

Abstract
Despite substantial attention from theoreticians, the evolutionary mechanisms that drive intra- and interspecific variation in the mutation rate remain unclear. It has often been argued that mutation rates associated with the major replicative polymerases have been driven down to their physiological limits, defined as the point at which further enhancement in replication fidelity incurs a cost in terms of reproductive output, but no evidence in support of this argument has emerged for cellular organisms. Here, it is suggested that the lower barrier to mutation rate evolution may ultimately be defined not by molecular limitations but by the power of random genetic drift. As the mutation rate is reduced to a very low level, a point will eventually be reached at which the small advantage of any further reduction is overwhelmed by the power of drift. This hypothesis is consistent with a number of observations, including the inverse relationship between the per-site mutation rate and genome size in microbes, the negative scaling between the per-site mutation rate and effective population size in eukaryotes, and the elevated error rates associated with less frequently deployed polymerases and repair pathways.
Keywords: antimutator, genome evolution, molecular evolution, mutation rate, mutator, random genetic drift
Introduction
Although evolutionary biologists have explained many attributes of biodiversity in terms of extrinsic adaptive challenges, much less is known about the intrinsic mechanisms that define the tempo and mode of evolutionary change itself. The origins of the substantial divergence in error rates associated with different polymerases, nucleotide pool sanitizers, and repair enzymes within and among species remain especially unclear. Developing such an understanding is critical to establishing a general theory for the constraints on evolutionary processes in divergent phylogenetic lineages.
Without genetic variation, there can be no evolution, so a nonzero mutation rate is essential for adaptive progress in the face of a changing environment. However, because the vast majority of mutations with effects on fitness are deleterious, special conditions are required for positive selection to promote increases in the mutation rate. Although both theoretical work and empirical work demonstrate that strong mutator alleles can sometimes rise to high frequencies by hitchhiking with linked beneficial mutations in asexual populations (Sniegowski et al. 1997; Taddei et al. 1997; Oliver et al. 2000; Tenaillon et al. 2001; Hall and Henderson-Begg 2006), such a mechanism has limited explanatory power for mutation rates in sexual species. This is because almost all mutations in recombining genomes will be either unlinked or loosely linked to the locus responsible for their production and hence remain in statistical association with a mutator allele for only two to a few generations. Even in the case of asexuality, unless mutator alleles revert to a nonmutator state at high rates (Taddei et al. 1997), recurrent hitchhiking effects are expected to eventually lead to the extinction of mutator lineages as a permanent deleterious mutation load develops between bouts of adaptive substitution (Gerrish et al. 2007).
These general considerations have led to a common view, dating to Sturtevant (1937), that natural selection will typically drive mutation rates to the minimum possible level (Kimura 1967; Dawson 1999; Baer et al. 2007). This lower limit has usually been thought to be rendered nonzero by fitness costs associated with excessive investment in replication fidelity, for example, a reduction in reproductive rates resulting from slow genome replication. Because a structural trade-off exists between the rate of polymerization and the investment in proofreading (Bessman et al. 1974; Loh et al. 2007, 2010; Tian et al. 2008), such costs must exist at some level. However, there is no direct evidence that time constraints on genome replication are substantial enough to influence the mutation rate, except perhaps in viruses (Furió et al. 2005, 2007). In prokaryotes, for example, genome replication constitutes just a small fraction of the overall energy budget (Ingraham et al. 1983), rates of DNA replication are more than 10-fold faster than those for messenger RNA elongation (Cox 2004), cells often experience nested genome replication events before dividing (Casjens et al. 1998), and there is no discernible association between growth rate and genome size (Vieira-Silva et al. 2010). Yet, the fact that prokaryotes with reduced mutation rates can be obtained experimentally (Tröbner and Piechocki 1984; Fijalkowska et al. 1993; Schaaper 1998; Galán et al. 2007; Loh et al. 2007) suggests that the accuracy of the replication machinery is less than what is achievable at the molecular level, although arguments have been made that pleiotropic constraints often result in declines in some types of fidelity in response to increases in others (Drake 1993; Bebenek et al. 2005).
An additional but unexplored barrier to mutation rate reduction is the reduced efficiency of selection for weakly advantageous antimutator alleles. As the mutation rate is driven to lower and lower levels by selection, a point must eventually be reached where the advantage of any further increase in replication fidelity is smaller than the power of random genetic drift (Lynch 2008, 2010). The goal here is to evaluate the extent to which such an intrinsic barrier can provide an adequate explanation for the patterns of mutation rates known to have evolved in natural populations. To analyze this problem, the situation involving a single mutation rate modifier in an effectively infinite asexual population (where drift plays a negligible role relative to selection) will first be explored, with attention then being expanded to the effects of multiple mutator states, finite population size, and recombination. Throughout, it is assumed that the only selective pressure on the mutation rate is the statistical association of excess deleterious mutations with mutator alleles. The expectations of the resultant theory are then shown to be consistent with empirical observations on the replication machinery and associated error rates in various lineages.
Results
An Effectively Infinite Asexual Population
Consider a modifier allele m that magnifies the genome-wide mutation rate to deleterious alleles by an amount ΔU. Individuals are assumed to be diploid, but provided the selective disadvantage of mutator heterozygotes is much greater than the rate of origin of mutator alleles, mutator homozygotes are expected to be extremely rare, and the population can be effectively treated as containing two classes of individuals, MM and Mm. Genotype MM is converted to Mm at rate 2μ, where μ is the mutation rate per M gene copy, whereas reversion of the latter to MM occurs at rate ν. The dynamics of the frequency of Mm individuals can be written as
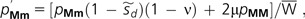 |
(1) |
where is the selective disadvantage of Mm individuals and the mean population fitness relative to an MM fitness of 1.0. The equilibrium frequency of the mutator genotype is then This expression and all the remaining results of this section also apply to a haploid asexual population if μ is substituted for 2μ.
The central remaining issue is the magnitude of the selective disadvantage resulting from the accumulation of excess deleterious mutations on Mm relative to MM backgrounds. Because new Mm individuals are derived recurrently from relatively mutation-free MM genotypes, whereas their descendants stochastically accumulate new deleterious mutations, the pool of Mm individuals will be heterogeneous with respect to fitness. Letting U be the genome-wide deleterious mutation rate in the MM background, deleterious mutations arise at rate U + ΔU within Mm individuals, and we assume that each mutation arises at a unique site (ensuring that all mutations remain heterozygous in a diploid species). With all mutations assumed to have a fixed deleterious effect s, the fitness function is defined to be W(n) = (1 − s)n, where n is the number of deleterious mutations in an individual.
To obtain a time-averaged measure of the selective disadvantage of mutator individuals, an estimate of the average excess number of deleterious mutations residing on Mm backgrounds is required. With new mutations arising randomly, under the multiplicative fitness model, the number of excess mutations arising follows a Poisson distribution with the mean cumulative number being fractionally reduced by (1 − s) by selection and increased by ΔU by mutation each generation (Haigh 1978). Assuming these features are closely approximated from the time of appearance of a new Mm genotype, the average buildup of excess mutations is then described by
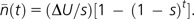 |
(2) |
Relative to the situation in MM individuals, which at equilibrium carry an average U/s deleterious mutations, the mean selective disadvantage of a mutator genotype t generations after introduction is
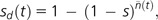 |
(3) |
so that the average selective disadvantage over the expected life span of an Mm sublineage is:
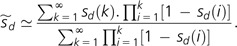 |
(4) |
This expression follows from the fact that an expected fraction [1 − sd(i)] of the descendants of a newly arisen cohort of Mm individuals survives the ith generation, with the cumulative product giving the survivorship of the cohort through time. Further simplification is achieved by approximating powers of (1 − s) as exponentials,
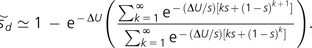 |
(5) |
Although this expression ignores the variation in n within mutator and nonmutator genotypes, it yields a very close approximation to the average selection coefficient determined from the mean genotypic fitnesses of a fully characterized infinite population iterated to equilibrium (fig. 1). For ΔU≪ s, which is likely to be the usual case given that to 0.01 (Lynch and Walsh 1998), a newly arisen sublineage of mutators generally achieves its excess equilibrium mutation load, ΔU/s, before being purged from the population, and that is, the selective disadvantage of the mutator is independent of the effects of the mutations it produces. However, highly aggressive mutator alleles are relatively rapidly eliminated (as a consequence of their substantial indirect mutation load; Johnson 1999a), resulting in
FIG. 1.—

Properties of a mutator allele m in an infinite asexual population, assuming a mutation rate to mutator genotype Mm of 2μ = 10 − 6, and a negligible back mutation rate. Results were obtained by a set of recursion equations that tracked the distributions of deleterious mutation numbers within MM and Mm individuals, until the population achieved mutation–selection equilibrium. Left: Equilibrium frequency of the Mm mutator heterozygotes (dashed line) and average selective disadvantage of the Mm genotype at equilibrium (solid line obtained from recursions; dotted line from eq. (4)). Right: Population average mutation rate at equilibrium.
At mutation–selection equilibrium, the increase in the mean genome-wide deleterious mutation rate resulting from the recurrent production of the mutator allele is Assuming a low level of mutator allele reversion, (), provided ΔU≪s, , that is, half the harmonic mean of the diploid mutation rate to mutator alleles and the elevation in the genome-wide deleterious mutation rate per such mutation, which approaches 2μ if ΔU≫2μ. An evaluation of a more extensive model with an array of mutator classes (differing by a constant multiplicative factor) demonstrates that this expression still closely approximates the inflation of if ΔU is taken to be the mutation rate difference between the two least mutagenic classes, which is where nearly all of the population resides. Thus, weak mutator alleles generally result in only a small increase in the population average mutation rate, which is independent of the effects of the induced deleterious mutations.
Finite Asexual Populations
Although the preceding results imply that the mutation rate will evolve to the minimum possible level in an infinite population, they are best viewed as providing a conceptual basis for determining the influence that finite population size has on the minimum mutation rate achievable by selection (ignoring, for the time being, any counterselection that might be associated with a cost to replication fidelity). Because represents the absolute magnitude of selection operating against a cohort of mutator alleles in an infinite population, the capacity of natural selection to further reduce a prevailing genome-wide mutation rate will be limited unless there are accessible antimutator alleles with larger than the power of drift, defined as ∼1/Na in asexuals, where Na is the asexual effective population size. Although the degree to which mutations operating on highly refined replication/repair loci can produce alleles with ΔU > 1/Na is uncertain, it is clear that ΔU cannot exceed U itself.
To evaluate the extent to which a population evolves to such a barrier, stochastic computer simulations were performed on clonal populations in which the genome-wide deleterious mutation rate was subject to modification through an unbounded range of alternative states, with the mutation rates (U) of adjacent allelic classes differing by a constant factor, 1 + λ. This has the effect of ΔU = λU becoming progressively smaller as U declines, which must occur with diminishing room for improvement. New deleterious mutations arose in the various mutation rate backgrounds in a Poisson fashion, and genotypic fitnesses were entirely a function of the number of deleterious mutations carried, using the multiplicative fitness function noted above. Each mutation rate class was subject to stepwise mutational conversion to the two immediately adjacent classes, with the overall rate of such conversion being proportional to the class-specific U, and a fraction fd of such events being in the direction of higher U. Drift was imposed by multinomial sampling of the pool of expected genotype frequencies following each generation of selection and mutation, as in the classical Wright–Fisher discrete-generation model.
Because mutations that either increase or decrease the mutation rate are allowed for in this model, regardless of the starting conditions, the population average mutation rate () gradually converges to a quasi-steady-state level dictated by the effective population size (fig. 2). If the population is initiated at a high genomic mutation rate, antimutators with a substantial selective advantage are produced relatively quickly, and rapidly declines toward the quasi-equilibrium. The latter state is attained when the mean mutation rate has become so low that the production of antimutators with an associated ΔU greater than 1/Na is no longer possible. At this point, slight further reductions in may still result from the fortuitous increase of a very weak antimutator by drift, but subsequent increases in will also occur as weak (and effectively neutral) mutator alleles arise.
FIG. 2.—

Sample evolutionary trajectories for the average genome-wide deleterious mutation rate () in finite asexual populations with different monomorphic starting conditions. Results are shown for two different population sizes and two different selection coefficients against deleterious mutations, with 48 mutation rate classes differing by a factor of 1.111 between adjacent classes and with the mutation rate to mutator/antimutator genotypes being equal to 0.02 times the total genome-wide mutation rate to deleterious alleles at fitness loci. Mutations to antimutators were relatively rare (10% of the total; fd = 0.1). Note that the quasi-steady-state predictions for (given by the horizontal gray lines) are simply the points at which ΔU = 0.1U is equal to 1/N.
The time to converge on the quasi-equilibrium from above is especially prolonged in populations of large size because of the progressively reduced rate of production of antimutators as the population approaches a lower and lower mutation rate state. In contrast, if the population is initiated at a very low mutation rate, the mean mutation rate increases toward the quasi-equilibrium but does so slowly because the rate of production of mutator genotypes is low. This gradual rise of the average mutation rate is not a reflection of selection for an optimal mutation rate, as all mutations are deleterious, but a passive outcome of an upward mutational bias toward the production of mutator versus antimutator alleles (fd > 0.5).
As anticipated from the theoretical results presented above, the quasi-equilibrium is essentially independent of the effects of mutations on fitness (s), depending only on the effects of mutator/antimutators on the mutation rate itself, ΔU (fig. 2). Moreover, is driven to lower levels in populations with larger size because of the greater efficiency of selection.
The Temporal Scale of Mutation Rate Evolution
Although the argument laid out above provides a heuristic basis for understanding the limits to what selection can accomplish with replication fidelity, it is of interest to have a more quantitative picture of the rate at which the drift barrier is approached, and the degree to which the mutation rate itself is subject to drift via the fixation of sufficiently mild antimutator/mutator alleles in the vicinity of the quasi-equilibrium. With large numbers of potential mutation rate states but an unknown distribution of effects in real organisms, a complete understanding of these issues is not yet possible. However, motivated by the observation that populations generally reside in a nearly monomorphic state with rare and relatively rapid excursions to adjacent states (fig. 2), some insight can be gained by considering the rates of transition between adjacent pure states via the fixation of derived alleles. To achieve quantitative expressions for the waiting times for such transitions, it is useful to subdivide the process into two phases: 1) the arrival time of newly derived antimutator/mutator alleles and 2) the time for such alleles to progress to fixation.
A key to understanding the transition process in an asexual population is the fact that newly derived mutator/antimutator genotypes are only likely to go to fixation if they arise in the most fit background, as all other fitness classes are destined to eventual loss, assuming the population is large enough to avoid progressive mutational deterioration. The latter condition requires that the ratio of the power of selection to the power of drift, sNa, be much greater than one (Gordo and Charlesworth 2000). Provided this condition is fulfilled, a population will approach a selection–mutation balance in which the fraction of individuals contained within the best (deleterious mutation free) class is e − U/s (Haigh 1978), so the long-term effective population size is approximately Na = Ne − U/s, where N is the actual number of adults. If U≪s at the mutation rate barrier, as it is in figure 2, then e − U/s≃1, and the effective population size is near the expectation based on the effective number of adults (Na≃N). However, if U at the barrier is such that e − U/s≪1, the relevant drift barrier (1/Na) will be much greater than 1/N. In the following, it is assumed that Na≫1, for if this is not the case, the highest fitness classes will be progressively lost by Muller's ratchet, eventually leading to population extinction via mutational meltdown (Lynch and Gabriel 1990; Lynch et al. 1993).
Letting μ be the mutation rate from the current mutation rate to a derived antimutator genotype, a population arrives at a state containing one or more such alleles with evolutionary potential at rate φa≃1 − e − 2Naμ, which is just the rate of origin of new sustainable alleles (2Naμ) for 2Naμ≪1, and approaches one for 2Naμ≫1. Given such a starting point, the probability of fixation of the derived antimutator allele in an asexual population is:
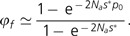 |
(6) |
This is the standard diffusion approximation for the fixation probability for an advantageous mutation, with two modifications: s* = ΔU + 2μ (where ΔU is positive for an antimutator), and p0 = (1/Na) + [2μ(Na − 1)/Na]≃(1/Na) + 2μ. The modified selection coefficient (s*) includes the rate of production of derived alleles 2μ, as this might be nontrivial (relative to ΔU) near the drift barrier. The modified starting frequency of the derived allele (p0) allows for the fact that when 2Naμ > 1, more than a single copy of the derived genotype can appear per generation. The mean time to arrive at a state at which the derived antimutator is destined to fixation is then:
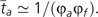 |
(7) |
To obtain the additional time to fixation, we employ the following reasoning. Newly derived alleles with small influences on the mutation rate will be subject to relatively strong stochastic processes in their early phase of establishment. However, once a weak antimutator allele has arisen to sufficiently high frequency, here denoted as p*, it can be promoted by selection in an effectively deterministic fashion. From a modification of the standard fixation equation for a beneficial allele, the frequency to which a beneficial mutation must rise in order to have a 0.9 probability of fixation is:
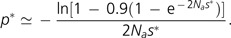 |
(8) |
The mean time to fixation of an antimutator is then approximated by subdividing the dynamics into an early phase of drift until the derived allele first reaches frequency p*, and a subsequent phase of deterministic selection. Modifying from Kimura and Ohta (1969), the mean time to drift to frequency p* from starting frequency p0, conditional on eventual fixation, is:
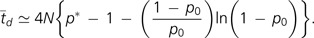 |
(9) |
The subsequent deterministic time is approximated by assuming that the antimutator simply expands exponentially to frequency 1.0 once it has reached the critical frequency,
 |
(10) |
The average number of generations required for a transition from a monomorphic ancestral state to fixation of a derived antimutator allele is then:
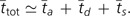 |
(11) |
The results for the arrival time for advantageous antimutator alleles () are readily extended to deleterious mutator alleles by allowing ΔU be negative in the latter case. The total time to fixation of a mutator (conditional on fixation, despite its disadvantage) can be assumed to be equal to that for an antimutator with the same absolute selective advantage, based on prior work (Nei and Roychoudhury 1973; Maruyama 1974; Taylor et al. 2006).
The sets of expressions for both mutators and antimutators yield good approximations to the mean transition times obtained by computer simulations of the stochastic process, provided sNa > 1 (fig. 3). (When sNa < 1, the effective population size is actually elevated relative to Ne − U/s because mutator/antimutator alleles in multiple classes of individuals have the potential for fixation when the permanent maintenance of the best class by selection is no longer guaranteed.) To obtain a fuller understanding of the dynamics of mutation rate evolution, the preceding formulations might be applied in a transition matrix approach for the various mutation rate classes, although in the absence of explicit information on the rates of origin of mutator/antimutator alleles, the issue is not pursued here. Nevertheless, the preceding results help clarify two key issues with respect to the constraints on mutation rate evolution.
FIG. 3.—

Mean times (in generations) for transitions from one fixed mutation rate state to another, given for three asexual population sizes. Upwardly bowed curves refer to derived mutators, and downward curves to antimutators. Data points are the averages of 500 stochastic simulations, whereas the curved lines are the expectations based on the theory in the text.
First, the lower bound on the mutation rate depends not just on the magnitude of ΔU that is accomplishable for antimutator alleles but also on the net rate of mutational production of such alleles. The latter is treated here as μ under the assumption that the antimutator undergoes no back mutation while en route to fixation, although μ should be viewed more generally as the difference between forward and backward mutation rates to such an allele. Provided |ΔU|≫2μ, both mutators and antimutators are potentially driven by indirect selection on the associated deleterious mutation load. Such selection is ineffective if |ΔU|≪(1/Na) (below the drift barrier), but otherwise the fixation of mutator alleles is strongly impeded, the more so in very large populations. In contrast, when |ΔU|≪2μ, both mutators and antimutators behave in an effectively neutral fashion (regardless of population size), being driven only by directional mutation pressure.
Second, although the success or failure of a mutator/antimutator depends on the magnitude of ΔU, rather than on the specific value of U for the derived allele, the absolute value of U in the ancestral genotype does influence the fates of derived alleles through its influence on the number of individuals (Na) in the restricted class with evolutionary potential. Although smaller Na prolongs the arrival times of potentially fixable mutator/antimutator alleles, it also reduces the time to drift to fixation. The latter effect tends to dominate, so that populations with small Na (either because of small N or large U) undergo more frequent transitions between mutation rate classes (fig. 3).
Sexual Populations
As first pointed out by Kimura (1967), the magnitude of indirect selection opposing mutator alleles is greatly diminished in sexual populations because the statistical associations with induced deleterious mutations are rapidly removed by recombination. As loci are no longer inherited as a unit in recombining species, it is more appropriate to evaluate mutational properties on a per-locus basis. Considering a modifier allele m that magnifies the mutation rate to deleterious alleles by an amount Δu at a locus directly influencing fitness, Kimura (1967) showed that in an infinite population, an equilibrium will rapidly be reached where the input of new deleterious alleles is balanced by their disassociation from m by recombination, at which point the selective disadvantage of the mutator allele is approximately sΔu/(s + r − sr), where s is the heterozygous effect of a deleterious mutation and r the recombination rate between the two (fitness and fidelity) loci. To obtain the total selection coefficient against the mutator, , this expression must be summed over all loci affecting fitness. For the case of no recombination (r = 0), the result from the preceding section, , is recovered (where Uh is now the haploid genome-wide deleterious mutation rate), whereas with free recombination (r = 0.5), which is closely approximated by when (the usual situation; Lynch and Walsh 1998).
This result, which has been obtained in a number of different ways (Kondrashov 1995; Dawson 1999; Johnson 1999a), shows that free recombination reduces the magnitude of indirect selection experienced by a mutator allele by a factor of This is because the deleterious mutation load associated with a mutator quickly reaches a mutation–recombination equilibrium rather than building up to the much larger mutation–selection equilibrium. With free recombination, a mildly deleterious mutation becomes disassociated from the mutator in just two generations on average. A mutator allele will then have an elevated probability of being associated with a deleterious mutation of 2Δu, the overall indirect fitness reduction being , with the denominator accounting for the slight reduction in frequency of associated deleterious alleles resulting from selection.
Because some mutations will be linked with the mutator locus, will be a slight underestimate of the selective disadvantage of a mutator in a sexual population. However, given that most organisms contain five or more chromosomes, the vast majority of mutations induced by a mutator allele will generally arise on different chromosomes (a fraction 1 − n − 1 with n chromosomes of equal length) or distant from the mutator when on the same chromosome. More precise expressions that account for finite chromosome numbers and integrate over positions of linked mutations show that is generally no more than (Johnson 1999b; Lynch 2008). Because the excess equilibrium load of a mutator allele is established within just a few generations of its appearance in a sexual population, these expressions provide a very close approximation to the time-averaged selective disadvantage of a segregating mutator allele at all but enormous (and unrealistic) ΔUh.
Recalling ΔU = 2ΔUh as the diploid genome-wide increase in the deleterious mutation rate, and noting that the power of drift in a diploid sexual population is 1/(2Ne), where Ne is the effective population size, the expected drift barrier to the downward evolution of the mutation rate in a sexual species is ΔU < 1/(2Nes), in contrast to 1/Na for an asexual population. As in figure 2, this prediction is well supported by simulations of a replication fidelity locus with an array of alleles differing in the mutation rate by a constant factor (and acting in an additive fashion to define the genotypic mutation rates), with deleterious mutations assumed to arise in a freely segregating background.
Discussion
Although the focus here has been primarily on deleterious mutation accumulation, the net forces in favor of a newly arisen antimutator allele in an asexual population can be viewed as the sum s*≃ΔU + (μ − ν) + sp, where ΔU is the reduction in the genome-wide deleterious mutation rate, (μ − ν) the net mutation pressure in the direction of antimutator alleles, and sp the pleiotropic selective effect of the antimutator allele (independent of the reduced mutation load). When this summed quantity is smaller than the power of drift, the ability of natural selection to further reduce the mutation rate will be strongly diminished. In the context of an antimutator allele, ΔU is positive by definition, whereas (μ − ν) is likely to be negative (assuming it is more difficult to produce antimutator alleles than disrupt them). Under the assumption that there is a physiological cost to high replication fidelity, sp would be negative. However, conditions in which sp is positive can be envisioned, for example, in multicellular organisms, where the rate of germline replication is unlikely to be a limiting factor in reproduction, a reduced somatic mutation rate may be highly advantageous (Lynch 2008).
As the mutation rate is pushed to a lower and lower level by selection, the effects of further molecular refinements of the replication/repair apparatus must become progressively diminished, and ΔU will necessarily become smaller, (μ − ν) likely more negative, and sp smaller (and potentially negative) as well, eventually leading to the inability of selection to diminish the mutation rate any further in the face of random genetic drift. Although the ways in which these three components vary with U and their relative quantitative contributions are unknown, and may differ among phylogenetic lineages, this overall theoretical construct yields qualitative predictions that have the potential to explain several previously disconnected observations.
First, consider Drake's (1991) contention that an inverse relationship exists between the mutation rate per nucleotide site and genome size in microbes, such that the genome-wide mutation rate remains constant across taxa. This original conjecture was based on rather limited data, but a more recent analysis (Lynch 2010) continues to support the general pattern for bacteriophage and prokaryotes, although because of the limited range of bacterial genome sizes, the overall pattern remains highly dependent on the inclusion of bacteriophage in the analysis. Can Drake's contention can be explained on theoretical grounds? Imagine a series of mutation rate states such that U0 is the minimum possible rate, and with multiplicative increases among states such that Ui = (1 + λ)Ui − 1, or ΔU = λUi − 1. The theory implies that the minimum selectable genome-wide deleterious mutation rate satisfies ΔU + (μ − ν) + sp = 1/Na (for an asexual population), or in other words,
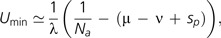 |
(12a) |
where the composite term (μ − ν + sp) is the implied value of the sum at the boundary that satisfies Umin. Thus, the theory predicts an approximate inverse scaling between the total genome-wide deleterious mutation rate and the effective size of an asexual population. Noting that the minimum mutation rate per nucleotide site (umin) is equal to Umin/(ϕdG), where ϕd is the fraction of mutations with deleterious effects and G the number of nucleotide sites per genome, the lower bound to the per-site mutation rate is predicted to be:
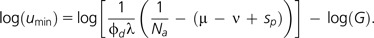 |
(12b) |
Thus, for a group of organisms with comparable effective population sizes, the theory predicts an inverse relationship between the mutation rate per nucleotide site (umin) and total genome size (G), with species-specific Umin being distributed around the regression to a degree that depends on variation in the composite parameter in large brackets. Although the latter quantity must be subject to variation, it is plausible that the degree of variation among microbes is small relative to that in G. For example, with few exceptions, microbial genomes are about 95% coding DNA, so φd is likely to be roughly constant. In addition, microbial populations clearly vary in absolute population sizes (N), but a case can be made that once N exceeds a very large size (as it does in microbes), the effective population size is no longer limited by absolute numbers but by the physical structure of the genome, that is, by the selective interference that results from linked sites on chromosomes (Lynch 2007, 2010). Possibly, most microbes are near this limit.
In contrast, eukaryotic effective population sizes vary by a few orders of magnitude, with multicellular species typically having much smaller Ne (Lynch 2007, 2010). The existing data on such species suggest that u (per generation) scales with roughly the −0.6 power of Ne, although the true scaling could be as extreme as − 1.0 (Lynch 2010). Thus, keeping in mind that it is unclear whether the composite term (μ − ν + sp) contributes substantially to the overall pressure on the mutation rate in eukaryotes, it can at least be stated that the theory is qualitatively consistent with the negative scaling between u and Ne of cellular species. (For sexual species, the right side of equation (12a) is only modified by a factor of 1/s, which does not alter the predicted negative scaling between u and Ne.)
Second, most genomes contain two or more “error-prone” polymerases, often used in replication across bulky lesions in the DNA and often elicited in periods of cellular stress. In vitro studies indicate that the error rates of these enzymes are typically 10- to 10,000-fold higher than those for polymerases involved in genome replication (fig. 4). (In vivo error rates could be lower than those summarized in figure 4, although it is unlikely that the qualitative pattern would be altered.) Because it is unlikely that such polymerases are molecularly constrained to be so inaccurate, many investigators have argued that natural selection has promoted stress-induced mutation as a strategy to facilitate adaptive evolution during challenging periods (Radman et al. 2000; Rosenberg 2001; Tenaillon et al. 2001; Earl and Deem 2004; Foster 2007; Galhardo et al. 2007). However, with the vast majority of mutations being deleterious, it is unclear that there is any net long-term advantage for elevated levels of stress-induced mutation, and the preceding results provide an alternative explanation that eliminates the need to invoke such an argument.
FIG. 4.—

In vitro estimates of rates of base misincorporation by polymerases in four species groups (given as averages from multiple estimates from independent studies in Supplementary Material online). Rates are averaged over all nucleotide contexts, and for the error-prone polymerases, some sites are replicated at fidelity rates considerably lower (and others considerably higher) than the average. Note that for Escherichia coli, Pol I is used to replace the small RNA primers that initiate replication, and Pol III is the major replicative polymerase. For eukaryotes, Pol α is used to extend the RNA primers to a DNA length sufficient for Pol δ to take over, and Pols δ and ε are the major replicative polymerases (one for the leading and the other for the lagging strand). Data are limited for archaeal polymerases.
As implied by equation (12b), the error rate of a polymerase is expected to scale inversely with the number of nucleotide transactions engaged in per generation. Thus, there is no reason to invoke selection for evolvability to explain the error-prone nature of the polymerases involved in stress-induced mutagenesis. Rather, such a pattern is expected to be a natural outcome of the reduction in the efficiency of selection operating on infrequently invoked enzymes. This argument does not deny the critical role of error-prone polymerases in the elimination of DNA damage, nor does it deny the possibility that induced mutagenesis occasionally plays a role in survival/adaptation in extreme times. The hypothesis that mutation rates should naturally evolve to higher levels with enzymes involved in fewer replication events is also consistent with the fact that polymerases involved in the replacement of replication initiation primers have higher error rates than those involved in bulk polymerization (fig. 4) and that the primases that lay down the RNA primers initially involved in replication but subsequently replaced are extraordinarily error prone (Zhang and Grosse 1990; Sheaff and Kuchta 1994; Kuchta and Stengel 2010).
Third, for the reasons just noted, the error rates of pathways downstream in the stages of genome replication are expected to be elevated relative to those for the initial polymerization machinery, as a smaller number of nucleotide sites will remain to be serviced. Thus, it is worth noting that the in vitro proofreading error rates associated with the major replication polymerases in Escherichia coli and Saccharomyces cerevisiae, the only species for which data exist, are much higher than those in the initial polymerization step (Bebenek et al. 1990; Cai et al. 1995; Bloom et al. 1997; Shimizu et al. 2002; Hashimota et al. 2003; Shcherbakova et al. 2003; Fortune et al. 2005; Nick McElhinny et al. 2007; Pursell et al. 2007; McCulloch et al. 2009). Observations on the still further downstream mismatch repair (MMR) pathway are also consistent with theoretical expectations. In E. coli, the in vitro error rate of the major replicative polymerase is approximately 10 − 6 per base incorporation (fig. 4), whereas the in vivo error rate associated with MMR in this and most other species of eubacteria is in the range of 0.01 to 0.05 (e.g., Schaaper and Dunn 1998; Prudhomme et al. 1991; Schaaper 1993; Fujii et al. 1999; Oliver et al. 2000; Richardson and Stojilkovic 2001; Rossolillo and Albertini 2001; Young and Ornston 2001; Mérino et al. 2002; Shaver and Sniegowski 2003; Prunier and Leclercq 2005). (Here, the MMR error rate is simply defined as the fraction of errors emerging after polymerase action that are not eliminated by MMR.) Similarly, in the yeast S. cerevisiae, the in vitro error rate of the major replicative polymerase is approximately 5×10 − 5 (fig. 4), whereas that for MMR is approximately 0.025 (Prolla et al. 1994; Johnson et al. 1996; Marsischky et al. 1996; Sia et al. 1997; Harrington and Kolodner 2007); and in mammals, the respective rates are approximately 10 − 5 (fig. 4) and 0.05 (Bhattacharyya et al. 1995; Glaab and Tindall 1997; Tindall et al. 1998; Umar et al. 1998; Baross-Francis et al. 2001; Xu et al. 2001; Zhang et al. 2002; Dobrovolsky et al. 2003; Hegan et al. 2006). Thus, for the limited systems for which data are available, error rates associated with MMR are three to four orders of magnitude greater than those associated with the initial stages of polymerization. Qualitatively, such a reduction is consistent with the theory in the sense that during replication, the MMR pathway operates at only the 10 − 6 to 10 − 5 fraction of genomic sites that emerge with errors following the initial stages of polymerization, while also engaging in other repair processes in nonreplicating DNA.
Fourth, the theory predicts that evolved mutation rates should be elevated in recombining species relative to asexual taxa with the same effective population sizes, by a factor equal to approximately the inverse of the average selective disadvantage of a new mutation, which is typically in the range of 0.001–0.01 (Lynch and Walsh 1998). More generally, if a species switches from obligate outcrossing to obligate asexual reproduction, selection is expected to reduce the mutation rate if which suggests that truly asexual species with very large population sizes will likely harbor particularly accurate replication systems. In the absence of accurate information on Na/Ne for any such lineages, it is currently difficult to address this matter in a confident way. It might be argued that the reduced rate of mutation in prokaryotes relative to eukaryotes is qualitatively consistent with this hypothesis, as prokaryotes are commonly viewed as being asexual. However, indirect evidence suggests a comparable amount of recombination in prokaryotes and eukaryotes when scaled to the mutation rate (Lynch 2007).
It has been suggested that a bacterial endosymbiont inhabiting aphids, and thought to have Na/Ne≪1 and essentially no recombination, has a mutation rate approximately 10× that in free-living bacterial species (Moran et al. 2009). As the two derived life history changes are expected to have conflicting effects on the mutation rate, the theory suggests that the reduction in Ne has had a greater effect than the reduction in the recombination rate, although this interpretation is clouded by the fact that an absence of recombination is expected to induce a reduction in Ne via hitchhiking effects. A related point concerns the argument that increased sensitivity to amino acid-altering mutations in thermophilic bacteria results in the evolution of a reduced base-substitution mutation rate (Friedman et al. 2004). As noted above, in nonrecombining species, the magnitude of selection on the mutation rate is independent of the fitness effects of mutations, which only become a factor when the recombination rate is much greater than the average deleterious effect of mutations. Thus, the validity of the interpretation of Friedman et al. (2004) depends on the degree to which the genomes of thermophilic bacteria are inherited in a clonal fashion.
Finally, it should be emphasized that the theory yields predictions on how the per-generation mutation rate is expected to evolve in response to deleterious mutation load. Although large multicellular organisms exhibit substantially higher per-generation germline mutation rates than do single-celled eukaryotes (Lynch 2010), consistent with the theory, the former also experience multiple germline cell divisions per generation. As the per-generation mutation rate is a consequence of the net accumulation of mutations throughout germline development, the theory predicts that the mutation rate per germline cell division will scale inversely with the number of such divisions per generation. Although the evolution of multicellularity results in a reduction in Ne, which is predicted to encourage an increase in the mutation rate per generation, if the number of germline cell divisions per generation is large enough, some multicellular species may actually evolve lower mutation rates per germline cell division than the per-generation rates observed in microbes. This seems to be the case for several well-studied model species, including humans, which exhibit mutation rates per germline cell division that are comparable with or lower than mutation rates in E. coli and yeast (Lynch 2010). The results in figure 4 suggest that such low rates may be achieved by a reduction in intracellular activities (e.g., metabolism) leading to premutations rather than by an increase in the accuracy of the replication/repair machinery.
Supplementary Material
Supplementary material is available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The author thanks M. Ackerman, C. Baer, J. Drake, T. Kunkel, and P. Sniegowski for their helpful comments. This work was supported by the National Institute of Health (grant R01 GM036827 to M.L. and W. K. Thomas), National Science Foundation (grant EF-0827411 to M.L.), and US Department of Defense (grant ONRBAA10-002 to M.L., P. Foster, H. Tang, and S. Finkel).
References
- Baer CF, Miyamoto MM, Denver DR. Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat Rev Genet. 2007;8:619–631. doi: 10.1038/nrg2158. [DOI] [PubMed] [Google Scholar]
- Baross-Francis A, Makhani N, Liskay RM, Jirik FR. Elevated mutant frequencies and increased C: G→T: A transitions in Mlh1-/- versus Pms2-/- murine small intestinal epithelial cells. Oncogene. 2001;20:619–625.. doi: 10.1038/sj.onc.1204138. [DOI] [PubMed] [Google Scholar]
- Bebenek A, Carver GT, Kadyrov FA, Kissling GE, Drake JW. Processivity clamp gp45 and ssDNA-binding-protein gp32 modulate the fidelity of bacteriophage RB69 DNA polymerase in a sequence-specific manner, sometimes enhancing and sometimes compromising accuracy. Genetics. 2005;169:1815–1824. doi: 10.1534/genetics.104.037630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebenek K, Joyce CM, Fitzgerald MP, Kunkel TA. The fidelity of DNA synthesis catalyzed by derivatives of Escherichia coli DNA polymerase I. J Biol Chem. 1990;265:13878–13887. [PubMed] [Google Scholar]
- Bessman MJ, Muzyczka N, Goodman MF, Schnaar RL. Studies on the biochemical basis of spontaneous mutation. II. The incorporation of base and its analogue into DNA by wild type, mutator and antimutator DNA polymerases. J Mol Biol. 1974;88:409–421. doi: 10.1016/0022-2836(74)90491-4. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya NP, et al. Molecular analysis of mutations in mutator colorectal carcinoma cell lines. Hum Mol Genet. 1995;4:2057–2064. doi: 10.1093/hmg/4.11.2057. [DOI] [PubMed] [Google Scholar]
- Bloom LB, et al. Fidelity of Escherichia coli DNA polymerase III holoenzyme. The effects of beta, gamma complex processivity proteins and epsilon proofreading exonuclease on nucleotide misincorporation efficiencies. J Biol Chem. 1997;272:27919–27930. doi: 10.1074/jbc.272.44.27919. [DOI] [PubMed] [Google Scholar]
- Cai H, Yu H, McEntee K, Kunkel TA, Goodman MF. Purification and properties of wild-type and exonuclease-deficient DNA polymerase II from Escherichia coli. J Biol Chem. 1995;270:15327–15335. doi: 10.1074/jbc.270.25.15327. [DOI] [PubMed] [Google Scholar]
- Casjens S. The diverse and dynamic structure of bacterial genomes. Annu Rev Genet. 1998;32:339–377. doi: 10.1146/annurev.genet.32.1.339. [DOI] [PubMed] [Google Scholar]
- Cox RA. Quantitative relationships for specific growth rates and macromolecular compositions of Mycobacterium tuberculosis, Streptomyces coelicolor A3(2) and Escherichia coli B/r: an integrative theoretical approach. Microbiology. 2004;150:1413–1426. doi: 10.1099/mic.0.26560-0. [DOI] [PubMed] [Google Scholar]
- Dawson KJ. The dynamics of infinitesimally rare alleles, applied to the evolution of mutation rates and the expression of deleterious mutations. Theor Popul Biol. 1999;55:1–22. doi: 10.1006/tpbi.1998.1375. [DOI] [PubMed] [Google Scholar]
- Dobrovolsky VN, et al. Pms2 deficiency results in increased mutation in the Hprt gene but not the Tk gene of Tk (+/-) transgenic mice. Mutagenesis. 2003;18:365–370. doi: 10.1093/mutage/geg007. [DOI] [PubMed] [Google Scholar]
- Drake JW. A constant rate of spontaneous mutation in DNA-based microbes. Proc Natl Acad Sci U S A. 1991;88:7160–7164. doi: 10.1073/pnas.88.16.7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW. General antimutators are improbable. J Mol Biol. 1993;229:8–13. doi: 10.1006/jmbi.1993.1002. [DOI] [PubMed] [Google Scholar]
- Earl DJ, Deem MW. Evolvability is a selectable trait. Proc Natl Acad Sci U S A. 2004;101:11531–11536. doi: 10.1073/pnas.0404656101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijalkowska IJ, Dunn RL, Schaaper RM. Mutants of Escherichia coli with increased fidelity of DNA replication. Genetics. 1993;134:1023–1030. doi: 10.1093/genetics/134.4.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune JM, et al. Saccharomyces cerevisiae DNA polymerase δ: high fidelity for base substitutions but lower fidelity for single- and multi-base deletions. J Biol Chem. 2005;280:29980–29987. doi: 10.1074/jbc.M505236200. [DOI] [PubMed] [Google Scholar]
- Foster PL. Stress-induced mutagenesis in bacteria. Crit Rev Biochem Mol Biol. 2007;42:373–397. doi: 10.1080/10409230701648494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman R, Drake JW, Hughes AL. Genome-wide patterns of nucleotide substitution reveal stringent functional constraints on the protein sequences of thermophiles. Genetics. 2004;167:1507–1512. doi: 10.1534/genetics.104.026344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, et al. DNA replication errors produced by the replicative apparatus of Escherichia coli. J Mol Biol. 1999;289:835–850. doi: 10.1006/jmbi.1999.2802. [DOI] [PubMed] [Google Scholar]
- Furió V, Moya A, Sanjuán R. The cost of replication fidelity in an RNA virus. Proc Natl Acad Sci U S A. 2005;102:10233–10237. doi: 10.1073/pnas.0501062102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furió V, Moya A, Sanjuán R. The cost of replication fidelity in human immunodeficiency virus type 1. Proc Biol Sci. 2007;274:225–230. doi: 10.1098/rspb.2006.3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galán JC, et al. Mutation rate is reduced by increased dosage of mutL gene in Escherichia coli K-12. FEMS Microbiol Lett. 2007;275:263–269. doi: 10.1111/j.1574-6968.2007.00902.x. [DOI] [PubMed] [Google Scholar]
- Galhardo RS, Hastings PJ, Rosenberg SM. Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol Biol. 2007;42:399–435. doi: 10.1080/10409230701648502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrish PJ, Colato A, Perelson AS, Sniegowski PD. Complete genetic linkage can subvert natural selection. Proc Natl Acad Sci U S A. 2007;104:6266–6271. doi: 10.1073/pnas.0607280104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaab WE, Tindall KR. Mutation rate at the hprt locus in human cancer cell lines with specific mismatch repair-gene defects. Carcinogenesis. 1997;18:1–8. doi: 10.1093/carcin/18.1.1. [DOI] [PubMed] [Google Scholar]
- Gordo I, Charlesworth B. The degeneration of asexual haploid populations and the speed of Muller's ratchet. Genetics. 2000;154:1379–1387. doi: 10.1093/genetics/154.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigh J. The accumulation of deleterious genes in a population—Muller's ratchet. Theor Popul Biol. 1978;14:251–267. doi: 10.1016/0040-5809(78)90027-8. [DOI] [PubMed] [Google Scholar]
- Hall LM, Henderson-Begg SK. Hypermutable bacteria isolated from humans—a critical analysis. Microbiology. 2006;152:2505–2514. doi: 10.1099/mic.0.29079-0. [DOI] [PubMed] [Google Scholar]
- Harrington JM, Kolodner RD. Saccharomyces cerevisiae Msh2-Msh3 acts in repair of base-base mispairs. Mol Cell Biol. 2007;27:6546–6554. doi: 10.1128/MCB.00855-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Shimizu K, Nakashima N, Sugino A. Fidelity of DNA polymerase δ holoenzyme from Saccharomyces cerevisiae: the sliding clamp proliferating cell nuclear antigen decreases its fidelity. Biochemistry. 2003;42:14207–14213. doi: 10.1021/bi0348359. [DOI] [PubMed] [Google Scholar]
- Hegan DC, et al. Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6. Carcinogenesis. 2006;27:2402–2408. doi: 10.1093/carcin/bgl079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingraham JL, Maaløe O, Neidhardt FC. Growth of the bacterial cell. Sunderland (MA): Sinauer Associates; 1983. [Google Scholar]
- Johnson RE, Kovvali GK, Prakash L, Prakash S. Requirement of the yeast MSH3 and MSH6 genes for MSH2-dependent genomic stability. J Biol Chem. 1996;271:7285–7288. doi: 10.1074/jbc.271.13.7285. [DOI] [PubMed] [Google Scholar]
- Johnson T. The approach to mutation-selection balance in an infinite asexual population, and the evolution of mutation rates. Proc R Soc Lond B. 1999a;266:2389–2397. doi: 10.1098/rspb.1999.0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T. Beneficial mutations, hitchhiking and the evolution of mutation rates in sexual populations. Genetics. 1999b;151:1621–1631. doi: 10.1093/genetics/151.4.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. On the evolutionary adjustment of spontaneous mutation rates. Genet Res. 1967;9:23–34. [Google Scholar]
- Kimura M, Ohta T. The average number of generations until fixation of a mutant gene in a finite population. Genetics. 1969;61:763–771. doi: 10.1093/genetics/61.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov AS. Modifiers of mutation-selection balance: general approach and the evolution of mutation rates. Genet Res. 1995;66:53–70. [Google Scholar]
- Kuchta RD, Stengel G. Mechanism and evolution of DNA primases. Biochim Biophys Acta. 2010;1804:1180–1189. doi: 10.1016/j.bbapap.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh E, Choe J, Loeb LA. Highly tolerated amino acid substitutions increase the fidelity of Escherichia coli DNA polymerase I. J Biol Chem. 2007;282:12201–12209. doi: 10.1074/jbc.M611294200. [DOI] [PubMed] [Google Scholar]
- Loh E, Salk JJ, Loeb LA. Optimization of DNA polymerase mutation rates during bacterial evolution. Proc Natl Acad Sci U S A. 2010;107:1154–1159. doi: 10.1073/pnas.0912451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. The origins of genome architecture. Sunderland (MA): Sinauer Associates; 2007. [Google Scholar]
- Lynch M. The cellular, developmental, and population-genetic determinants of mutation-rate evolution. Genetics. 2008;180:933–943. doi: 10.1534/genetics.108.090456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. Evolution of the mutation rate. Trends Genet. 2010;26:345–352. doi: 10.1016/j.tig.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Gabriel W. Mutation load and the survival of small populations. Evolution. 1990;44:1725–1737. doi: 10.1111/j.1558-5646.1990.tb05244.x. [DOI] [PubMed] [Google Scholar]
- Lynch M, Walsh JB. Genetics and analysis of quantitative traits. Sunderland (MA): Sinauer Associates; 1998. [Google Scholar]
- Lynch RB, Butcher D, Gabriel W. Mutational meltdowns in asexual populations. Heredity J. 1993;84:339–344. doi: 10.1093/oxfordjournals.jhered.a111354. [DOI] [PubMed] [Google Scholar]
- Marsischky GT, Filosi N, Kane MF, Kolodner R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996;10:407–420. doi: 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- Maruyama T. The age of an allele in a finite population. Genet Res. 1974;23:137143. doi: 10.1017/s0016672300014750. [DOI] [PubMed] [Google Scholar]
- McCulloch SD, Kokoska RJ, Garg P, Burgers PM, Kunkel TA. The efficiency and fidelity of 8-oxo-guanine bypass by DNA polymerases δ and η. Nucleic Acids Res. 2009;37:2830–2840. doi: 10.1093/nar/gkp103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mérino D, Réglier-Poupet H, Berche P, Charbit A European Listeria Genome Consortium. A hypermutator phenotype attenuates the virulence of Listeria monocytogenes in a mouse model. Mol Microbiol. 2002;44:877–887. doi: 10.1046/j.1365-2958.2002.02929.x. [DOI] [PubMed] [Google Scholar]
- Moran NA, McLaughlin HJ, Sorek R. The dynamics and time scale of ongoing genomic erosion in symbiotic bacteria. Science. 2009;323:379–382. doi: 10.1126/science.1167140. [DOI] [PubMed] [Google Scholar]
- Nei M, Roychoudhury AK. Probability of fixation and mean fixation time of an overdominant mutation. Genetics. 1973;74:371–380. doi: 10.1093/genetics/74.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Stith CM, Burgers PMJ, Kunkel TA. Inefficient proofreading and biased error rates during inaccurate DNA synthesis by a mutant derivative of Saccharomyces cerevisiae DNA polymerase δ. J Biol Chem. 2007;282:2324–2332. doi: 10.1074/jbc.M609591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver A, Cantón R, Campo P, Baquero F, Blázquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–1254. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- Prolla TA, Christie DM, Liskay RM. Dual requirement in yeast DNA mismatch repair for MLH1 and PMS1, two homologs of the bacterial mutL gene. Mol Cell Biol. 1994;14:407–415. doi: 10.1128/mcb.14.1.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudhomme M, Méjean V, Martin B, Claverys JP. Mismatch repair genes of Streptococcus pneumoniae: HexA confers a mutator phenotype in Escherichia coli by negative complementation. J Bacteriol. 1991;173:7196–7203. doi: 10.1128/jb.173.22.7196-7203.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prunier AL, Leclercq R. Role of mutS and mutL genes in hypermutability and recombination in Staphylococcus aureus. J Bacteriol. 2005;187:3455–3464. doi: 10.1128/JB.187.10.3455-3464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pursell ZF, Isoz I, Lundström EB, Johansson E, Kunkel TA. Regulation of B family DNA polymerase fidelity by a conserved active site residue: characterization of M644W, M644L and M644F mutants of yeast DNA polymerase ε. Nucleic Acids Res. 2007;35:3076–3086. doi: 10.1093/nar/gkm132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radman M, Taddei F, Matic I. Evolution-driving genes. Res Microbiol. 2000;151:91–95. doi: 10.1016/s0923-2508(00)00122-4. [DOI] [PubMed] [Google Scholar]
- Richardson AR, Stojiljkovic I. Mismatch repair and the regulation of phase variation in Neisseria meningitidis. Mol Microbiol. 2001;40:645–655. doi: 10.1046/j.1365-2958.2001.02408.x. [DOI] [PubMed] [Google Scholar]
- Rosenberg SM. Evolving responsively: adaptive mutation. Nat Rev Genet. 2001;2:504–515. doi: 10.1038/35080556. [DOI] [PubMed] [Google Scholar]
- Rossolillo P, Albertini AM. Functional analysis of the Bacillus subtilis y shD gene, a mutS paralogue. Mol Gen Genet. 2001;264:809–818. doi: 10.1007/s004380000370. [DOI] [PubMed] [Google Scholar]
- Schaaper RM. Base selection, proofreading, and mismatch repair during DNA replication in Escherichia coli. J Biol Chem. 1993;268:23762–23765. [PubMed] [Google Scholar]
- Schaaper RM. Antimutator mutants in bacteriophage T4 and Escherichia coli. Genetics. 1998;148:1579–1585. doi: 10.1093/genetics/148.4.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaper RM, Dunn RL. Effect of Escherichia coli dnaE antimutator mutants on mutagenesis by the base analog N4-aminocytidine. Mutat Res. 1998;402:23–28. doi: 10.1016/s0027-5107(97)00278-9. [DOI] [PubMed] [Google Scholar]
- Shaver AC, Sniegowski PD. Spontaneously arising mutL mutators in evolving Escherichia coli populations are the result of changes in repeat length. J Bacteriol. 2003;185:6076–6082. doi: 10.1128/JB.185.20.6076-6082.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova PV, et al. Unique error signature of the four-subunit yeast DNA polymerase ε. J Biol Chem. 2003;278:43770–43780. doi: 10.1074/jbc.M306893200. [DOI] [PubMed] [Google Scholar]
- Sheaff RJ, Kuchta RD. Misincorporation of nucleotides by calf thymus DNA primase and elongation of primers containing multiple noncognate nucleotides by DNA polymerase alpha. J Biol Chem. 1994;269:19225–19231. [PubMed] [Google Scholar]
- Shimizu K, et al. Fidelity of DNA polymerase ε holoenzyme from budding yeast Saccharomyces cerevisiae. J Biol Chem. 2002;277:37422–37429. doi: 10.1074/jbc.M204476200. [DOI] [PubMed] [Google Scholar]
- Sia EA, Kokoska RJ, Dominska M, Greenwell P, Petes TD. Microsatellite instability in yeast: dependence on repeat unit size and DNA mismatch repair genes. Mol Cell Biol. 1997;17:2851–2858. doi: 10.1128/mcb.17.5.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski PD, Gerrish PJ, Lenski RE. Evolution of high mutation rates in experimental populations of E. coli. Nature. 1997;387:703–705. doi: 10.1038/42701. [DOI] [PubMed] [Google Scholar]
- Sturtevant AH. Essays on evolution. I. On the effects of selection on mutation rate. Quart Rev Biol. 1937;12:464–476. [Google Scholar]
- Taddei F, et al. Role of mutator alleles in adaptive evolution. Nature. 1997;387:700–702. doi: 10.1038/42696. [DOI] [PubMed] [Google Scholar]
- Taylor C, Iwasa Y, Nowak MA. A symmetry of fixation times in evolutionary dynamics. J Theor Biol. 2006;243:245–251. doi: 10.1016/j.jtbi.2006.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon O, Taddei F, Radman M, Matic I. Second-order selection in bacterial evolution: selection acting on mutation and recombination rates in the course of adaptation. Res Microbiol. 2001;152:11–16. doi: 10.1016/s0923-2508(00)01163-3. [DOI] [PubMed] [Google Scholar]
- Tian W, Hwang YT, Hwang CB. The enhanced DNA replication fidelity of a mutant herpes simplex virus type 1 DNA polymerase is mediated by an improved nucleotide selectivity and reduced mismatch extension ability. J Virol. 2008;82:8937–8941. doi: 10.1128/JVI.00911-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tindall KR, et al. Complementation of mismatch repair gene defects by chromosome transfer. Mutat Res. 1998;402:15–22. doi: 10.1016/s0027-5107(97)00277-7. [DOI] [PubMed] [Google Scholar]
- Tröbner W, Piechocki R. Selection against hypermutability in Escherichia coli during long term evolution. Mol Gen Genet. 1984;198:177–178. doi: 10.1007/BF00328720. [DOI] [PubMed] [Google Scholar]
- Umar A, et al. Functional overlap in mismatch repair by human MSH3 and MSH6. Genetics. 1998;148:1637–1646. doi: 10.1093/genetics/148.4.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira-Silva S, Touchon M, Rocha EP. No evidence for elemental-based streamlining of prokaryotic genomes. Trends Ecol Evol. 2010;25:319–320. doi: 10.1016/j.tree.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Xu XS, Narayanan L, Dunklee B, Liskay RM, Glazer PM. Hypermutability to ionizing radiation in mismatch repair-deficient, Pms2 knockout mice. Cancer Res. 2001;61:3775–3780. [PubMed] [Google Scholar]
- Young DM, Ornston LN. Functions of the mismatch repair gene mutS from Acinetobacter sp. strain ADP1. J Bacteriol. 2001;183:6822–6831. doi: 10.1128/JB.183.23.6822-6831.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Lloyd R, Bowden G, Glickman BW, de Boer JG. Thymic lymphomas arising in Msh2 deficient mice display a large increase in mutation frequency and an altered mutational spectrum. Mutat Res. 2002;500:67–74. doi: 10.1016/s0027-5107(01)00297-4. [DOI] [PubMed] [Google Scholar]
- Zhang SS, Grosse F. Accuracy of DNA primase. J Mol Biol. 1990;216:475–479. doi: 10.1016/0022-2836(90)90370-2. [DOI] [PubMed] [Google Scholar]