

Abstract
Background
There is a large amount of inconsistency in gene structure annotations of bacterial strains. This inconsistency is a frustrating impedance to effective comparative genomic analysis of bacterial strains in promising applications such as gaining insights into bacterial drug resistance.
Results
Here, we propose CAMBer as an approach to support comparative analysis of multiple bacterial strains. CAMBer produces what we called multigene families. Each multigene family reveals genes that are in one-to-one correspondence in the bacterial strains, thereby permitting their annotations to be integrated. We present results of our method applied to three human pathogens: Escherichia coli, Mycobacterium tuberculosis and Staphylococcus aureus.
Conclusions
As a result, more accurate and more comprehensive annotations of the bacterial strains can be produced.
Background
Large amounts of genomic information are currently being generated, including whole-genome sequences of multiple strains of many bacterial species. The availability of these sequences provides exciting opportunities and applications for comparative genomic analysis of multiple bacterial strains. For example, comparative genomic analysis of the avirulent H37Ra and virulent H37Rv strains of M. tuberculosis provides insights into the virulence and pathogenesis of M. tuberculosis[1]. As another example, comparative genomic analysis of three linezolid-resistant S. pneumoniae strains identified three mutations and the associated genes involved in antibiotic resistance [2]. As a last example, an ingenious comparative genomic analysis of susceptible and resistant strains of M. tuberculosis and M. smegmatis found that the only gene commonly affected in all three resistant strains encodes atpE, thereby uncovering the mode of action of the novel class of compound Diarylquinoline to be the inhibition of the proton pump of M. tuberculosis ATP synthase [3].
These impressive results were achieved by integrating and connecting information generated during the sequencing of multiple distinct strains of the bacteria species mentioned. In order to repeat these past successes, there is a need for a general annotation consensus, as the physical and functional annotations of the strains of the same bacteria differ significantly in some cases. As an extreme case of the problem, the strains of E. coli reportedly have only 20% of their genes in common [4]. One cause for the inconsistency of gene annotations is sequencing errors. For example, surprised by the higher similarity between H37Ra and CDC1551 M. tuberculosis strains than that between H37Ra and H37Rv, Zheng et al. [1] re-sequenced the relevant loci in H37Rv and discovered a mere 6 out of 85 of the variations were genuine and the rest were sequencing errors [1]. A second cause for gene annotation inconsistency is gene structure prediction errors. For example, when Wakaguri et al. determined the entire sequences of 732 cDNAs in T. gondii to evaluate earlier annotated gene models of T. gondii at the complete full-length transcript level, they found that 41% of the gene models contained at least one inconsistency [5]. Also, a persistent weakness of gene structure prediction methods is the accuracy of start codon assignment [6], giving rise to a significant amount of gene annotation inconsistency from the resulting gene size variations. Another cause for the inconsistency of gene annotations is the inability to put genes from different strains into correct gene families. For example, the extreme case of E. coli is probably due to the simple-minded BLAST reciprocal pairwise comparison that was used in [4] to identify genes belonging to the same gene family. This strategy may identify as few as 15% of genes that are known to have evolutionary relationship; a more sophisticated strategy based on linking by intermediate sequences—a strategy that we also adopt—may increase the ability to recognize genes evolutionary relationship by 70% [7].
This is a frustrating state of affairs for both biologists and bioinformaticians. Therefore, we require structured, exhaustive, comparative databases. While broad-based, web-technology-enabled community annotation has been proposed as a solution to the problem [8], it is feasible only for species having a large interested research community. Unfortunately, this may not be the case for many bacterial strains such as M. Tuberculosis due to, for example, insufficient profit opportunity [9]. Another well-known effort is the Fellowship for Interpretation of Genomes [10], which has developed and successfully applied a tool called SEED [11] to support functional annotations of bacterial strains, based on a concept of integrating annotations among multiple bacterial strains in a so-called “subsystem” or gene-family-centric manner. SEED [11] provides functions for navigating and annotating genes such as identifying similar genes from other organisms and comparing their neighborhoods. These functionalities allow users to investigate how a given gene relates to other genes and permit them to update and extend the annotation database via a web interface. However, this process is not automated and the functionalities are more customized for gene function annotation than for gene structure annotation.
Therefore, we should explore the development of alternative approaches and technologies that integrate, connect, and produce consensus gene annotations to support comparative analysis of multiple bacterial strains. We have designed CAMBer to support comparative analysis of multiple bacterial strains. CAMBer approaches the problem as follows. First, we use intermediate sequences—a tactic originally proposed for enhancing FASTA’s ability to detect evolutionary relationship [7]—to link multiple annotations on a gene. We call the resulting structure a multigene. Next, multigenes are linked by BLAST edges between their elements into a consolidation graph. Multigenes in the same connected component of the graph are proposed to form a family. Finally, we use genomic context information—a tactic originally proposed for enhancing gene function prediction [12]—to refine the consolidation graph. This way we obtain more multigene families where the multigenes in each family are in one-to-one relationship in the bacterial strains considered. These resulting multigene families can be used to support more detailed comparative analysis of multiple bacterial strains for detecting sequencing error, identifying mutations for drug resistance, and other purposes.
In the remainder of this paper, we present the details of CAMBer and our results on M. tuberculosis, S. aureus and E. coli. A preliminary version of CAMBer was described in [13]. The current paper differs from the preliminary version by (i) a more careful analysis and handling of noise due to short possibly erroneous annotations, (ii) testing on more species, (iii) demonstrating scalability on a much larger set of strains, (iv) an analysis of core vs pangenomes, and (v) a substantially revised CAMBer software release—CAMBer is available at http://bioputer.mimuw.edu.pl/camber and can now be run on computer clusters powered by Sun Grid Engine.
Methods
We present here the details of our approach. We assume that we have a set of bacterial strains whose genomes have been sequenced and annotated. The goal is to arrive at revised annotations of the strains which arise from projecting an annotation of one strain onto the annotations of another. Furthermore, we focus on Translation Initiation Site (TIS) annotations. In this operation, we do not remove the original TIS in the second strain, but rather add new TISs suggested by the annotations of the first one. In particular, we may arrive at new annotated genes in the second strain. In this way, we naturally arrive at the concept of a multigene which is just a gene with possibly several TISs.
More precisely: Given an annotation A in strain S1 and let x be an ORF which according to A encodes a gene in S1. We run BLAST with query x against the sequence of a genome of a strain S2. Let y' be a hit in S2 returned by BLAST to the query x and let y be the sequence obtained from y' by extending it to the nearest stop codon (in the 3’ direction on the same strand as y'). We call y an acceptable hit with respect to x if the following five conditions are satisfied:
• y starts with one of the appropriate start codons: ATG, GTG, TTG.
• The BLAST hit y' has aligned beginning of the query x with the beginning of y'.
• The e-value score of the BLAST hit from x to y' is below a given threshold et (typically it is set to 10 –10 or 10 –20).
• The ratio of the length of y to x is less than 1 + pt and greater than 1 – pt, where pt is a given threshold (typically 0.2 or 0.3). This condition is imposed in order to keep similar lengths of related sequences.
• The percent of identity of the hit (calculated as the number of identities divided by the query length times 100) is above a length-dependent threshold given by the HSSP curve [14]. The curve was originally designed for amino-acid queries, in our case we use the formula:
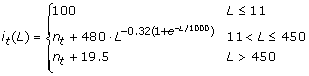 |
where L is the floor of the number of aligned nucleotide residues divided by 3. Typically nt is set to 30.5% or 50.5%.
So, assuming that we use BLAST with default parameters, our method has three specific parameters: e-value threshold et, length tolerance threshold pt, and length-depended percent of identity threshold implied by nt.
It follows from the definition above that an acceptable hit y may overlap a gene annotated in S2 in the same frame, sharing the same stop codon, but having a different TIS. As mentioned above, this gives rise to the notion of a multigene. Different TISs in a multigene g give rise to different putative genes. We call them elements of g. Obviously genes can be viewed as multigenes with just one element.
Therefore, we have two types of gene structure annotations in the rest of this paper. The first type of annotations are the original annotations (of genes) given along with the input sequenced genomes. The second type of annotations are the predicted annotations (of multigenes) putatively transferred from one genome to another in the multigene construction and closure processes.
Consolidation graph
We compute iteratively a closure of annotations which is based on the above described operation of taking acceptable hits. Initially, as step zero, we take original annotations which are furnished with the genomic sequences. Assume that at step i ≥ 0 we have an annotation 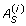 associated with each strain S.
associated with each strain S.
Annotation 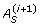 in the step i + 1 is obtained by taking all acceptable hits in S for the queries ranging over all genes annotated in
in the step i + 1 is obtained by taking all acceptable hits in S for the queries ranging over all genes annotated in 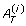 , for every other strain T. This process stops when no new acceptable hit is obtained. This process generalizes a proven strategy for identifying more homologs by linking intermediate sequences [7].
, for every other strain T. This process stops when no new acceptable hit is obtained. This process generalizes a proven strategy for identifying more homologs by linking intermediate sequences [7].
Having computed the closure we can construct now a consolidation graph G. Its nodes are all multigenes obtained during the process of computing the closure. There is an edge from a multigene g to a multigene g' if one of the elements of g' is obtained as an acceptable hit with respect to one of the elements of g. Figure 1 illustrates the process of computing the closure, as well as a construction of the consolidation graph.
Figure 1.

Schema of the method Schema of our method to represent the structure of multigenes. For clarity of presentation only one step of the procedure is shown. Square brackets correspond to stop codons of annotated genes, while round brackets with a star correspond to start codons of annotated genes. Round brackets without a star correspond to putative genes indicated by our method (new elements of the multigene). a) Input annotations for strains indicate the initial state of the procedure. b) Dashed arrows indicate acceptable hits. The reader should notice a birth of a second element, rendering a multigene with two elements. c) Two examples of edges in the consolidation graph. Dots represent different elements of a multigene which is represented here as a rectangle. Edges connecting dots represent acceptable hits (we ignore directions here). Edges between rectangles represent edges of the consolidation graph.
Refinement of the consolidation graph
Connected components of a consolidation graph G represent multigene families with a common ancestor. Our next goal is refining the multigene homology relation represented by edges in G to obtain as many one-to-one homology classes as possible, i.e. having at most one multigene per strain in such a class. We call a connected component of G an anchor if it includes at most one multigene for every strain.
One-element anchors are called orphans. Non-anchors are the components which fail to be anchors. Obviously the definitions of anchors, orphans, and non-anchors apply to any graph with nodes being multigenes from various strains.
Multigenes in the same anchor are potentially orthologous to each other. In contrast, a non-anchor contains at least two multigenes that are potentially non-orthologous. Genomic context information has been successfully used to clarify gene relationships and improve gene function prediction [12]. So, we propose exploiting genomic context information to analyse and decompose non-anchors into smaller connected subgraphs that can emerge as anchors in the resulting refined consolidation graph.
Our construction of the refinement proceeds in stages. At each stage we carry a graph which is a subgraph of the graph from the previous stage. At stage 0, the original consolidation graph G is used as the initial input graph G(0).
Suppose we have at stage i a graph G(i). We restrict this graph by performing the following test on each pair (g,g') of multigenes originating from strains S1 and S2, connected by an edge in G(i) which belongs to a non-anchor component of G(i). Let a be the nearest left neighbor multigene of g in S1 which belongs to an anchor of G(i) containing a multigene from S2. Let b be the nearest right neighbor multigene of g in S1 which belongs to an anchor of G(i) containing a multigene from S2. In similar way define left (a') and right (b') neighbors of g' in S2. Assuming that all four multigenes a, a', b, b' exist, we keep the edge connecting g and g' in G(i+1) if either (a, a') and (b,b') (see Figure 2 (a)), or (a, b') and (b, a') (see Figure 2 (b)) are edges in G(i), i.e. the corresponding pairs are in the same anchors of G(i). If at least one of the multigenes a, a', b, b' does not exist, the edge connecting g and g' in G(i+1) is not copied from G(i). The procedure stops when no edge is removed from the current graph. We call the resulting graph a refinement of G. Figure 2 (c) shows a situation when we have to retain two edges, leading to a cluster with unresolved one-to-one relationship. These cases may get resolved later when more anchors are obtained.
Figure 2.

Refinement procedure One step of the refinement procedure. Rectangles denote multigenes which belong to non-anchors in the current stage. Rhombus denotes a multigene which is already in an anchor at this stage. Edges connecting rectangles (dashed and solid) are edges of the graph of the current stage. Edges connecting rhombuses are the anchor edges. ‘YES’ means that the edge is keep for the next stage, while ‘NO’ means it is omitted. Parts a) and b) illustrate two situations when we can select one of the edges and leaving out the other. Part c) illustrates the situation when we cannot make such a decision, leading to an unresolved cluster. Both edges are kept in the graph of the next stage. Such a cluster may be resolved at a later stage. Other cases which lead to omitting the edges are possible too.
Time complexity
The most time consuming operation in the closure procedure is running BLAST. We denote by blast() the BLAST running time. Let k be the number of all considered strains and let n be the maximal number of annotated genes in the genomes under consideration. For each strain during the closure operation we use every identified or annotated ORF only once. Assuming that the number of newly discovered multigenes does not grow fast, we can estimate the total time of the procedure as k2 * n * blast().
Now, we estimate time complexity of one iteration in the refinement procedure. Again, let k be the number of all considered strains and let n be the maximal number of identified multigenes among all strains. Denote by m the number of non-anchors in the consolidation graph. Additionally, let p denote the maximal number of multigenes for one strain among all non-anchor components. In order to find the nearest left and right neighbors of a multigene in linear time we first sort all of them. This takes time k ∗ n ∗ log n. Since we have at most 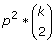 edges to check for support of the neighboring anchors (checking for support may take time at most n), for each of the m non-anchors, it follows that the estimated total time to resolve all of the m non-anchors is
edges to check for support of the neighboring anchors (checking for support may take time at most n), for each of the m non-anchors, it follows that the estimated total time to resolve all of the m non-anchors is 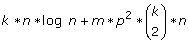 .
.
Results and Discussion
Our approach, called CAMBer, was applied to 9 M. tuberculosis (MTB), 22 S. aureus and 41 E. coli strains. It was ran with the following parameters: et = 10–10, pt = 0.3 and nt = 30.5%. In our earlier work [13], we used the constant percent of identity threshold (=50%), but finally we decided to use length-dependent percent of identity as we obtained much fewer suspicious very-short predictions. The input datasets comprise nucleotide genome sequences and gene structure annotations of protein-coding genes of the strains in each case study. However, annotations for pseudo genes were filtered. The input datasets were generally taken from GenBank [15], with the exception of six M. tuberculosis strains. The input datasets for three of these strains came from the Broad Institute database; while the remaining three came from the supplementary material of [16].
Mycobacterium tuberculosis case study
Tuberculosis is still a major cause of deaths worldwide, in particular due to still poorly-understood mechanisms of drug resistance. The first fully sequenced M. tuberculosis strain was H37Rv in 1998 and since then several new strains have been sequenced [1,16-18].
Table 1 gives details of the strains. We notice that there is substantial variance (left box plot in Figure 3) in the number of originally annotated genes. This is probably due to different gene-finding tools and methodologies being applied by different labs, rather than the real genomic composition.
Table 1.
M. tuberculosis dataset
| strain ID | source | resist. | # of genes | lab. |
|---|---|---|---|---|
| H37Rv | NC_000962 | DS | 3988(26) | S |
| H37Ra | NC_009525 | DS | 4034(22) | C |
| F11 | NC_009565 | DS | 3941(5) | B |
| KZN 4207(T) | PLoS One. [16] | DS | 3902(47) | T |
| KZN 4207(B) | Broad Institute | DS | 3996(4) | B |
| KZN 1435 | Broad Institute | MDR | 4059(10) | B |
| KZN V2475 | PLoS One. [16] | MDR | 3893(3792) | T |
| KZN 605 | Broad Institute | XDR | 4024(26) | B |
| KZN R506 | PLoS One. [16] | XDR | 3902(46) | T |
Details for input strains for the M. tuberculosis case study. The first number in column called ’# of genes’ corresponds to the number of annotated genes, the second (in brackets) corresponds to the number of genes excluded in the study due to unusual start or stop codons or sequence length not divisible by three. In order to avoid ambiguity in naming the same strain sequenced by two labs we introduce an additional suffix (T or B). Characters in last column, called ’lab.’, describe the sequencing laboratories: B - The Broad Institute, T - Texas A&M University, C - Chinese National Human Genome Center at Shanghai, S - Sanger Institute.
Figure 3.

M. tuberculosis, before and after the closure Left: deviation from mean (=3957) in numbers of annotated protein coding genes (KZN V2475 is omitted, because only 101 genes have correct annotation due to a shift in the annotated gene coordinates). Right: deviation from mean (=4146) in numbers of multigenes after unification by the closure procedure. The same scale is used for both charts. Level 0 in the Y axis corresponds to the mean value.
It is quite remarkable that variance in the number of predicted multigenes after the closure is much smaller (right box plot in Figure 3). The reader may also compare the corresponding data presented in Tables 1 and 2. Table 2 shows for each strain the distribution of multigenes with respect to the number of elements (TISs). By far the largest group in each strain are one-element multigenes. Also, Figure 4 shows that the predicted multigenes are quite even distributed in terms of gene length.
Table 2.
Statistics of multigene start sites after the closure procedure for the M. tuberculosis case study. M. tuberculosis, multigene start sites statistics Multigene start sites statistics after the closure procedure.
| # of multigenes with | ||||||
|---|---|---|---|---|---|---|
| 5 elt. | 4 elt. | 3 elt. | 2 elt. | 1 elt. | total | |
| F11 | 1 | 6 | 68 | 605 | 3475 | 4155 |
| H37Ra | 1 | 5 | 66 | 607 | 3488 | 4167 |
| H37Rv | 1 | 6 | 66 | 602 | 3483 | 4158 |
| KZN 605 | 1 | 6 | 68 | 602 | 3457 | 4134 |
| KZN 1435 | 1 | 6 | 69 | 597 | 3472 | 4145 |
| KZN 4207(T) | 1 | 6 | 70 | 600 | 3463 | 4140 |
| KZN R506 | 1 | 6 | 70 | 602 | 3459 | 4138 |
| KZN V2475 | 1 | 6 | 70 | 601 | 3461 | 4139 |
| KZN 4207(B) | 1 | 5 | 69 | 602 | 3465 | 4142 |
Figure 4.

M. tuberculosis, distribution of gene lengths Histograms of gene lengths in logarithmic scale (base = 10) for all M. tuberculosis taken together. The x-axis is quantified into ranges of length 0.1. Black dotted line presents the distribution of annotated gene lengths, blue solid line shows the distribution of multigene lengths, red dashed line presents the distribution of length of multigenes with no annotated elements.
The careful reader may have also noticed that the same strain (KZN 4207) sequenced in two labs has quite different numbers of annotated genes (3902 vs. 3996); but after consolidation we have for these two genomes almost the same number of multigenes (4140 vs. 4142).
This case study shows that the method can also be applied to completely unannotated genomes, yielding an initial annotation of a newly sequenced genome. For example, due to a shift in annotation coordinates for the strain KZN V2475 we removed most of the gene annotations (after the shift). Using our method, we were able to annotate 4139 multigenes in the genome.
After refinement of the consolidation graph, the number of connected components rose from 4177 to 4287, but size of the largest component dropped from 127 (there are two such components in the consolidation graph) to 15 (only one such component after refinement). Also the maximal number of multigenes in one strain and in one non-anchor dropped from 17 in the consolidation graph to 3 in the refined consolidation graph.
It is interesting to compare the two largest components of the consolidation graph. As mentioned above they have in total 127 multigenes, each strain having between 12 and 17 multigenes in these non-anchors. What is remarkable here is that H37Rv, having 16 multigenes in each of the two components, has all of these 32 genes annotated in the Tuberculist database (http://tuberculist.epfl.ch/) as transposons which belong to the same insertion element (IS6110). Even though these two non-anchors were not successfully resolved by the refinement procedure, the resulting non-anchors (four obtained from each of the original two large non-anchors in the consolidation graph) are pretty small: at most two multigenes per strain.More precisely, each of the original non-anchors was split by the refinement procedure into 34 subclusters (4 non-anchors, and 30 anchors with 9 orphans).
The consolidation graph contains 4177 connected components, with only 43 components (about 1%) being non-anchors and 48 being orphans. After the refinement procedure we obtained slightly more connected components (4287), but the number of non-anchors substantially dropped to 22 (Table 3). Figure 5 gives another point of view for the refinement procedure results.
Table 3.
Statistics of the connected components before and after refinement for the M. tuberculosis case study. M. tuberculosis, before and after refinement Statistics of the connected components before and after refinement.
| # of connected components before refinement | # of connected components after refinement | |
|---|---|---|
| all connected components | 4177 | 4287 |
| non-anchors | 43 | 22 |
| anchors | 4134 | 4265 |
| orphans | 48 | 68 |
| core anchors | 3943 | 4012 |
| core connected components | 3985 | 4030 |
Figure 5.

M. tuberculosis, distribution of connected components Histogram of the number of connected components (y-axis) shared by a particular number of strains (x-axis). For better clarity only numbers of connected components after the refinement procedure are shown.
With this approach we were also able to discover five cases of gene fusion/fission in the investigated genomes which seems pretty unusual for such closely related strains. We leave the analysis of this phenomenon for further study.
See additional file 1 for detailed summary of the case study results.
Staphylococcus aureus case study
Since penicillin was introduced for S. aureus treatment in 1943, penicillin resistance has become common among S. aureus isolates [19]. Two meticillin-resistant strains (N315 and Mu50) are the first fully sequenced S. aureus genomes [20].
Genome sequences and annotations of 22 fully sequenced strains were used in our study. At the time of writing, these were the only available S. aureus strains with “completed” sequencing status. Table 4 presents details of the strains.
Table 4.
S. aureus dataset. Details for input strains for the S. aureus case study. The first number in column called ’# of genes’ corresponds to the number of annotated genes, the second (in brackets) corresponds to the number of genes excluded in the study due to unusual start or stop codons or sequence length not divisible by three.
| strain ID | source (GenBank ID) | # of genes | genome length | lab. |
|---|---|---|---|---|
| TW20 0582 | FN433596 | 2769(5) | 3043210 | Welcome Trust Sanger Institute |
| JKD6008 | CP002120 | 2680(0) | 2924344 | Monash University |
| JH9 | CP000703 | 2769(5) | 2906700 | US DOE Joint Genome Institute |
| JH1 | CP000736 | 2680(0) | 2906507 | US DOE Joint Genome Institute |
| MRSA252 | BX571856 | 2697(0) | 2902619 | Sanger Institute |
| Mu3 | AP009324 | 2746(0) | 2880168 | Juntendo University |
| Newman | AP009351 | 2655(5) | 2878897 | Juntendo University |
| Mu50 | BA000017 | 2699(63) | 2878529 | Juntendo University |
| USA300 TCH1516 | CP000730 | 2624(0) | 2872915 | Baylor College of Medicine |
| USA300 FPR3757 | CP000255 | 2699(61) | 2872769 | University of California, San Francisco |
| ST398 S0385 | AM990992 | 2657(0) | 2872582 | University Medical Centre Utrecht |
| ED133 | CP001996 | 2560(0) | 2832478 | University of Edinburgh |
| ED98 | CP001781 | 2699(0) | 2824404 | University of Edinburgh |
| 04-02981 | CP001844 | 2653(2) | 2821452 | Robert Koch Institute |
| NCTC 8325 | CP000253 | 2661(0) | 2821361 | University of Oklahoma Health Sciences Center |
| MW2 | BA000033 | 2650(59) | 2820462 | NITE |
| N315 | BA000018 | 2892(0) | 2814816 | Juntendo University |
| JKD6159 | CP002114 | 2632(6) | 2811435 | University of Melbourne |
| COL | CP000046 | 2593(59) | 2809422 | TIGR |
| TCH60 | CP002110 | 2555(1) | 2802675 | Baylor College of Medicine |
| MSSA476 | BX571857 | 2672(1) | 2799802 | Sanger Institute |
| RF122 | AJ938182 | 2673(0) | 2742531 | University of Minnesota |
In this medium-size case study most of the results and corollaries are similar to the M. tuberculosis case study. However, we highlight below three interesting observations. The first observation is that there is a much large number of short predicted multigenes compared to the number of short original annotated genes, as shown in Figure 6. This contrasts sharply with the situation for M. tuberculosis depicted in Figure 4. This means that in S. aureus, many strains have short original annotations that are annotated to one of them but not to other strains, even though highly homologous regions exist in other strains. This suggests possible higher occurrence of annotation errors in short genes of S. aureus, especially in strains like NCTC8325; see Figure 7. The second observation is that the computing of the closure took 8 iterations, which is similar to the much larger study of E. coli (8 iterations) and more than the M. tuberculosis case study (3 iterations). The third observation is that the maximal number of TISs in a multigene is 13 (see Table 5 for more details), where for E. coli it is 9 and for M. tuberculosis 5. As in the other case studies we observe uneven distribution in the number of original annotated genes; see Figure 7. To assess the degree of unevenness we calculated the mean absolute difference in counts coming from two neighboring strains, where strains are ordered in decreasing order of the size of their genomes. It is 78 for the original annotation curve vs. 70 for the curve constructed after the closure operation, which further drops to 29 after post-processing removal of multigenes shorter than 200 nucleotides. This inconsistency was probably caused by different gene-finding methodologies applied by different labs. Curves like those presented in Figure 7 allow us also to estimate which labs were more conservative and which were more liberal when calling a given ORF a gene. For example, we observe a big peak in the number of original annotated genes for the strain NCTC8325, suggesting that this is perhaps the case of a more liberal annotation. Indeed, we investigated the number of connected components with multigenes present in all strains but have original annotations in only one strain. It turned out that there are only 7 strains that contribute at least one such connected component, of which the strain NCTC8325 contributes the highest number (22), with the second strain being USA300 TCH1516 (18). All other strains contributed less than 4 such components. An example of a strain with a rather conservative annotation is USA300 FPR3757, as can be clearly seen from a dip of the curve in Figure 7.
Figure 6.

S. aureus, distribution of gene lengths Histograms of gene lengths in logarithmic scale (base = 10) for all S. aureus strains taken together. The x-axis is quantified into ranges of length 0.1. Black dotted line presents the distribution of annotated gene lengths, blue solid line shows the distribution of multigene lengths, red dashed line presents the distribution of length of multigenes with no annotated elements.
Figure 7.

S. aureus, before and after the closure This plot presents numbers of annotated genes and numbers of the multigenes after the closure procedure applied to S. aureus strains. On the x-axis strains are listed (from left to right) in descending order of their genome length. The blue line annotated and the red line closure present respectively the number of annotated genes and the number of multigenes (after the closure) for each strain. The green line removal of <200 presents the number of multigenes after the closure and after applied post-processing of removal multigenes shorter than 150 nucleotides length.
Table 5.
Statistics of multigene start sites after the closure procedure for the S. aureus case study. S. aureus, multigene start sites statistics Multigene start sites statistics after the closure procedure.
| # of multigenes with | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 13 elt. | 10 elt. | 9 elt. | 8 elt. | 7 elt. | 6 elt. | 5 elt. | 4 elt. | 3 elt. | 2 elt. | 1 elt. | total | |
| TW20 | 0 | 0 | 1 | 0 | 1 | 3 | 9 | 45 | 224 | 823 | 2183 | 3289 |
| JKD6008 | 0 | 0 | 1 | 0 | 1 | 2 | 8 | 44 | 218 | 827 | 2058 | 3159 |
| JH9 | 0 | 0 | 0 | 1 | 0 | 0 | 10 | 42 | 240 | 805 | 2052 | 3150 |
| MRSA252 | 9 | 0 | 0 | 0 | 1 | 2 | 8 | 44 | 207 | 805 | 1970 | 3046 |
| Mu3 | 0 | 0 | 0 | 1 | 0 | 0 | 12 | 39 | 235 | 789 | 2032 | 3108 |
| Newman | 0 | 0 | 1 | 0 | 1 | 2 | 12 | 46 | 231 | 818 | 2089 | 3200 |
| Mu50 | 0 | 0 | 0 | 1 | 0 | 0 | 12 | 39 | 234 | 788 | 2033 | 3107 |
| USA300 TCH1516 | 0 | 0 | 1 | 0 | 1 | 2 | 12 | 49 | 237 | 815 | 2020 | 3137 |
| USA300 FPR3757 | 0 | 0 | 1 | 0 | 1 | 2 | 12 | 49 | 239 | 813 | 2016 | 3133 |
| ST398 | 0 | 0 | 1 | 0 | 0 | 0 | 6 | 39 | 198 | 768 | 2017 | 3029 |
| ED133 | 0 | 0 | 1 | 0 | 0 | 1 | 9 | 41 | 212 | 762 | 1946 | 2972 |
| ED98 | 0 | 0 | 0 | 1 | 0 | 0 | 11 | 38 | 235 | 769 | 1974 | 3028 |
| 04-02981 | 0 | 0 | 0 | 1 | 0 | 0 | 11 | 40 | 236 | 778 | 1967 | 3033 |
| NCTC8325 | 0 | 0 | 1 | 0 | 1 | 2 | 11 | 44 | 228 | 799 | 2044 | 3130 |
| MW2 | 0 | 0 | 0 | 0 | 0 | 3 | 11 | 45 | 230 | 790 | 1948 | 3027 |
| N315 | 0 | 0 | 0 | 1 | 0 | 0 | 12 | 40 | 234 | 765 | 1947 | 2999 |
| JKD6159 | 6 | 1 | 0 | 0 | 0 | 0 | 9 | 38 | 208 | 760 | 1880 | 2902 |
| COL | 0 | 0 | 1 | 0 | 1 | 2 | 13 | 49 | 234 | 785 | 1964 | 3049 |
| TCH60 | 4 | 1 | 1 | 0 | 0 | 1 | 8 | 48 | 192 | 776 | 1936 | 2967 |
| MSSA476 | 0 | 0 | 0 | 0 | 0 | 3 | 11 | 42 | 225 | 780 | 1933 | 2994 |
| RF122 | 0 | 0 | 0 | 0 | 0 | 5 | 8 | 40 | 186 | 706 | 1905 | 2850 |
It is rather expected that most of the inconsistencies concern short genes, leading to a sudden increase in the number of short multigenes after the closure procedure; see Figure 6. Therefore, it is interesting to investigate the cases where new long multigenes are predicted after the closure. There are in total 31 connected components with multigenes of length at least 300 nucleotides which were originally annotated in less than half of the strains. Two of them have multigenes in all 22 strains with only one originally annotated element. More precisely these two connected components were contributed by genes SAOUHSC_00630 and SAOUHSC_01489 annotated in NCTC8325. Both these genes are overlapped by genes which have original annotations in all remaining strains, which suggests that these two genes were perhaps incorrectly annotated.
We also checked the structure of annotations for highly overlapping multigenes as another source of possible inconsistencies in genome annotations. For each strain we searched for pairs of highly overlapping multigenes (after the closure) belonging to core anchors (i.e., anchors with elements in every strain). Here, we define a pair of multigenes as highly overlapping when the length of the overlap is at least 50% of the length of the shorter multigene in the pair. The number of identified pairs of multigenes in one strain varies from 17 to 20, depending on the strain. As it can be expected, strains with more liberal annotations have higher number of annotated overlapping multigene pairs. In particular, NCTC8325 has 7 pairs of multigenes where both multigenes in the pair have at least one original annotated element; ST398 has 5 such pairs; and ED98 has 4. On the other hand, RF122, USA300 FPR3757, Newman, N315 and 8 other strains do not have any such highly overlapping pair of annotated multigenes.
Table 6 presents statistics of the refinement procedure. After the closure procedure we obtained 273 (around 5%) non-anchors in the consolidation graph, of which the refinement procedure split 210 and completely resolving 175 of them. The refinement procedure yielded 4 new anchors with multigenes in all strains. Figure 8 gives another perspective on the refinement procedure results. See additional file 2 for detailed summary of the case study results.
Table 6.
Statistics of the connected components before and after refinement for the S. aureus case study. S. aureus, before and after refinement Statistics of the connected components before and after refinement.
| # of connected components before refinement | # of connected components after refinement | |
|---|---|---|
| all connected components | 4737 | 5528 |
| non-anchors | 273 | 107 |
| anchors | 4464 | 5421 |
| orphans | 839 | 1373 |
| core anchors | 2115 | 2119 |
| core connected components | 2156 | 2146 |
Figure 8.

S. aureus, distribution of connected components Histogram of the number of connected components (y-axis) shared by a particular number of strains (x-axis).
Escherichia coli case study
Escherichia coli is the most well-studied prokaryotic organism and has been used in numerous research studies as a model organism. The strain K-12 MG1655 became the first fully sequenced E. coli genome in 1997 [21].
We perform the analysis on E. coli to test scalability of CAMBer and check stability of the results on a large dataset. In our case study, we use genome sequences and annotations of 41 fully sequenced strains deposited in NCBI. At the time of writing, these were the only available E. coli strains with “completed” status. Table 7 presents details of the strains.
Table 7.
E. coli dataset
| strain ID | source (GenBank ID) | # of genes | genome length | lab. |
|---|---|---|---|---|
| O26:H11 11368 | AP010953 | 5363(4) | 5697240 | University of Tokyo |
| O157:H7 EC4115 | CP001164 | 5315(0) | 5572075 | J. Craig Venter Institute |
| O157:H7 EDL933 | AE005174 | 5348(10) | 5528445 | University of Wisconsin |
| O157:H7 TW14359 | CP001368 | 5263(6) | 5528136 | University of Washington |
| O157:H7 Sakai | BA000007 | 5360(5) | 5498450 | GIRC |
| O103:H2 12009 | AP010958 | 5053(4) | 5449314 | University of Tokyo |
| O55:H7 CB9615 | CP001846 | 5014(0) | 5386352 | Nankai University |
| O111:H 11128 | AP010960 | 4971(4) | 5371077 | University of Tokyo |
| 042 | FN554766 | 4792(18) | 5241977 | Welcome Trust Sanger Institute |
| CFT073 | AE014075 | 5378(4) | 5231428 | University of Wisconsin |
| ED1a | CU928162 | 4914(4) | 5209548 | Genoscope |
| UMN026 | CU928163 | 4825(4) | 5202090 | Genoscope |
| 55989 | CU928145 | 4762(4) | 5154862 | Institute Pasteur and Genoscope |
| ETEC H10407 | FN649414 | 4696(3) | 5153435 | Welcome Trust Sanger Institute |
| IAI39 | CU928164 | 4731(7) | 5132068 | Genoscope |
| ABU 83972 | CP001671 | 4793(6) | 5131397 | Georg-August-University Goettingen |
| IHE3034 | CP001969 | 4757(3) | 5108383 | IGS |
| APEC O1 | CP000468 | 4467(3) | 5082025 | Iowa State University |
| SMS-3-5 | CP000970 | 4742(3) | 5068389 | TIGR |
| UTI89 | CP000243 | 5066(13) | 5065741 | Washington University |
| S88 | CU928161 | 4695(3) | 5032268 | Genoscope |
| UM146 | CP002167 | 4650(0) | 4993013 | MBRI |
| E24377A | CP000800 | 4755(0) | 4979619 | TIGR |
| O127:H6 E2348/69 | FM180568 | 4553(4) | 4965553 | Sanger Institute |
| 536 | CP000247 | 4629(2) | 4938920 | University of Goettingen |
| W | CP002185 | 4478(4) | 4900968 | AIBN/KRIBB |
| SE11 | AP009240 | 4679(0) | 4887515 | Kitasato Institute for Life Sciences |
| O83:H1 NRG 857C | CP001855 | 4429(13) | 4747819 | Public Health Agency of Canada Laboratory for Foodborne Zoonoses |
| ATCC 8739 | CP000946 | 4180(7) | 4746218 | US DOE Joint Genome Institute |
| SE15 | AP009378 | 4338(0) | 4717338 | Kitasato University |
| IAI1 | CU928160 | 4353(4) | 4700560 | Genoscope |
| K-12 substr. DH10B | CP000948 | 4125(5) | 4686137 | University of Wisconsin-Madison |
| K-12 substr. W3110 | AP009048 | 4225(9) | 4646332 | Nara Institute of Science and Technology |
| HS | CP000802 | 4383(3) | 4643538 | TIGR |
| K-12 substr. MG1655 | U00096 | 4144(7) | 4639675 | University of Wisconsin-Madison |
| DH1 | CP001637 | 4159(4) | 4630707 | US DOE Joint Genome Institute |
| BL21-Gold(DE3)pLysS | CP001665 | 4208(8) | 4629812 | US DOE Joint Genome Institute |
| BW2952 | CP001396 | 4083(5) | 4578159 | TEDA School of Biological Sciences and Biotechnology |
| BL21(DE3) BL21 | AM946981 | 4227(4) | 4570938 | Austrian Center for Biopharmaceutical Technology |
| B REL606 | CP000819 | 4158(6) | 4558953 | International E. coli B Consortium |
| BL21(DE3) | CP001509 | 4181(23) | 4558947 | Korea Research Institute of Bioscience and Biotechnology |
Details for input strains for the E. coli case study. The first number in column called ’# of genes’ corresponds to the number of annotated genes, the second (in brackets) corresponds to the number of genes excluded in the study due to unusual start or stop codons or sequence length not divisible by three.
Figure 8 presents a distribution of gene (original annotation) and multigene (after applying our closure procedure) counts for the 41 strains. Strains in this plot occur (from left to right) in decreasing order of sizes of their genomes. We observe that the curve based on the original annotations is quite bumpy, which reflects incongruence of annotations made by different labs. This observation is supported by computing an average absolute difference in counts coming from two neighboring strains: it is 152.1 for the original annotation curve vs. 95.6 for the curve constructed after the closure operation; and it is only 64 after post-processing removal of multigenes shorter than 200 nucleotides was applied.
We have also analyzed the distribution of sizes of the newly predicted multigenes. Figure 9 presents these distributions for all E. coli strains taken together. The striking feature is that most of the newly predicted multigenes are pretty short, around 200 nucleotides. Of course each such newly predicted multigene must have a witness coming from an original annotation in another strain. This distribution suggests that annotations of short genes may be a possible source of annotation errors. It also suggests one should remove very short multigenes from global considerations. The distribution after removal is flatter, resembling closer to the distribution for original annotated genes, as shown in Figure 10.
Figure 9.

E. coli, distribution of gene lengths Histograms of gene lengths in logarithmic scale (base = 10) for all E. coli strains taken together. The x-axis is quantified into ranges of length 0.1. Black dotted line presents the distribution of annotated gene lengths, blue solid line shows the distribution of multigene lengths, red dashed line presents the distribution of length of multigenes with no annotated elements.
Figure 10.

E. coli, before and after the closure This plot presents numbers of annotated genes and numbers of the multigenes after the closure procedure applied to E. coli strains. On the x-axis strains are listed (from left to right) in descending order of their genome length. The blue line annotated and the red line closure present respectively the number of annotated genes and the number of multigenes (after the closure) for each strain. The green line removal of < 200 presents the number of multigenes after the closure and after applied post-processing removal of multigenes shorter than 200 nucleotides length.
It is also interesting to investigate which strains had the most liberal annotations of genes. This can be seen by considering connected components which have an element in each strain, but only one gene in such a component has original annotation. Such a situation suggests that the lab which was annotating this strain annotated the ORF as a gene, while other labs did not, even though the corresponding ORF was present in genomes that the other labs were working on. The top 5 most liberal annotations were obtained for CFT073 (37 components), E24377A (22 components), O157-H7 EC4115 (13 components), UTI89 (12 components), and IAI1 (10 components). For the rest of the strains, the number of such components was smaller than 8. In total, there were 22 strains of E. coli which contributed components described above. Adopting a similar approach as in the S. aureus case study we performed the analysis of annotations for highly overlapping multigenes viewed as another source of inconsistencies in genome annotations. In the case of E. coli strains, the number of highly overlapping pairs of multigenes varies in strains from 167 to 172.
Again, strains with local maxima on the curve of annotated genes (see Figure 10) tend to have a higher number of pairs of highly overlapping multigenes with both multigenes annotated. In particular, CFT073 has 86, UTI89 has 76, and E24377A has 30. On the other hand, APECO1 has only one such pair.
Even though there are known cases of functional genes with untypical start codons, we decided to restrict our attention to the three typical start codons (ATG, GTG, CTG), hoping that it does not influence our results in a substantial way. However, it is interesting to follow the fate of genes which have untypical start codons in some strains. For example, the first fully sequenced E. coli strain (K-12 MG1655) has annotated two protein-coding genes with untypical start codons. The first gene is infC, encoding IF3 translation initiation factor. As discussed in [22], this untypical start codon (ATT) may be in use for self-regulation. Interestingly, using CAMBer, we revealed that annotations for 25 (i.e., more than half) of the studied strains have annotated a shorter version of the gene (435 nucleotides instead of 543) with the GTG start codon. The second gene, htgA (synonym htpY ), is involved in heat shock response. The possible explanation for the untypical start codon (CTG) was discussed in [23]. Using CAMBer, we identified 7 strains which annotated this gene with a different TIS. Six of them have annotated 495 nucleotides as gene length and one 486. In both cases, GTG was selected as the start codon. It is possible that some other start codons may also be used in E. coli[21].
In this case study the maximal number of TIS in a multigene is 9; see Table 8 for more details. Interestingly, it is less than for S. aureus — the medium-size case study; see Table 5. Table 9 presents statistics of the refinement procedure. After the closure procedure we obtained 1176 non-anchors, of which we were able to split 934 using the refinement procedure, 689 of them we resolved completely into anchors. The refinement procedure produced only two new anchors with multigenes in all strains. Most of the connected components obtained were small, in particular, the number of orphans doubled; see Figure 11.
Table 8.
Statistics of multigene start sites after the closure procedure for the E. coli case study. E. coli, multigene start sites statistics Multigene start sites statistics after the closure procedure.
| # of multigenes with | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 9 elt. | 8 elt. | 7 elt. | 6 elt. | 5 elt. | 4 elt. | 3 elt. | 2 elt. | 1 elt. | total | |
| O26-H11-11368 | 7 | 7 | 4 | 20 | 57 | 213 | 631 | 1793 | 4310 | 7042 |
| O157-H7-EC4115 | 13 | 13 | 7 | 14 | 62 | 157 | 624 | 1857 | 4226 | 6973 |
| O157-H7-EDL933 | 10 | 13 | 5 | 18 | 46 | 143 | 617 | 1831 | 4242 | 6925 |
| O157-H7-TW14359 | 14 | 12 | 7 | 13 | 58 | 151 | 616 | 1836 | 4195 | 6902 |
| O157-H7-Sakai | 14 | 8 | 5 | 16 | 49 | 152 | 600 | 1826 | 4198 | 6868 |
| O103-H2-12009 | 0 | 28 | 3 | 16 | 53 | 162 | 583 | 1704 | 4078 | 6627 |
| O55-H7-CB9615 | 0 | 4 | 10 | 12 | 44 | 156 | 564 | 1722 | 3950 | 6462 |
| O111-H-11128 | 35 | 7 | 1 | 18 | 54 | 154 | 565 | 1686 | 3970 | 6490 |
| 042 | 0 | 2 | 2 | 11 | 28 | 138 | 538 | 1598 | 3791 | 6108 |
| CFT073 | 6 | 2 | 4 | 8 | 33 | 161 | 534 | 1721 | 3836 | 6305 |
| ED1a | 1 | 4 | 0 | 11 | 24 | 144 | 524 | 1577 | 3957 | 6242 |
| UMN026 | 0 | 3 | 7 | 9 | 29 | 139 | 539 | 1556 | 3719 | 6001 |
| 55989 | 0 | 2 | 3 | 11 | 37 | 146 | 559 | 1605 | 3766 | 6129 |
| ETECH10407 | 1 | 3 | 2 | 11 | 36 | 143 | 549 | 1589 | 3809 | 6143 |
| IAI39 | 22 | 5 | 2 | 4 | 43 | 149 | 508 | 1619 | 3566 | 5918 |
| ABU83972 | 0 | 3 | 3 | 7 | 29 | 140 | 530 | 1662 | 3736 | 6110 |
| IHE3034 | 0 | 1 | 2 | 9 | 32 | 144 | 563 | 1644 | 3712 | 6107 |
| APECO1 | 0 | 1 | 2 | 12 | 29 | 145 | 542 | 1675 | 3705 | 6111 |
| SMS-3-5 | 3 | 0 | 5 | 8 | 24 | 116 | 500 | 1515 | 3586 | 5757 |
| UTI89 | 1 | 1 | 2 | 9 | 30 | 147 | 561 | 1655 | 3658 | 6064 |
| S88 | 0 | 2 | 3 | 9 | 33 | 149 | 550 | 1658 | 3678 | 6082 |
| UM146 | 1 | 1 | 1 | 8 | 28 | 137 | 528 | 1590 | 3640 | 5934 |
| E24377A | 0 | 1 | 2 | 6 | 31 | 125 | 516 | 1502 | 3656 | 5839 |
| O127-H6-E234869 | 0 | 3 | 2 | 8 | 15 | 169 | 471 | 1474 | 3618 | 5760 |
| 536 | 1 | 0 | 2 | 8 | 21 | 135 | 510 | 1560 | 3546 | 5783 |
| W | 0 | 1 | 2 | 6 | 27 | 112 | 483 | 1492 | 3636 | 5759 |
| SE11 | 0 | 3 | 0 | 9 | 32 | 119 | 505 | 1467 | 3625 | 5760 |
| O83-H1-NRG857C | 0 | 1 | 2 | 7 | 23 | 117 | 489 | 1503 | 3427 | 5569 |
| ATCC8739 | 0 | 1 | 3 | 6 | 26 | 106 | 491 | 1431 | 3468 | 5532 |
| SE15 | 0 | 1 | 1 | 10 | 22 | 111 | 467 | 1445 | 3366 | 5423 |
| IAI1 | 0 | 1 | 1 | 5 | 29 | 121 | 484 | 1442 | 3428 | 5511 |
| K12-DH10B | 0 | 3 | 1 | 6 | 23 | 98 | 457 | 1475 | 3504 | 5567 |
| K12-W3110 | 0 | 3 | 1 | 6 | 25 | 100 | 458 | 1467 | 3471 | 5531 |
| HS | 0 | 0 | 1 | 7 | 24 | 121 | 480 | 1439 | 3429 | 5501 |
| K12-MG1655 | 0 | 3 | 1 | 6 | 25 | 97 | 463 | 1455 | 3473 | 5523 |
| DH1 | 0 | 3 | 1 | 6 | 25 | 97 | 458 | 1453 | 3447 | 5490 |
| B-REL606 | 0 | 3 | 2 | 5 | 24 | 99 | 511 | 1472 | 3389 | 5505 |
| BW2952 | 0 | 3 | 1 | 7 | 25 | 97 | 453 | 1447 | 3421 | 5454 |
| BL21-Gold-DE3 | 0 | 2 | 1 | 5 | 25 | 98 | 497 | 1460 | 3388 | 5476 |
| BL21-DE3 | 0 | 2 | 1 | 5 | 25 | 100 | 497 | 1461 | 3362 | 5453 |
| BL21-DE3-BL21 | 0 | 2 | 1 | 5 | 25 | 100 | 497 | 1461 | 3370 | 5461 |
Table 9.
Statistics of the connected components before and after refinement for the E. coli case study. E. coli, before and after refinement Statistics of the connected components before and after refinement.
| # of connected components before refinement | # of connected components after refinement | |
|---|---|---|
| all connected components | 13973 | 20257 |
| non-anchors | 1176 | 563 |
| anchors | 12797 | 19694 |
| orphans | 3637 | 8380 |
| core anchors | 2963 | 2979 |
| core connected components | 3089 | 3084 |
Figure 11.

E.coli, distribution of connected components Histogram of the number of connected components (y-axis) shared by a particular number of strains (x-axis).
See additional file 3 for detailed summary of the case study results.
Core genome vs. pangenome
Finally, we computed core genome and pangenome for the family of E. coli strains using our concept of a multigene and compared the result to the core genome and pangenome computed along the lines described in [4]. The latter paper considered 61 strains, many of them not having the sequencing status of “completed”. Our set of strains is not a subset of the 61 strains mentioned above since there were some newly published strains (e.g., E. coli UM146, published in January 2011). For this reason, we had to repeat the computations as described in [4] for our set of strains.
As in [4] we call two genes homologous if the percent of identity is at least 50% covering at least 50% of the longer gene. We order all strains with respect to decreasing size of their genomes. We start with the strain having the largest genome, initializing both the pangenome and the core genome equal to the set of all genes of that strain. In the n-th step, we put a gene of the n-th strain into the pangenome if it is not homologous to any of the genes of the previously considered strains. We also remove a gene from the core genome when it not homologous to any of the genes of the n-th strain.
We run two experiments on our set of strains: one which relies on the original gene annotations, as it was done in [4], and another one which relies on previously pre-computed multigenes. Figure 12 shows the dynamics of change in gene numbers both for pangenome and core genome. It shows that as the number of strains increases both methods asymptotically converge to a pangenome size of around 13 000 genes. This suggests that the notion of a pangenome is quite robust when considering a large number of strains. On the other hand, there is a consistent difference between sizes of the core genome computed for the original annotations vs. pre-computed multigenes. For the latter method the core genome is substantially larger than for the former, resulting in an increase of the percentage with respect to pangenome from 18% to 25%. The analogous percentage for the 61 strains considered in [4] was reported in that paper as only 6%, but the computation was relying on original annotations.
Figure 12.

E. coli, core genome vs pangenome Core vs. pangenome plots of 41 E. coli strains calculated using original annotations and multigene annotations, predicted by CAMBer. Strains are sorted (from left to right) in descending order of their genome sizes. Violet and green (coregenome-annot and pangenome-annot) lines connect cumulative numbers of core and pangenome sizes using annotated genes, while red and blue (coregenome-multi and pangenome-multi) lines connect cumulative numbers of core and pangenome sizes using multigenes after the closure procedure. The proportion of core genome to pangenome size has risen from 18% to 25% after the closure.
We also performed a similar analysis on M. tuberculosis and S. aureus strain families. Figures 13 and 14 present results for M. tuberculosis and S. aureus respectively. The conclusions are similar as for E. coli. The size of pangenome computed using both methods converges, as the number of considered strains increases. On the other hand size of the core genome shows a consistent difference for both methods. As a result the proportion of the size of core genome with respect to the pangenome substantially depends on the chosen method, yielding higher score for the method based on pre-computed multigenes. The increase is from 42% to 52% for S. aureus and from 88% to 96% for M. tuberculosis.
Figure 13.

M. tuberculosis, core genome vs pangenome Core vs. pangenome plots of 9 M. tuberculosis strains calculated using original annotations and multigene annotations, predicted by CAMBer. Strains are sorted (from left to right) in descending order of their genome sizes. Violet and green (coregenome-annot and pangenome-annot) lines connect cumulative numbers of core and pangenome sizes using annotated genes, while red and blue (coregenome-multi and pangenome-multi) lines connect cumulative numbers of core and pangenome sizes using multigenes after the closure procedure. The strain KZN V2475 was excluded due to wrong annotation, caused by a shift in gene coordinates. The proportion of core genome to pangenome size has risen from 88.5% to 96.1% after the closure.
Figure 14.

S. aureus, core genome vs pangenome Core vs. pangenome plots of 22 S. aureus strains calculated using original annotations and multigene annotations, predicted by CAMBer. Strains are sorted (from left to right) in descending order of their genome sizes. Violet and green (coregenome-annot and pangenome-annot) lines connect cumulative numbers of core and pangenome sizes using annotated genes, while red and blue (coregenome-multi and pangenome-multi) lines connect cumulative numbers of core and pangenome sizes using multigenes after the closure procedure. The proportion of core genome to pangenome size has risen from 42% to 52% after the closure.
Conclusions
As the number of sequenced genomes of closely related bacterial strains grows, there is a need to join and consolidate different annotations of genomes. It turns out that annotations of related strains are often inconsistent in declaring Translation Initiation Sites (TIS) for the corresponding homologous genes. They also sometimes miss a gene in a segment which sequence-wise is very similar to a segment in the genome of another species which is declared as a gene. We propose in this paper a methodology which consists in collecting all possible different TISs, as well as genes which are present sequence-wise in a strain but whose annotation is missing. We believe this is the right approach toward correcting annotations.
To achieve this goal we constructed a consolidation graph which is based on the concept called here a multigene. Multigene is an entity which combines all different TISs derived from sequence comparisons with genes annotated in other strains, or genes which were already established as multigenes. The transitive closure of this operation on all genomes of interest gives the space of multigenes. Multigenes serve as nodes of the consolidation graph. Each TIS in a multigene gives rise to a gene which we called an element of the multigene. All elements of a given multigene share the same stop codon. Each multigene with more than one element can be viewed as a task of deciding on the right TIS. Such a decision may have to involve some wet lab experiments or consideration of ESTs or 5’ cDNAs [5]. This issue is not discussed in the present paper. So conceptually a multigene corresponds to a gene in which a TIS is yet to be determined (hopefully by selecting one of the listed start sites).
Why does genome alignment not give similar results as the consolidation graph? The main reason is that in genome alignment one works with sequences which are fragments of genomes without paying any attention to functional genetic elements. In this way one discovers genomic areas of high similarity. Even though postprocessing is often performed, by considering functional genomic elements and the homology relationship between genes or revised genes, gene annotation is not always correctly reconstructed. Moreover, pairwise genome alignment approaches may also miss homologous fragments that can only be linked by intermediate sequences [7]. In contrast, in the consolidation graph we start with annotated genes and close up iteratively with the sequences which come out as significant BLAST hits to the queries already obtained in this analysis. There is a caveat to this iteration process however. In particular, when the input contains a conserved genomic region that is incorrectly annotated as a gene in one strain, CAMBer may fish out homologous regions from other strains and propagate the incorrect gene structure annotation to them. Connected components of the consolidation graph naturally define sets of multigenes which might be called multigene families. This concept of a multigene family is rather new, since in the multigene family construction we did not rely exclusively on given annotations. It turns out that these multigene families can be used to reconstruct a one-to-one homology relation for most of the genes. This procedure we call refinement. For this we start off with families which consist of at most one multigene from each strain. These we called anchors. Then we extend the one-to-one homology relation by considering a genome position of genes, which were not yet related by the one-to-one relationship, with respect to the anchors. This method leaves unresolved only very few small families which presumably should be further curated manually. The one-to-one relationship can be used, among other things, in deciding which multiple alignments should be considered for detection of possible mutations, or even detection of possible sequencing errors.
The methodology above was illustrated with three case studies on 9 Mycobacterium tuberculosis, 22 Staphylococcus aureus and 41 Escherichia coli strains. It is evident from the results presented in this paper that genome annotations done in different labs were not congruent to each other. After performing the consolidation, variance in the total gene count is much smaller than before, suggesting that the revised annotations could lead to a more coherent view of functional elements in various strains.
Analyzing CAMBer results, we find out that most of the inconsistencies are related to short genes. Moreover, we find huge disagreement in annotations of highly overlapping ORF’s, located in different reading frames (possibly on the opposite strand). Comparing annotations of pairs of highly overlapping multigenes belonging to core anchors, we found many inconsistencies in them. For example, the S. aureus strain NCTC8325 has originally annotated both highly overlapping multigenes in 7 such pairs, whereas 10 out of 22 strains have no such a pair at all. This observation suggests that an analysis of overlapping genes should use annotations with caution. The issue of possibly missing annotations in the case of overlapping genes was previously mentioned in [24].
The M. tuberculosis case study showed that CAMBer can also be applied to completely unannotated genomes, yielding an initial annotation of a newly sequenced genome. This case may be illustrated with strain KZN V2475. Presumably due to a shift in annotation coordinates most genes of this strain have clearly incorrect annotations (see the corresponding entry in Table 1). For this reason we have discarded the originally published annotation for this strain and run CAMBer on the remaining annotated strains plus unannotated KZN V2475. As can be seen in Table 2 we were able to retrieve annotations for KZN V2475 which look quite similar in terms of statistics as annotations for the other strains.
We computed the core genome and pangenome for M. tuberculosis, S. aureus and E. coli strain families using two approaches: one that relies on originally annotated genes (along the lines of [4]) and another which uses our notion of a multigene. Interestingly, both methods give similar results for pangenome, but they significantly differ on the core genome, with the latter method producing larger result. Both these observations hold true for all three case studies. The proportion of the core genome size to the pangenome size increases from 18% to 25% for E. coli, from 42% to 52%; for S. aureus, and from 88% to 96% for M. tuberculosis, when switching from the former to the latter method. We suggest that the method based on pre-computed multigenes, as it is done by CAMBer, gives a more reliable estimate of the core genome. However, it is probably the case that the number of strains in this study of M. tuberculosis and S. aureus is too small to correctly approximate the proportions, so we expect that the actual proportions will turn smaller.
This experiment showed also the good scalability of CAMBer. We ran our largest case study on 41 E. coli strains on a cluster of 17 computer nodes using Sun Grid Engine technology to spread the computations. Most time consuming were blast computations, which took around two days. We also found that it took around 9 hours to compute the closure and the consolidation graph assuming precomputed blasts. In the E. coli case study computations of the refinement took around 2 hour. We also ran the S. aureus case study on the same cluster. It took around 4 hours to compute the closure assuming precomputed blasts, and around 1 hour to compute the refinement. However, we did the case study on M. tuberculosis—which is much smaller—using a single computer with 16 cores, 3000 MHz, 64 GB RAM. It took about 10 hours to compute the consolidation graph (including time consuming blast computations) and only several minutes to perform the refinement procedure.
All the above statistics for the computational experiments suggest that CAMBer may be a useful utility in comparing and revising annotations of closely related bacterial genomes.
Input data, software used in the paper (written in Python), and detailed xls files with results of the case study experiments are available at http://bioputer.mimuw.edu.pl/camber.
Competing interests
The authors declare that they have no competing interests.
Authors contributions
All authors contributed to design of the method, analysis of results and writing of the manuscript. MW wrote software and performed experiments. All authors read and approved the final manuscript.
Supplementary Material
summary of the M. tuberculosis case study results An Excel file containing the summary of CAMBer results (after refinement) for the M. tuberculosis case study.
summary of the S. aureus case study results An Excel file containing the summary of CAMBer results (after refinement) for the S. aureus case study.
summary of the E. coli case study results An Excel file containing the summary of CAMBer results (after refinement) for the E. coli case study.
Contributor Information
Michal Wozniak, Email: m.wozniak@mimuw.edu.pl.
Limsoon Wong, Email: wongls@comp.nus.edu.sg.
Jerzy Tiuryn, Email: tiuryn@mimuw.edu.pl.
Acknowledgments
This work is partially supported by Polish Ministry of Science and Higher Education grant no. N N301 065236 and Singapore Ministry of Education Tier-2 grant MOE2009-T2-2-004.
This article has been published as part of BMC Genomics Volume 12 Supplement 2, 2011: Selected articles from the IEEE International Conference on Bioinformatics and Biomedicine 2010. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2164/12?issue=S2.
References
- Zheng H, Lu L, Wang B, Pu S, Zhang X, Zhu G, Shi W, Zhang L, Wang H, Wang S, Zhao G, Zhang Y. Genetic basis of virulence attenuation revealed by comparative genomic analysis of Mycobacterium tuberculosis strain H37Ra versus H37Rv. PLoS One. 2008;3(6):e2375. doi: 10.1371/journal.pone.0002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Lupien A, Gingras H, Wasserscheid J, Dewar K, Légaré D, Ouellette M. Genome sequencing of linezolid-resistant Streptococcus pneumoniae mutants reveals novel mechanisms of resistance. Genome Research. 2009;19(7):1214–1223. doi: 10.1101/gr.089342.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andries K, Verhasselt P, Guillemont J, Göhlmann HWH, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307(5707):223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- Lukjancenko O, Wassenaar TM, Ussery DW. Comparison of 61 sequenced Escherichia coli genomes. Microb Ecol. 2010;60(4):708–720. doi: 10.1007/s00248-010-9717-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakaguri H, Suzuki Y, Sasaki M, Sugano S, Watanabe J. Inconsistencies of genome annotations in apicomplexan parasites revealed by 5’-end-one-pass and full-length sequences of oligo-capped cDNAs. BMC Genomics. 2009;10:312. doi: 10.1186/1471-2164-10-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boneca IG, de Reuse H, Epinat JC, Pupin M, Labigne A, Moszer I. A revised annotation and comparative analysis of Helicobacter pylori genomes. Nucleic Acids Research. 2003;31(6):1704–1714. doi: 10.1093/nar/gkg250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Teichmann SA, Hubbard T, Chothia C. Intermediate sequences increase the detection of homology between sequences. Journal of Molecular Biology. 1997;273:349–354. doi: 10.1006/jmbi.1997.1288. [DOI] [PubMed] [Google Scholar]
- Schlueter SD, Wilkerson MD, Huala E, Rhee SY, Brendel V. Community-based gene structure annotation. Trends Plant Sci. 2005;10:9–14. doi: 10.1016/j.tplants.2004.11.002. [DOI] [PubMed] [Google Scholar]
- van den Boogaard J, Kibiki GS, Kisanga ER, Boeree MJ, Aarnoutse RE. New drugs against tuberculosis: Problems, progress, and evaluation of agents in clinical development. Antimicrob Agents Chemother. 2009;53(3):849–862. doi: 10.1128/AAC.00749-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Begley T, B R M. et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic acids research. 2005;33(17):5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disz T, Akhter S, D C. et al. Accessing the SEED genome databases via Web services API: Tools for programmers. BMC Bioinformatics. 2010;11:319+. doi: 10.1186/1471-2105-11-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Rogozin IB, Kondrashov AS, Koonin EV. Genome alignment, evolution of prokaryotic genome organization and prediction of gene function using genomic context. Genome Research. 2001;11:356–372. doi: 10.1101/gr.GR-1619R. [DOI] [PubMed] [Google Scholar]
- Wozniak M, Wong L, Tiuryn J. CAMBer: an approach to support comparative analysis of multiple bacterial strains. Proceedings of the International Conference on Bioinformatics and Biomedicine: 18-21 December 2010; Hong Kong. 2010. pp. 121–126.
- Rost B. Twilight zone of protein sequence alignments. Protein engineering. 1999;12(2):85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2011;39(Database issue):D32–D37. doi: 10.1093/nar/gkq1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioerger TR, Koo S, No EG, Chen X, Larsen MH, Jacobs WR Jr., Pillay M, Sturm AW, Sacchettini JC. Genome analysis of multi- and extensively-drug-resistant tuberculosis from KwaZulu-Natal, South Africa. PLoS ONE. 2009;4(11):e7778. doi: 10.1371/journal.pone.0007778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann RD, Alland D, Eisen JA. et al. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. Journal of Bacteriology. 2002;184(19):5479–5490. doi: 10.1128/JB.184.19.5479-5490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole S, Brosch R, Parkhill J. et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393(6685):537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Chambers HF. The changing epidemiology of Staphylococcus aureus? Emerging infectious diseases. 2001;7(2):178–182. doi: 10.3201/eid0702.010204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Aoki K, Nagai Y, Lian J, Ito T, Kanamori M, Matsumaru H, Maruyama A, Murakami H, Hosoyama A, Mizutani-Ui Y, Takahashi NK, Sawano T, Inoue R, Kaito C, Sekimizu K, Hirakawa H, Kuhara S, Goto S, Yabuzaki J, Kanehisa M, Yamashita A, Oshima K, Furuya K, Yoshino C, Shiba T, Hattori M, Ogasawara N, Hayashi H, Hiramatsu K. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet. 2001;357(9264):1225–1240. doi: 10.1016/S0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. The complete genome sequence of Escherichia coli K-12. Science. 1997;277(5331):1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Sacerdot C. et al. Sequence of a 1.26-kb DNA fragment containing the structural gene for E. coli initiation factor IF3: presence of an AUU initiator codon. EMBO. 1982;1(3):311–315. doi: 10.1002/j.1460-2075.1982.tb01166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiakas D, Georgopoulos C, Raina S. The Escherichia coli heat shock gene htpY: mutational analysis, cloning, sequencing, and transcriptional regulation. Journal of Bacteriology. 1993;175(9):2613–2624. doi: 10.1128/jb.175.9.2613-2624.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabath N, Price N, Graur D. A potentially novel overlapping gene in the genomes of Israeli acute paralysis virus and its relatives. Virology Journal. 2009;6:144+. doi: 10.1186/1743-422X-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
summary of the M. tuberculosis case study results An Excel file containing the summary of CAMBer results (after refinement) for the M. tuberculosis case study.
summary of the S. aureus case study results An Excel file containing the summary of CAMBer results (after refinement) for the S. aureus case study.
summary of the E. coli case study results An Excel file containing the summary of CAMBer results (after refinement) for the E. coli case study.