in a functionally similar manner to HLA-B-restricted CTLs and contribute to the in vivo CTL response, although HLA-B-restricted CTLs were found to be stronger overall. Finally, Makadzange et al.61 characterized HLA-C-restricted CTLs in an HIV-infected cohort and found that these CTLs contributed up to 54% of the total CTL response. In vitro analyses of these HLA-C-restricted CTLs demonstrated surface markers and activities to be phenotypically identical to HLA-A- or HLA-B-derived CTLs.61
The HIV variants that alter CTL epitopes allow the virally infected cell to evade CTL recognition and their existence provides evidence that CTLs exert anti-viral pressure in vivo. These substitutions sometimes result in a fitness cost for the virus that leads to lower viral loads.62,63 Recently, two studies reported HIV escape mutants within CTL epitopes restricted to two different HLA-C alleles (HLA-Cw*-03 and HLA-Cw*1202).64,65 In the study by Honeyborne et al.64 fitness constraints resulting from escape mutations were compensated for by mutations elsewhere within the Gag protein. The study by Honda et al.65 described CTLs directed against HLA-Cw*1202-restricted Pol epitopes that effectively suppressed HIV replication in vitro and that resulted in escape mutations in vivo. Interestingly, the HLA-Cw*1202 allele used in their analysis is in linkage disequilibrium with the -35CC allele.46 These data suggest that HLA-C-restricted CTLs may contribute to the control of viral replication in vivo.
Evolution of HIV Nef in response to HLA-C allele variation
Although the -35C allele is significantly associated with lower HIV viral load set point and slower disease progression than the -35T allele, the distributions overlap. This would suggest that some of the alleles associated with the -35C SNP may not induce an effective CTL response. Alternatively, the virus may compensate by evolving new mechanisms to evade the immune system. To examine this, Specht et al. identified subjects with the protective -35CC genotype but that exhibited high viral loads. They isolated HIV nef alleles and asked whether Nef had acquired the ability to down-modulate HLA-C in these patients.47 Interestingly, none of the Nef variants that they isolated affected HLA-C expression levels. These data suggest that there may be a substantial fitness cost to the virus if HLA-C levels are further reduced, which may be explained by the role of HLA-C as a ligand for inhibitory KIRs on NK cells (Fig. 4).
Surprisingly, Nef proteins isolated from patients with both the -35CC genotype and high viral loads had evolved to more effectively perform other Nef functions beneficial to the virus. For example, they were more efficient at down-modulating the HIV co-receptors CD4 and CXCR4, the T-cell co-stimulatory molecule CD28, and MHC class II invariant chain Ii than Nef proteins isolated from people with the -35TT genotype.47 Hence, in some people, the benefit of higher HLA-C levels may be overcome by the evolution of a generally more pathogenic virus.
Conclusion
Infection with HIV has a variable disease course and the host factors that influence rapid versus slow disease progression are not well understood. Individuals with high HLA-C expression may benefit from more effective recognition and lysis of HIV-infected cells by HLA-C-restricted CTLs (Fig. 4). Additionally, higher HLA-C expression in cross-presenting APCs may allow continued naive CD8+ T-cell priming during the chronic phase of infection, which would allow the development of new CTL responses to variant epitopes restricted to a variety of MHC-I allotypes (Fig. 3). The benefit of high HLA-C expression may be limited by the inhibitory effect of HLA-C on the NK cell (and possibly CTL) response. Elevated HLA-C expression may increase the risk of some types of infections or malignancies that require an effective NK cell response for control.66 The relative expression level of HLA-C, which has both costs and benefits, is an important phenotype that may evolve in response to pressure from pathogens.
Acknowledgments
K.L.C. and D.A.K. are supported by grants from the National Institutes of Health and the National Institute of Allergy and Infectious Diseases.
Disclosures
The authors have no conflicts of interest.
References
- 1.The International HIVCS. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–7. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fellay J, Shianna KV, Ge D, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–7. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fellay J, Ge D, Shianna KV, et al. Common genetic variation and the Control of HIV-1 in humans. PLoS Genet. 2009;5:e1000791. doi: 10.1371/journal.pgen.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams EJ, Parham P. Species-specific evolution of MHC class I genes in the higher primates. Immunol Rev. 2001;183:41–64. doi: 10.1034/j.1600-065x.2001.1830104.x. [DOI] [PubMed] [Google Scholar]
- 5.Hughes AL, Nei M. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature. 1988;335:167–70. doi: 10.1038/335167a0. [DOI] [PubMed] [Google Scholar]
- 6.Older Aguilar AM, Guethlein LA, Adams EJ, Abi-Rached L, Moesta AK, Parham P. Coevolution of killer cell Ig-like receptors with HLA-C to become the major variable regulators of human NK cells. J Immunol. 2010;185:4238–51. doi: 10.4049/jimmunol.1001494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moretta A, Bottino C, Vitale M, Pende D, Biassoni R, Mingari MC, Moretta L. Receptors for HLA class-I molecules in human natural killer cells. Annu Rev Immunol. 1996;14:619–48. doi: 10.1146/annurev.immunol.14.1.619. [DOI] [PubMed] [Google Scholar]
- 8.Blais M-E, Dong T, Rowland-Jones SL. HLA-C as a mediator of natural killer and T-cell activation: spectator or key player? Immunology. 2011;133:1–7. doi: 10.1111/j.1365-2567.2011.03422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snary D, Barnstable CJ, Bodmer WF, Crumpton MJ. Molecular structure of human histocompatibility antigens: the HLA-C series. Eur J Immunol. 1977;7:580–5. doi: 10.1002/eji.1830070816. [DOI] [PubMed] [Google Scholar]
- 10.McCutcheon JA, Gumperz J, Smith KD, Lutz CT, Parham P. Low HLA-C expression at cell surfaces correlates with increased turnover of heavy chain mRNA. J Exp Med. 1995;181:2085–95. doi: 10.1084/jem.181.6.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neisig A, Melief CJM, Neefjes J. Reduced cell surface expression of HLA-C molecules correlates with restricted peptide binding and stable TAP interaction. J Immunol. 1998;160:171–9. [PubMed] [Google Scholar]
- 12.Kulkarni S, Savan R, Qi Y, et al. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature. 2011;472:495–8. doi: 10.1038/nature09914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neefjes JJ, Ploegh HL. Allele and locus-specific differences in cell surface expression and the association of HLA class I heavy chain with β2-microglobulin: differential effects of inhibition of glycosylation on class I subunit association. Eur J Immunol. 1988;18:801–10. doi: 10.1002/eji.1830180522. [DOI] [PubMed] [Google Scholar]
- 14.Setini A, Beretta A, De Santis C, et al. Distinctive features of the α1-domain a helix of HLA-C heavy chains free of β2-microglobulin. Hum Immunol. 1996;46:69–81. doi: 10.1016/0198-8859(96)00011-0. [DOI] [PubMed] [Google Scholar]
- 15.Shimizu Y, DeMars R. Production of human cells expressing individual transferred HLA-A,-B,-C genes using an HLA-A,-B,-C null human cell line. J Immunol. 1989;142:3320–8. [PubMed] [Google Scholar]
- 16.Schaefer MR, Williams M, Kulpa DA, Blakely PK, Yaffee AQ, Collins KL. A novel trafficking signal within the HLA-C cytoplasmic tail allows regulated expression upon differentiation of macrophages. J Immunol. 2008;180:7804–17. doi: 10.4049/jimmunol.180.12.7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anfossi N, Doisne J-M, Peyrat M-A, et al. Coordinated expression of Ig-like inhibitory MHC class I receptors and acquisition of cytotoxic function in human CD8+ T cells. J Immunol. 2004;173:7223–9. doi: 10.4049/jimmunol.173.12.7223. [DOI] [PubMed] [Google Scholar]
- 18.Alter G, Altfeld M. NK cells in HIV-1 infection: evidence for their role in the control of HIV-1 infection. J Intern Med. 2009;265:29–42. doi: 10.1111/j.1365-2796.2008.02045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carrington M, Martin MP, van Bergen J. KIR-HLA intercourse in HIV disease. Trends Microbiol. 2008;16:620–7. doi: 10.1016/j.tim.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khakoo SI, Thio CL, Martin MP, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science. 2004;305:872–4. doi: 10.1126/science.1097670. [DOI] [PubMed] [Google Scholar]
- 21.Lopez-Vazquez A, Rodrigo L, Martinez-Borra J, et al. Protective effect of the HLA-Bw4I80 epitope and the killer cell immunoglobulin-like receptor 3DS1 gene against the development of hepatocellular carcinoma in patients with hepatitis C virus infection. J Infect Dis. 2005;192:162–5. doi: 10.1086/430351. [DOI] [PubMed] [Google Scholar]
- 22.Martin AM, Freitas EM, Witt CS, Christiansen FT. The genomic organization and evolution of the natural killer immunoglobulin-like receptor (KIR) gene cluster. Immunogenetics. 2000;51:268–80. doi: 10.1007/s002510050620. [DOI] [PubMed] [Google Scholar]
- 23.Hsu KC, Chida S, Geraghty DE, Dupont B. The killer cell immunoglobulin-like receptor (KIR) genomic region: gene-order, haplotypes and allelic polymorphism. Immunol Rev. 2002;190:40–52. doi: 10.1034/j.1600-065x.2002.19004.x. [DOI] [PubMed] [Google Scholar]
- 24.Winter CC, Gumperz JE, Parham P, Long EO, Wagtmann N. Direct binding and functional transfer of NK cell inhibitory receptors reveal novel patterns of HLA-C allotype recognition. J Immunol. 1998;161:571–7. [PubMed] [Google Scholar]
- 25.Andersson S, Fauriat JA, Malmberg HG, Ljunggren HG, Malmberg KJ. KIR acquisition probabilities are independent of self-HLA class I ligands and increase with cellular KIR expression. Blood. 2009;114:95–104. doi: 10.1182/blood-2008-10-184549. [DOI] [PubMed] [Google Scholar]
- 26.Anfossi N, André P, Guia S, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25:331–42. doi: 10.1016/j.immuni.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 27.Yawata M, Yawata N, Draghi M, Little A-M, Partheniou F, Parham P. Roles for HLA and KIR polymorphisms in natural killer cell repertoire selection and modulation of effector function. J Exp Med. 2006;203:633–45. doi: 10.1084/jem.20051884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carneiro VL, Lemaire DC, Bendicho MT, et al. Natural killer cell receptor and HLA-C gene polymorphisms among patients with hepatitis C: a comparison between sustained virological responders and non-responders. Liver Int. 2010;30:567–73. doi: 10.1111/j.1478-3231.2010.02212.x. [DOI] [PubMed] [Google Scholar]
- 29.Middleton D, Curran M, Maxwell L. Natural killer cells and their receptors. Transpl Immunol. 2002;10:147–64. doi: 10.1016/s0966-3274(02)00062-x. [DOI] [PubMed] [Google Scholar]
- 30.Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. 1998;391:397–401. doi: 10.1038/34929. [DOI] [PubMed] [Google Scholar]
- 31.Herschhorn A, Marasco WA, Hizi A. Antibodies and lentiviruses that specifically recognize a T cell epitope derived from HIV-1 Nef protein and presented by HLA-C. J Immunol. 2010;185:7623–32. doi: 10.4049/jimmunol.1001561. [DOI] [PubMed] [Google Scholar]
- 32.Roeth JF, Williams M, Kasper MR, Filzen TM, Collins KL. HIV-1 Nef disrupts MHC-I trafficking by recruiting AP-1 to the MHC-I cytoplasmic tail. J Cell Biol. 2004;167:903–13. doi: 10.1083/jcb.200407031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wonderlich ER, Williams M, Collins KL. The tyrosine binding pocket in the adaptor protein 1 (AP-1) mu1 subunit is necessary for Nef to recruit AP-1 to the major histocompatibility complex class I cytoplasmic tail. J Biol Chem. 2008;283:3011–22. doi: 10.1074/jbc.M707760200. [DOI] [PubMed] [Google Scholar]
- 34.Le Gall S, Erdtmann L, Benichou S, Berlioz-Torrent C, Liu L, Benarous R, Heard J-M, Schwartz O. Nef interacts with the μ subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity. 1998;8:483–95. doi: 10.1016/s1074-7613(00)80553-1. [DOI] [PubMed] [Google Scholar]
- 35.Lifson JD, Nowak MA, Goldstein S, et al. The extent of early viral replication is a critical determinant of the natural history of simian immunodeficiency virus infection. J Virol. 1997;71:9508–14. doi: 10.1128/jvi.71.12.9508-9514.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Wolf F, Spijkerman I, Schellekens PT, et al. AIDS prognosis based on HIV-1 RNA, CD4+ T-cell count and function: markers with reciprocal predictive value over time after seroconversion. AIDS. 1997;11:1799–806. doi: 10.1097/00002030-199715000-00003. [DOI] [PubMed] [Google Scholar]
- 37.van Manen D, Kootstra NA, Boeser-Nunnink B, Handulle MAM, van't Wout A, Schuitemaker H. Association of HLA-C and HCP5 gene regions with the clinical course of HIV-1 infection. AIDS. 2009;23:19–28. doi: 10.1097/QAD.0b013e32831db247. [DOI] [PubMed] [Google Scholar]
- 38.Mellors JW, Rinaldo CR, Gupta P, White RM, Todd JA, Kingsley LA. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272:1167–70. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- 39.Klein MR, Miedema F. Long-term survivors of HIV-1 infection. Trends Microbiol. 1995;3:386–91. doi: 10.1016/s0966-842x(00)88984-2. [DOI] [PubMed] [Google Scholar]
- 40.Munoz A, Sabin CA, Phillips AN. The incubation period of AIDS. AIDS. 1997;11(Suppl. A):S69–76. [PubMed] [Google Scholar]
- 41.Concerted AoStAaDiE. Time from HIV-1 seroconversion to AIDS and death before widespread use of highly-active antiretroviral therapy: a collaborative re-analysis. Lancet. 2000;355:1131–7. [PubMed] [Google Scholar]
- 42.Veugelers PJ, Page KA, Tindall B, et al. Determinants of HIV disease progression among homosexual men registered in the Tricontinental Seroconverter Study. Am J Epidemiol. 1994;140:747–58. doi: 10.1093/oxfordjournals.aje.a117322. [DOI] [PubMed] [Google Scholar]
- 43.Han Y, Lai J, Barditch-Crovo P, Gallant JE, Williams TM, Siliciano RF, Blankson JN. The role of protective HCP5 and HLA-C associated polymorphisms in the control of HIV-1 replication in a subset of elite suppressors. AIDS. 2008;22:541–4. doi: 10.1097/QAD.0b013e3282f470e4. 10.1097/QAD.0b013e3282f470e4. [DOI] [PubMed] [Google Scholar]
- 44.Corrah TW, Goonetilleke N, Kopycinski J, Deeks SG, Cohen MS, Borrow P, McMichael A, Brackenridge S. A reappraisal of the relationship between the HIV-1-protective single nucleotide polymorphism 35kb upstream of the HLA-C gene and surface HLA-C expression. J Virol. 2011;85:3367–74. doi: 10.1128/JVI.02276-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stranger BE, Forrest MS, Clark AG, et al. Genome-wide associations of gene expression variation in humans. PLoS Genet. 2005;1:e78. doi: 10.1371/journal.pgen.0010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas R, Apps R, Qi Y, et al. HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat Genet. 2009;41:1290–4. doi: 10.1038/ng.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Specht A, Telenti A, Martinez R, et al. Counteraction of HLA-C-mediated immune control of HIV-1 by Nef. J Virol. 2010;84:7300–11. doi: 10.1128/JVI.00619-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, Farthing C, Ho DD. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. 1994;68:4650–5. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allen TM, Altfeld M, Geer SC, et al. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J Virol. 2005;79:13239–49. doi: 10.1128/JVI.79.21.13239-13249.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bansal A, Yue L, Conway J, et al. Immunological control of chronic HIV-1 infection: HLA-mediated immune function and viral evolution in adolescents. AIDS. 2007;21:2387–97. doi: 10.1097/QAD.0b013e3282f13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore CB, Johh M, James IR, Christiansen FT, Witt CS, Mallal SA. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science. 2002;296:1439–43. doi: 10.1126/science.1069660. [DOI] [PubMed] [Google Scholar]
- 52.Bhattacharya T, Daniels M, Heckerman D, et al. Founder effects in the assessment of HIV polymorphisms and HLA allele associations. Science. 2007;315:1583–6. doi: 10.1126/science.1131528. [DOI] [PubMed] [Google Scholar]
- 53.Leslie AJ, Pfafferott KJ, Chetty P, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med. 2004;10:282–9. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- 54.Brumme ZL, Brumme CJ, Heckerman D, et al. Evidence of differential HLA class I-mediated viral evolution in functional and accessory/regulatory genes of HIV-1. PLoS Pathog. 2007;3:e94. doi: 10.1371/journal.ppat.0030094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Novitsky V, Wang R, Margolin L, et al. Timing constraints of in vivo gag mutations during primary HIV-1 subtype C infection. PLoS ONE. 2009;4:e7727. doi: 10.1371/journal.pone.0007727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Treurnicht FK, Seoighe C, Martin DP, et al. Adaptive changes in HIV-1 subtype C proteins during early infection are driven by changes in HLA-associated immune pressure. Virology. 2010;396:213–25. doi: 10.1016/j.virol.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kiepiela P, Ngumbela K, Thobakgale C, et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med. 2007;13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- 58.Honeyborne I, Prendergast A, Pereyra F, et al. Control of human immunodeficiency virus type 1 is associated with HLA-B*13 and targeting of multiple gag-specific CD8+ T-cell epitopes. J Virol. 2007;81:3667–72. doi: 10.1128/JVI.02689-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adnan S, Balamurugan A, Trocha A, Bennett MS, Ng HL, Brander C, Yang OO. Nef interference with HIV-1-specific CTL antiviral activity is epitope specific. Blood. 2006;108:3414–19. doi: 10.1182/blood-2006-06-030668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rousseau CM, Daniels MG, Carlson JM, et al. HLA class I-driven evolution of human immunodeficiency virus type 1 subtype C proteome: immune escape and viral load. J Virol. 2008;82:6434–46. doi: 10.1128/JVI.02455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Makadzange AT, Gillespie G, Dong T, et al. Characterization of an HLA-C-restricted CTL response in chronic HIV infection. Eur J Immunol. 2010;40:1036–41. doi: 10.1002/eji.200939634. [DOI] [PubMed] [Google Scholar]
- 62.Kelleher AD, Long C, Holmes EC, et al. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J Exp Med. 2001;193:375–86. doi: 10.1084/jem.193.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martinez-Picado J, Prado JG, Fry EE, et al. Fitness cost of escape mutations in p24 gag in association with control of human immunodeficiency virus type 1. J Virol. 2006;80:3617–23. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Honeyborne I, Codoner FM, Leslie A, et al. HLA-Cw*03-restricted CD8+ T-cell responses targeting the HIV-1 gag major homology region drive virus immune escape and fitness constraints compensated for by intracodon variation. J Virol. 2010;84:11279–88. doi: 10.1128/JVI.01144-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Honda K, Zheng N, Murakoshi H, et al. Selection of escape mutant by HLC-C-restricted HIV-1 Pol-specific cytotoxic T lymphocytes carrying strong ability to suppress HIV-1 replication. Eur J Immunol. 2011;41:97–106. doi: 10.1002/eji.201040841. [DOI] [PubMed] [Google Scholar]
- 66.Martin M, Borecki I, Zhang Z, et al. HLA-Cw group 1 ligands for KIR increase susceptibility to invasive cervical cancer. Immunogenetics. 2010;62:761–5. doi: 10.1007/s00251-010-0477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]