

Abstract
Virus infections can result in a variety of cellular injuries, and these often involve the permeabilization of host membranes by viral proteins of the viroporin family. Prototypical viroporin 2B is responsible for the alterations in host cell membrane permeability that take place in enterovirus-infected cells. 2B protein can be localized at the endoplasmic reticulum (ER) and the Golgi complex, inducing membrane remodeling and the blockade of glycoprotein trafficking. These findings suggest that 2B has the potential to integrate into the ER membrane, but specific information regarding its biogenesis and mechanism of membrane insertion is lacking. Here, we report experimental results of in vitro translation-glycosylation compatible with the translocon-mediated insertion of the 2B product into the ER membrane as a double-spanning integral membrane protein with an N-/C-terminal cytoplasmic orientation. A similar topology was found when 2B was synthesized in cultured cells. In addition, the in vitro translation of several truncated versions of the 2B protein suggests that the two hydrophobic regions cooperate to insert into the ER-derived microsomal membranes.
INTRODUCTION
Virus infections can lead to a variety of cellular injuries, and usually these involve the restructuring of host membrane systems. Viroporins are a group of small virally encoded proteins that interact with cellular membranes to modify permeability and promote the release of viral particles. A typical feature exhibited by viroporins is the presence of at least one membrane-spanning helix anchoring the protein into membranes. After membrane insertion, their oligomerization creates hydrophilic channels or pores (22).
Poliovirus is the enterovirus prototype member of the Picornaviridae family. This small, nonenveloped, icosahedral virus possesses a single-stranded 7.5-kb positive-sense RNA genome that encodes a single polyprotein. Polyprotein processing by virus-encoded proteases yields the structural P1 region proteins that encapsidate viral RNA and the nonstructural P2 and P3 region proteins involved in the replication of the viral RNA and membrane permeabilization (2). Nonstructural 2B protein is one of the products generated on processing the P2 region (62). Viroporin 2B has been identified as one of the viral proteins responsible for the alterations in host cell membrane permeability that take place in enterovirus-infected cells. Different 2B proteins expressed in cells have been localized at the endoplasmic reticulum (ER), Golgi complex, and, to a lesser extent, to the plasma and mitochondrial membranes (18, 31, 49, 58). Biochemical and structural data indicate that viroporins form homo-oligomers that create pores in the ER and Golgi complex membranes (1, 16, 17, 30, 59). However, experimental data dealing with the mechanism of the membrane integration of the 2B product are lacking to date.
The poliovirus 2B viroporin protein is hydrophobic overall and rather small (97 amino acids). Hydrophobicity within the viroporin 2B sequence seems to cluster in two main regions (Fig. 1), one predicted to form a cationic amphipathic α-helix located between residues 35 and 55 (HR1) and a second, more hydrophobic, α-helix located between residues 61 and 81 (HR2), as previously suggested (1). Mutations in the amphipathic α-helix or the second hydrophobic region were shown to interfere with the ability of 2B to increase membrane permeability to promote virus release (3, 8, 58) and with viral RNA replication (57), indicating that the soundness of these regions is essential for viral infection. The amphipathic α-helices of several 2B proteins contain three lysine residues at similar positions, and the presence of an aspartic acid residue also is common in the hydrophobic HR2 region (15), suggesting an α-helical hairpin structure.
Fig. 1.

Membrane association of the chimeric viroporin 2B/P2 protein. At the top is a schematic representation of the poliovirus 2B/P2 chimeric protein (the fused P2 domain is shown in gray). Amino acid residues are shown (HR1 and HR2 are highlighted in gray boxes). The gel in the middle shows the segregation of [35S]Met/Cys-labeled viroporin 2B/P2 fusion protein into membranous and soluble fractions (untreated) and after alkaline wash (Alk. Ext.; sodium carbonate buffer) or urea treatments. P and S denote pellet and supernatant, respectively. In the graph at the bottom, to calculate the percentages of protein the signals present in each pellet and supernatant pair were summed and set to 100%. Data correspond to averages from at least three independent experiments; error bars show standard deviations.
Two views of the insertion process of α-helical hairpins into the membrane bilayer can be envisioned. One view postulates α-helical hairpin insertion to be a spontaneous process that does not require specific machinery (9, 19). The other supposes a role for the translocon, which is responsible for facilitating the translocation of secreted proteins across the membrane and insertion of membrane proteins into the lipid bilayer (27, 46), allowing en bloc α-helical hairpin insertion from a proteinaceous environment into the lipid bilayer (51).
In the present study, we find that viroporin 2B is an integral membrane protein that can be inserted into the ER membrane through the translocon. The in vitro translation of model integral membrane protein constructs in the presence of microsomal membranes initially suggested that when expressed separately, only the amphipathic helix (HR1) can span the membrane. However, the in vitro translation of truncated versions of the 2B protein carrying appropriate C-terminal reporter glycosylation tags further demonstrated that (i) the N-terminal hydrophobic domain may be stably inserted into the ER-derived microsomal membranes through the translocon, provided that an Ncyt/Clum topology is preserved (Ncy+, N-terminal end of the transmembrane [TM] segment is oriented toward the cytosol; Clum, C-terminal end of the TM segment is oriented toward the lumen); and (ii) within the complete 2B protein the two hydrophobic regions cooperate to insert into the ER membrane as a helical hairpin with an N-/C-terminal cytoplasmic orientation. In addition, a similar topology was adopted by viroporin 2B expressed in cultured cells under conditions leading to plasma membrane permeabilization.
MATERIALS AND METHODS
Enzymes and chemicals.
All enzymes (unless indicated otherwise) as well as plasmid pGEM1, the RiboMAX SP6 RNA polymerase system, and rabbit reticulocyte lysate (a cell-free translation system) were purchased from Promega (Madison, WI). The ER rough microsomes from dog pancreas were purchased from tRNA Probes (College Station, TX). To ensure consistent performance with minimal translational inhibition and background noise, microsomes have been isolated free from contaminating membrane fractions and stripped of endogenous membrane-bound ribosomes and mRNA. The [35S]Met/Cys and 14C-methylated markers were purchased from GE Healthcare. The restriction enzymes and endoglycosidase H (EndoH) were purchased from Roche Molecular Biochemicals. The DNA plasmid, RNA clean-up, and PCR purification kits were from Qiagen (Hilden, Germany). The PCR QuikChange mutagenesis kit was from Stratagene (La Jolla, CA). All oligonucleotides were purchased from Thermo (Ulm, Germany).
Computer-assisted analysis of viroporin 2B sequence.
The prediction of TM helices was done using up to 10 of the most common methods available on the Internet: DAS (14) (http://www.sbc.su.se/∼miklos/DAS), PHDhtm (47) (http://www.predictprotein.org/), MEMSAT3 (28) (http://bioinf.cs.ucl.ac.uk/psipred/), MEMSAT-SVM (43) (http://bioinf.cs.ucl.ac.uk/psipred/), SOSUI (26) (http://bp.nuap.nagoya-u.ac.jp/sosui/), TMHMM (29) (http://www.cbs.dtu.dk/services/TMHMM), TMPred (http://www.ch.embnet.org/software/TMPRED_form.html), ΔG Prediction Server (24, 25) (http://www.cbr.su.se/DGpred/), SPOCTOPUS (60) (http://octopus.cbr.su.se/), and TopPRED (12) (http://www.sbc.su.se/∼erikw/toppred2/).
DNA manipulations.
Plasmids encoding full-length 2B sequence (without a stop codon) were constructed by subcloning poliovirus serotype 1 (PV1) Mahoney strain 2B-encoding DNA (kindly provided by E. Wimmer, Stony Brook University) (56) into pGEM1 vector between the NcoI and NdeI restriction sites. This construct contained the P2 domain of the Escherichia coli leader peptidase (Lep) fused in frame at the C terminus, as described previously (41). Alternatively, we prepared templates for the in vitro transcription of the truncated 2B mRNA with a 3′ glycosylation tag. The truncated viroporin 2B sequence was prepared by the PCR amplification of a fragment of the pGEM1 plasmid. The 5′ primer was the same for all PCRs and had the sequence 5′-TTCGTCCAACCAAACCGACTC-3′. This primer was situated 210 bases upstream of the 2B translational start codon; thus, all amplified fragments contained the SP6 transcriptional promoter from pGEM1. The 3′ primers were designed to have approximately the same annealing temperature as the 5′ primer. They contained an optimized glycosylation tag followed by tandem translational stop codons, TAG and TAA, and annealed at specific positions to obtain the desired polypeptide length. PCR amplification comprised a total of 30 cycles with an annealing temperature of 52°C. The amplified DNA products were purified with the Qiagen PCR purification kit according to the manufacturer's protocol and verified on a 1% agarose gel.
In addition, the hydrophobic regions from 2B were introduced into the modified Lep sequence from the pGEM1 plasmid (24, 34) between the SpeI and KpnI sites using two double-stranded oligonucleotides with overlapping overhangs at the ends. The complementary oligonucleotide pairs first were annealed at 85°C for 10 min and then slowly cooled to 30°C, after which the two annealed double-stranded oligonucleotides were mixed, incubated at 65°C for 5 min, cooled slowly to room temperature, and ligated into the vector. The replacements of Lys 46 by Gly, Glu, Gln, and Arg in the LepHR1 construct were done using the QuikChange mutagenesis kit from Stratagene (La Jolla, CA) according to the manufacturer's protocol. All DNA manipulations were confirmed by the sequencing of plasmid DNAs.
Expression in vitro.
Full-length 2B DNA was amplified from 2B/P2 plasmid using a reverse primer with a stop codon at the end of the 2B sequence (2B-derived expressions), or the DNA derived from the pGEM1 plasmid was transcribed directly (2B/P2 construct). Alternatively, viroporin 2B was amplified fused to the first 50 amino acids from P2 using a reverse primer with tandem stop codons at the 3′ end (2B/50P2). The transcription of the DNA derived from the pGEM1 plasmid was done as described previously (61). Briefly, the transcription mixture was incubated at 37°C for 2 h. The mRNA products were purified with a Qiagen RNeasy clean-up kit and verified on a 1% agarose gel.
In vitro translation of in vitro-transcribed mRNA was performed in the presence of reticulocyte lysate, [35S]Met/Cys, and dog pancreas microsomes as described previously (21, 61). Lep constructs with HR-tested segments were transcribed and translated as previously reported (33, 34). For the posttranslational membrane insertion experiments, 2B-derived mRNAs were translated (37°C for 1 h) in the absence of rough microsomal membranes (RMs). Translation then was inhibited with cycloheximide (10 min at 26°C; 2 mg/ml final concentration), after which RMs were added and incubated for an additional hour at 37°C. In all cases, after translation membranes were collected by ultracentrifugation and analyzed by sodium-dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Finally, the gels were visualized on a Fuji FLA3000 phosphorimager with ImageGauge software.
For EndoH treatment, the translation mixture was diluted in 4 volumes of 70 mM sodium citrate (pH 5.6) and centrifuged (100,000 × g for 20 min at 4°C). The pellet was resuspended in 50 μl of sodium citrate buffer with 0.5% SDS and 1% β-mercaptoethanol, boiled for 5 min, and incubated for 1 h at 37°C with 0.1 mU of EndoH. The samples were analyzed by SDS-PAGE.
For the proteinase K protection assay, the translation mixture was supplemented with 1 μl of 50 mM CaCl2 and 1 μl of proteinase K (4 mg/ml) and then digested for 40 min on ice. The reaction was stopped by adding 1 mM phenylmethylsulfonyl fluoride (PMSF) before SDS-PAGE analysis.
Membrane sedimentation, alkaline wash, and urea treatments.
The translation mixture was diluted in 8 volumes of buffer A (35 mM Tris-HCl at pH 7.4 and 140 mM NaCl) for the membrane sedimentation, 4 volumes of buffer A supplemented with 100 mM Na2CO3 (pH 11.5) for the alkaline wash, and 4 or 8 M urea for urea treatments. The samples were incubated on ice for 30 min and clarified by centrifugation (10,000 × g for 20 min). Membranes were collected by the ultracentrifugation (100,000 × g for 20 min at 4°C) of the supernatant onto 50-μl sucrose cushions. Pellets (P) and supernatants (S) of the ultracentrifugation were analyzed by SDS-PAGE.
Phase separation in Triton X-114 solution.
The phase separation of integral membrane proteins using the detergent Triton X-114 was performed as described previously (10, 41). Triton X-114 (1%, vol/vol) was added to a translation mixture that previously had been diluted with 180 μl of phosphate-buffered saline (PBS). After mixing, the samples were incubated at 0°C for 1 h and overlaid onto 300 μl of PBS supplemented with 6% (wt/vol) sucrose and 1% (vol/vol) Triton X-114. After 10 min at 30°C, an organic droplet was obtained by centrifugation for 3 min at 1,500 × g. The resulting aqueous upper phase (AP; 200 μl) was collected, and the organic droplet at the bottom of the tube was diluted with PBS (organic phase [OP]). Both OP and AP were supplemented with sample buffer and boiled for 10 min prior to 12% (Fig. 2B) or 20% (Fig. 2C) SDS-PAGE analysis.
Fig. 2.
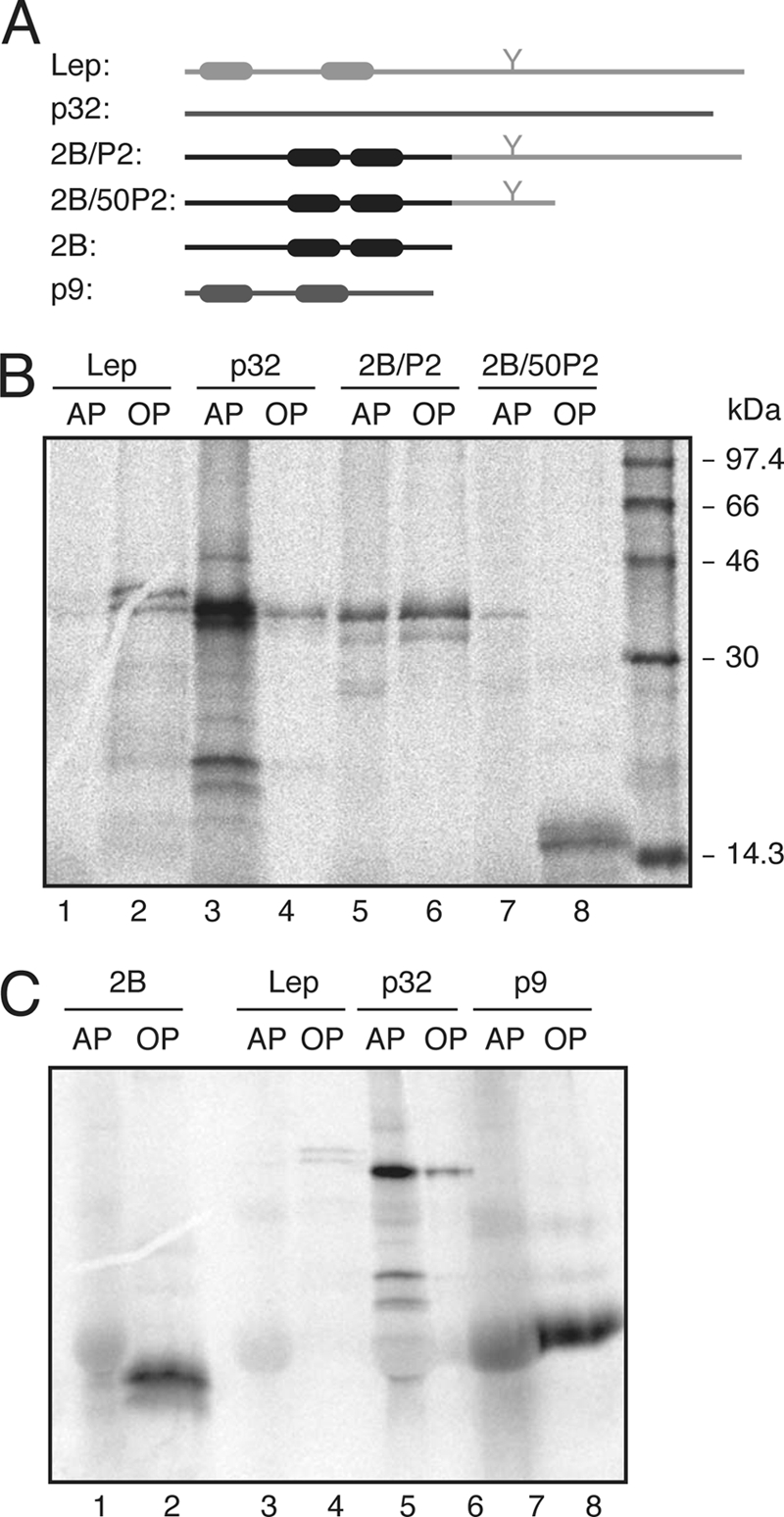
Triton X-114 partition of viroporin 2B and 2B-derived proteins. (A) Structural organization of the proteins used in the Triton X-114 partition experiments. (B) SDS-PAGE (12% polyacrylamide) analysis after Triton X-114 treatment of 2B/P2 (∼38 kDa) and 2B/50P2 (∼16 kDa) proteins. Integral membrane protein Lep (∼37 kDa; the nonglycosylated form) and peripheral PNRSV movement protein (∼32 kDa) were processed in parallel as control samples. (C) Phase separation of viroporin 2B. As a control for small integral membrane protein, Turnip crinkle virus p9 movement protein (∼9 kDa) was included. The gels used contained 20% polyacrylamide. AP and OP refer to aqueous and organic phases, respectively.
Transfection assay.
Baby hamster kidney (BHK) cells that stably express the T7 RNA polymerase (clone BSR-T7/5), designated BHKT7 (11), were used. Cells were grown at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5 or 10% fetal calf serum (FCS) and nonessential amino acids. BHKT7 cells additionally were treated with Geneticin G418 (Sigma) on every third passage at a final concentration of 2 mg/ml. Cells were transfected with 1 μg of plasmid pTM1-2B (3, 4) or the different constructs plus 2 μl of Lipofectamine per well in Opti-mem medium (Invitrogen) for 2 h at 37°C. After 2 h, Lipofectamine was removed and the cells were supplemented with fresh medium containing 5% FCS.
RESULTS
Viroporin 2B is an integral membrane protein.
Viroporin 2B amino acid sequence (Fig. 1) has been parsed to test the performance of several commonly used algorithms for predicting the topology of integral membrane proteins. As shown in Table 1, the predicted outcome showed great variability according to the methods used, likely due to the limited hydrophobicity of the membrane-associating regions of 2B. The membrane association properties of the full-length viroporin 2B were studied using an in vitro system that closely mimics the in vivo situation, in which cytosolic and membrane fractions of in vitro-translated [35S]Met/Cys-labeled 2B/P2 fusions in the presence of ER-derived microsomes were collected and analyzed by SDS-PAGE and autoradiography. In this system, the microsomes provide all of the membrane insertion and glycosylation components (i.e., the translocon machinery and the oligosaccharyltransferase enzyme). The reporter P2 domain is the extramembrane C-terminal domain from the bacterial leader peptidase (Lep) that carries an N-glycosylation site extensively used to report membrane translocation (Fig. 1, top). The viroporin 2B/P2 chimera was recovered mainly from the 100,000 × g pellet fraction (Fig. 1, untreated lanes) after the centrifugation of the microsome-containing translation reaction mixture, indicating that it could be either a membrane-associated protein or a lumenally translocated protein. The absence of glycosylation suggested that the chimeric protein was not translocated into the lumen of the microsomes. Nevertheless, to differentiate between these possibilities the translation reaction mixtures were washed with sodium carbonate (pH 11.5), which renders microsomes into membranous sheets, releasing the soluble luminal proteins (35, 41). As shown in Fig. 1, the 2B/P2 fusion appeared to be preferentially associated (approximately 58%; lanes 3 and 4) with the membranous pellet fraction, suggesting a tight association with membranes. Further treatment with 4 M urea demonstrated that approximately 88% of the protein was in the supernatant fraction (Fig. 1, lanes 5 and 6). More than 93% of the protein was extracted from the supernatant fraction by 8 M urea (Fig. 1, lanes 7 and 8), suggesting that 2B/P2 is released from the membrane environment when the secondary and tertiary structures of the protein are lost.
Table 1.
Computer analysis of viroporin 2B amino acid sequence
| Algorithm | Membrane protein | No. of TM segments (starting aa/ending aa) |
|---|---|---|
| DAS | Yes | 2 (45/53–64/71) |
| PHDhtm | Yes | 2 (41/57–64/72) |
| MEMSAT3 | Yes | 1 (44/63) |
| MEMSAT-SVM | Yes | 1 (40/55) |
| SOSUI | No | 0 |
| TMHMM | No | 0 |
| TMPred | Yes | 1 (58/78) |
| ΔG Prediction Server | Yes | 1 (61/82) |
| SPOCTOPUS | Yes | 1 (48/73) |
| TopPRED | Yes | 1 (38/58) |
The translation reaction mixtures also were treated with Triton X-114, a nonionic detergent that forms a separate organic phase into which membrane lipids and hydrophobic proteins are segregated from the aqueous phase, which contains nonintegral membrane proteins (10, 35). The 2B/P2 fusion protein (Fig. 2A) was detected in both the aqueous and organic phases (Fig. 2B, lanes 5 and 6), while a reduction of the P2 domain to its 50 N-terminal residues (Fig. 2A) led to the organic-phase detection of the chimera (Fig. 2B, lanes 7 and 8). Control analyses of Lep and Prune necrotic ring-spot virus (PNRSV) p32 movement protein showed, as previously demonstrated (35), organic- and aqueous-phase detections, respectively. Finally, viroporin 2B translated in the absence of fused domains was detected only in the organic phase (Fig. 2C, lanes 1 and 2), indicating that viroporin 2B is an integral membrane protein.
Insertion of the viroporin 2B hydrophobic regions into biological membranes.
We assayed the membrane insertion capacities of the viroporin 2B hydrophobic regions using an in vitro experimental system (24), which accurately reports the integration of transmembrane (TM) helices into microsomal membranes. This system uses ER-derived microsomal membranes and provides a sensitive way to detect the insertion or translocation of hydrophobic regions through the Sec translocon (25). An obvious advantage of this system is that the insertion assays are performed in the context of a biological membrane. The system is based on the cotranslational glycosylation performed by the oligosaccharyltransferase (OST) enzyme. OST adds sugar residues to an NX(S/T) consensus sequence (53), with X being any amino acid except proline (52), after the protein emerges from the translocon channel. The glycosylation of a protein region translated in vitro in the presence of microsomal membranes therefore indicates the exposure of this region to the OST active site on the luminal side of the ER membrane. In our first experimental assay (Fig. 3), a segment to be assayed (HR tested) is engineered into the luminal P2 domain of the integral membrane protein Lep from E. coli, where it is flanked by two acceptor sites (G1 and G2) for N-linked glycosylation (Fig. 3A). Both engineered glycosylation sites are to be used as membrane insertion reporters. The rationale behind using two glycosylation sites is that G1 will always be glycosylated due to its native luminal localization, while G2 will be glycosylated only on the translocation of the tested TM region through the microsomal membrane. Thus, single glycosylation indicates a correct TM integration (Fig. 3A, left), whereas double glycosylation reports the nonintegration capability of the tested HR segment (Fig. 3A, right). The single glycosylation of the molecule results in an increase in molecular mass of about 2.5 kDa relative to the observed molecular mass of Lep expressed in the absence of microsomes, and the mass is around 5 kDa in the case of double glycosylation.
Fig. 3.

Insertion of viroporin 2B hydrophobic regions 1 (HR1) and 2 (HR2) into microsomal membranes using Lep as a model protein. (A) Schematic representation of the leader peptidase (Lep) construct used to report insertion into the ER membrane of 2B HR1 and HR2. The HR under study is inserted into the P2 domain of Lep flanked by two artificial glycosylation acceptor sites (G1 and G2). The recognition of the HR by the translocon machinery as a TM domain locates only G1 in the luminal side of the ER membrane, preventing G2 glycosylation. The Lep chimera will be doubly glycosylated when the HR being tested is translocated into the lumen of the microsomes. (B) In vitro translation in the presence of membranes of the different Lep constructs. Constructs containing HR1 (residues 35 to 55; lanes 1 and 2) and HR2 (residues 61 to 81; lanes 3 and 4) were transcribed and translated in the presence of membranes. Control HRs were used to verify sequence translocation (trans.; lanes 5 and 6) and membrane integration (inser.; lanes 7 and 8). Bands of nonglycosylated protein are indicated by a white dot; singly and doubly glycosylated proteins are indicated by one and two black dots, respectively. (C) The HR sequence in each construct is shown together with the predicted ΔG apparent value, which was estimated using the ΔG prediction algorithm available on the Internet (http://dgpred.cbr.su.se/). Lysine residues in HR1 are shown in boldface. (D) In vitro translation of HR1-derived mutants at lysine 46.
The translation of the chimeric constructs harboring the predicted viroporin 2B hydrophobic regions as HR-tested segments resulted mainly in double-glycosylated forms (Fig. 3B, lanes 2 and 4), which is consistent with the translocation of these regions into the lumen of the ER. Control constructs with previously tested (37) translocation and integration sequences are shown in lanes 6 and 8, and they disclose the expected double and single glycosylation patterns, respectively (Fig. 3B). The permeabilization induced by an overlapping peptide library that spanned the complete viroporin 2B sequence mapped the cell plasma membrane-porating activity to the partially amphipathic HR1 domain (32). This region contains three lysine residues that would preclude TM disposition (Fig. 3C), especially in the case of lysine 46, since it would be located roughly in the center of the hydrophobic core of the lipid bilayer. Nevertheless, the replacement of this residue by glycine, glutamic acid, glutamine, or arginine renders glycosylation patterns consistent with the translocation of the hydrophobic region into the ER lumen (Fig. 3D). Taken together, these results suggest that an isolated HR1 segment does not span ER-derived membranes in an Nlum/Ccyt orientation.
Since native 2B does not have a cleavable signal sequence, it seems likely that HR1 acts as a signal-anchor sequence having an Ncyt/Clum orientation in the membrane (36). To test HR1 insertion in a reverse orientation, another Lep construct (Lep′) was used. In this Lep′ construct, HR1 replaces the second TM segment (H2) from Lep (Fig. 4A). The glycosylation acceptor site (G2′) located in the beginning of the P2 domain will be modified only if the HR1 segment inserts into the membrane, while the G1′ site, embedded in an extended N-terminal sequence of 24 amino acid residues, is always glycosylated. We found that HR1 significantly inserts into the membrane (up to 60% of the molecules) with the appropriate topology (Fig. 4B). The nature of the cytosolic/luminal domains was further examined by proteinase K (PK) digestions. Treatment with PK degrades domains of membrane proteins that protrude into the cytosol, but membrane-embedded or luminally exposed domains are protected. The addition of PK to a Lep′HR1 translation mixture (Fig. 4B, lane 4) rendered a protected, glycosylated HR1-P2 fragment, suggesting the proper insertion of HR1 sequence with an Ncyt/Clum orientation.
Fig. 4.
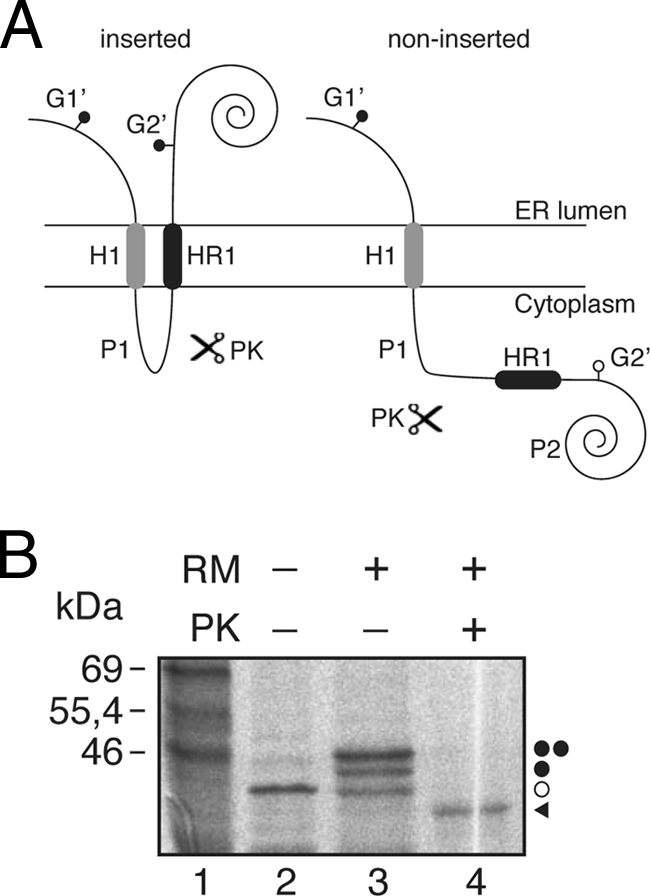
Insertion of HR1 into microsomal membranes using the Lep′ construct. (A) Schematic representation of the Lep-derived construct (Lep′). In this Lep′ construct (∼40 kDa; the nonglycosylated form), HR1 replaces the H2 domain from Lep. The glycosylation acceptor site (G2′) located in the beginning of the P2 domain will be modified only if HR1 inserts into the membrane, while the G1′ site, embedded in an extended N-terminal sequence of 24 amino acids, is always glycosylated. (B) In vitro translation in the presence of membranes. Bands of nonglycosylated protein are indicated by a white dot; singly and doubly glycosylated proteins are indicated by one and two black dots, respectively. The protected glycosylated HR1/P2 fragment (∼36.5 kDa) is indicated by a black triangle.
Viroporin 2B integrates into the ER membrane through the translocon with an N-terminal/C-terminal cytoplasmic orientation.
The microsomal in vitro system closely mimics the conditions of in vivo membrane protein assembly into the ER membrane. HR1 is properly recognized by the translocon as a TM segment out of its native context (Fig. 4). However, the presence of fused domains can influence its membrane insertion capacity. Hence, we next sought to investigate whether HR1 also could direct the integration into the ER membrane of the native 2B sequence (i.e., in the absence of nonviral fused domains) through the translocon.
Because N-glycosylation acceptor sites are absent from the viroporin 2B sequence, several modifications were prepared to determine the TM disposition of different 2B-derived proteins. First, to gain topological information, an N-glycosylation acceptor site was engineered at the hydrophilic N-terminal region of the 2B sequence by mutating glutamine 20 to an acceptor asparagine (…20NIS…; construct 2BNtGlyc). Second, we added a C-terminal N-glycosylation tag (CtGlyc; NSTMMM [the glycosylation sequon is in boldface]) that has been proven to be efficiently glycosylated (6). The first 60 residues of the viroporin 2B carrying an N-terminal glycosylation site were translated using C-terminal tags (Fig. 5A) either with an N-glycosylation acceptor site as a C-terminal tag (2BNt/Ct; Fig. 5B, lanes 1, 3, and 5) or with a nonacceptor site (2BNt/CtØ; Fig. 5B, lanes 2, 4, and 6). The lack of glycosylation at the N-terminal engineered acceptor site together with the efficient glycosylation observed only when using the C-terminal acceptor site (Fig. 5B, lane 3), as proven after EndoH treatment (Fig. 5B, lane 5), strongly indicates that HR1 in the viroporin context is acting as a noncleavable signal sequence and is properly recognized by the translocon machinery to be inserted into the membrane with its N terminus facing the cytoplasm (Ncyt/Clum topology). In addition, by blocking protein synthesis after 2BNt/Ct has been translated in the absence of membranes, we confirmed that the truncated version of 2B (2BNt/Ct) needs to be cotranslationally inserted into the ER membrane. As shown in Fig. 5C, 2BNt/Ct was glycosylated when microsomal membranes were added to the translation mixture cotranslationally. In contrast, when microsomal membranes were included posttranslationally (i.e., after translation had been inhibited by cycloheximide), the C-terminal acceptor site was not glycosylated (Fig. 5C, lane 3), thereby emphasizing that truncated 2B is integrated cotranslationally through the ER translocon.
Fig. 5.
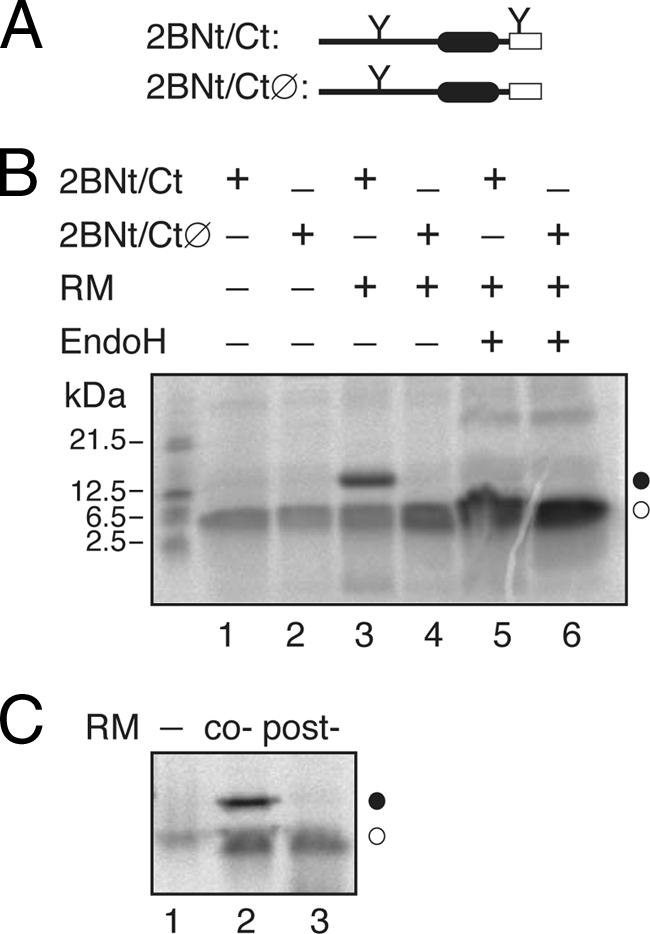
Membrane insertion of 60-mer viroporin 2B. (A) Structural organization of the 2B 60-mer truncated construct. (B) In vitro translations were performed in the presence (+) or in the absence of C-terminal glycosylation tag, RMs, and EndoH as indicated. (C) The 2BNt/Ct construct was translated in either the absence (lanes 1 and 3) or the presence (lane 2) of RMs. Lane 2, cotranslationally added microsomes. Lane 3, RMs were added posttranslationally (after 1 h of translation and 10 min of cycloheximide treatment; Post-), and incubation was continued for another 1 h.
Because of the low hydrophobicity and the relatively poor insertion propensity found in the Lep system for HR2 (Fig. 3), it is predicted that the 2B C-terminal region will be translocated into the ER lumen. However, 2B hydrophobic regions also are responsible for the membrane anchoring of the 2BC precursor, which performs its enzymatic and RNA binding activities in the cytosolic compartment. Thus, we speculated that some type of helix-helix interaction stabilizes the insertion of HR2 to keep an Ncyt/Ccyt 2B topology. It has been shown recently that in some cases the insertion of poorly hydrophobic regions depends on the presence of neighboring loops and/or TM segments (20), especially in the case of the preceding TM segment (23, 55). In our attempts to unravel the disposition of 2B in biological membranes, we focused on the insertion of truncated C-terminal reporter tag fusions. In Fig. 6 we show a series of experiments where HR1 and various lengths of downstream sequence were translated as truncated proteins with an N-glycosylation C-terminal tag (Fig. 6A). A mutant polypeptide truncated at the end of the hydrophilic loop between HR1 and HR2 (60-mer) is highly glycosylated (∼65%; Fig. 6B, lane 2), indicating that, similarly to what is shown in Fig. 5B, the C-terminal glycosylation tag has been translocated into the lumen of the ER, and thus HR1 is integrated into the ER membrane in a Ncyt/Clum orientation in this construct (Fig. 6D, left). The percentage of glycosylated truncated proteins is reported in Fig. 6C. Extending the 2B sequence to include roughly half of HR2 has a significant effect on this pattern (72-mer; Fig. 6B and 6C), suggesting some tendency of these truncated molecules to insert the C-terminal tag into the membrane. Moreover, extending the 2B sequence four residues (roughly one helical turn; 76-mer) (Fig. 6C) substantially diminished glycosylation (∼21%). The glycosylation level for the truncated protein shown in Fig. 6C cannot be explained by an increased hydrophobicity of the added amino acids, since the total free energy predicted (ΔGpred) for the 73CDAS76 sequence is 4.31 kcal/mol, where a positive value is indicative of extramembrane disposition (the calculation of ΔGpred was carried out using the scale of Hessa and collaborators [24]). Interestingly, the addition of four leucine residues (ΔGpred = −2.2 kcal/mol) instead of the 73CDAS76 sequence [76-mer(L4)] strongly precludes glycosylation (<3%) (Fig. 6C), demonstrating the clear hydrophobic effect of the leucine residues in this construct. This was further corroborated by extending the protein to include up to eight leucine residues [80-mer(L8)] (Fig. 6C). Finally, both the mutant truncated at the end of HR2 (80-mer) and the 2B full-length construct (2B/CtGlyc) had little effect on this pattern (∼17 and ∼10% glycosylation, respectively), indicating that the C terminus of the majority of these tagged proteins is cytosolic, and thus the HR2 sequence included in these constructs is integrated into the membrane in an Nlum/Ccyt orientation (Fig. 6D, right).
Fig. 6.
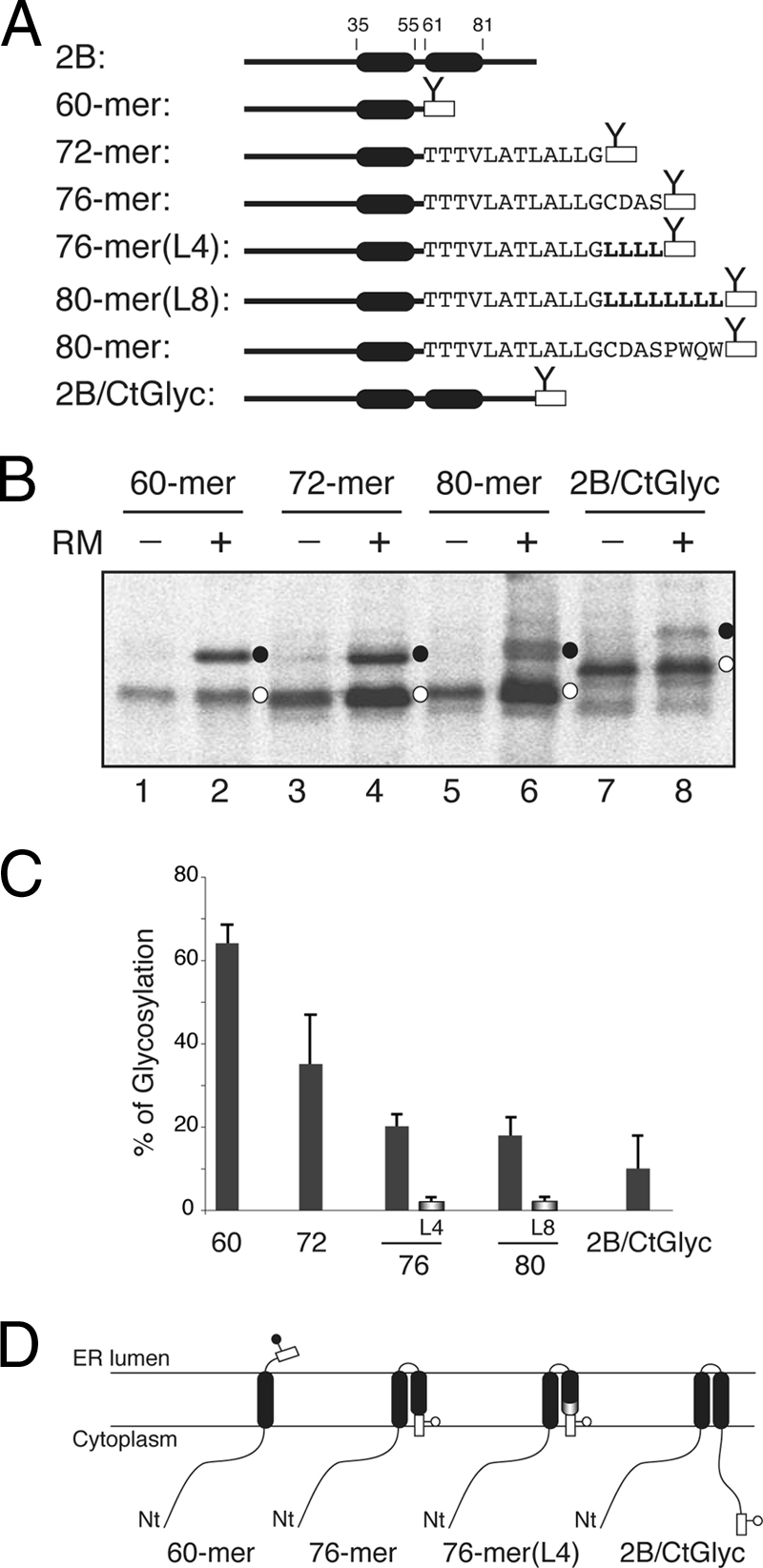
Effect of HR2 on 2B 60-mer insertion and topology. (A) Structural organization of full-length and truncated viroporin 2B constructs. (B) In vitro translation of truncated viroporin 2B 60-mer, 72-mer, 80-mer, and full-length (2B/CtGlyc) constructs in which a fused C-terminal N-glycosylation tag (rectangle) provides a simple readout for topology determination. The presence of RMs and nonglycosylated and glycosylated proteins (empty and black dots, respectively) is indicated. (C) In vitro glycosylation of truncated viroporin 2B-derived proteins. The level of glycosylation is quantified from SDS-PAGE gels by measuring the fraction of glycosylated (fg) versus glycosylated-plus-nonglycosylated (fng) molecules, using the equation p = fg/fg + fng. Data correspond to averages from at least four independent experiments, and error bars show standard deviations. (D) Topological models for 2B constructs. Nt, N terminus.
To confirm that the same topology is adopted by the full-length 2B protein, several 2B-derived constructs were prepared and their membrane disposition experimentally determined. The translation of the C-terminal-tagged 2B protein (2B/CtGlyc) (Fig. 7A) in the presence of RMs resulted in glycosylation in <8% of the molecules, as demonstrated by EndoH (a glycan-removing enzyme) treatment (Fig. 7B, lanes 1 to 3). The addition of the first 50 residues of the Lep P2 domain (2B/50P2) (Fig. 7A), which contains an N-glycosylation acceptor site as a topological reporter, yielded glycosylation in <5% of the viroporin 2B-derived molecules (Fig. 7B, lanes 4 to 6). Furthermore, when full-length Lep P2 domain was used as a reporter domain, the chimera was not glycosylated at all (2B/P2) (Fig. 7B, lanes 7 to 9). Finally, the in vitro translation of a construct harboring an N-terminal glycosylation acceptor site (2BNtGlyc, Fig. 7A) in the presence of RM only resulted in unmodified molecules (Fig. 7B, lanes 10 to 12). Taken together, these results suggest a preferential N-/C-terminal cytoplasmic orientation for viroporin 2B when expressed in the presence of ER-derived microsomal membranes.
Fig. 7.
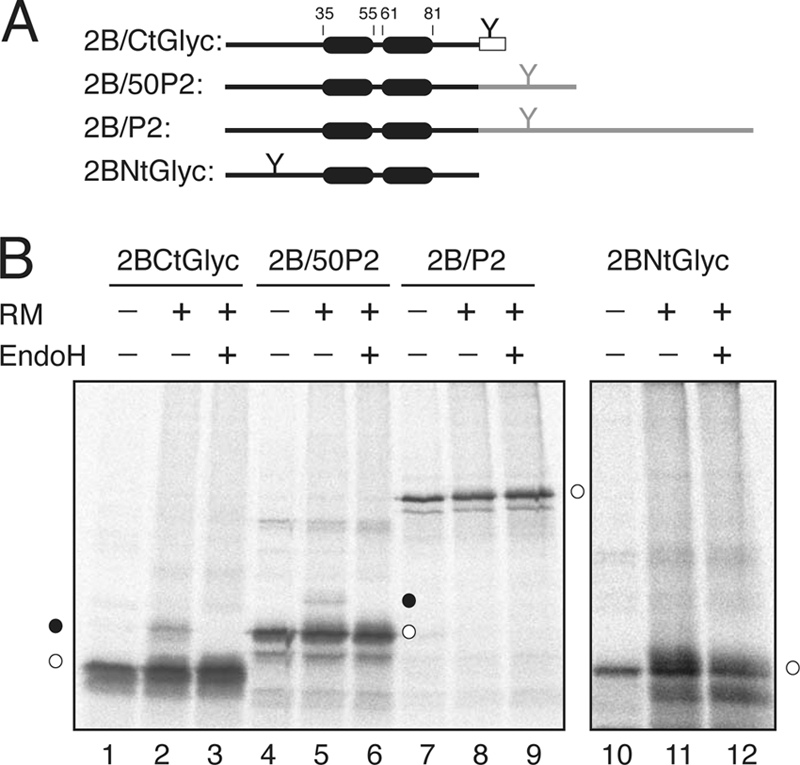
Insertion and topology of full-length viroporin 2B protein. (A) The structural organization of the 2B-derived constructs is shown at the top. The N-glycosylation site is highlighted by a Y-shaped symbol both when inserted in the protein sequence and when added as a C-terminal reporter tag (rectangles). (B) In vitro translation was performed in the presence (+) and in the absence (−) of RMs and EndoH as indicated. Nonglycosylated and singly glycosylated proteins are indicated by empty and black dots, respectively.
Viroporin 2B topology in mammalian cells.
To further assess the topology adopted by functional viroporin 2B in membranes, 2B variants containing designed N-glycosylation sites at different positions were expressed in cultured cells, and their plasma membrane-permeabilizing capacities were assessed (Fig. 8). For this purpose, the 2B variants were cloned in pTM1 vector and transfected in BHK cells that stably express the T7 RNA polymerase. These cells posses the machinery required for synthesizing the virion components and even to assemble infectious particles. As expected, 2B expression permeabilized BHK cells to the antibiotic hygromycin B (31) (Fig. 8B, lane 4). Although the synthesis of unmodified 2B is not detectable by radioactive labeling in the permeabilized cells (Fig. 8B, top), it can be detected by Western blotting using a specific 2B antibody (Fig. 8B, bottom). No glycosylation of 2B was observed when this protein bears the N-glycosylation site at the amino terminus, in the turn, or at the carboxy terminus of this viroporin (Fig. 8B, lanes 5 to 8). In addition, these two viroporin 2B variants retain their capacity to permeabilize cells to hygromycin B, suggesting that they are located at the membrane and exhibit the ability to alter membrane permeability. It should be noted that in the case of the N-glycosylation site located at the turn, although the location is luminal the absence of glycosylation is due to its proximity to the TM domains as previously reported (42, 44). In conclusion, the in vitro and in vivo assays consistently indicate that both the N and C termini of viroporin 2B face the cytoplasm, as displayed in Fig. 9A.
Fig. 8.
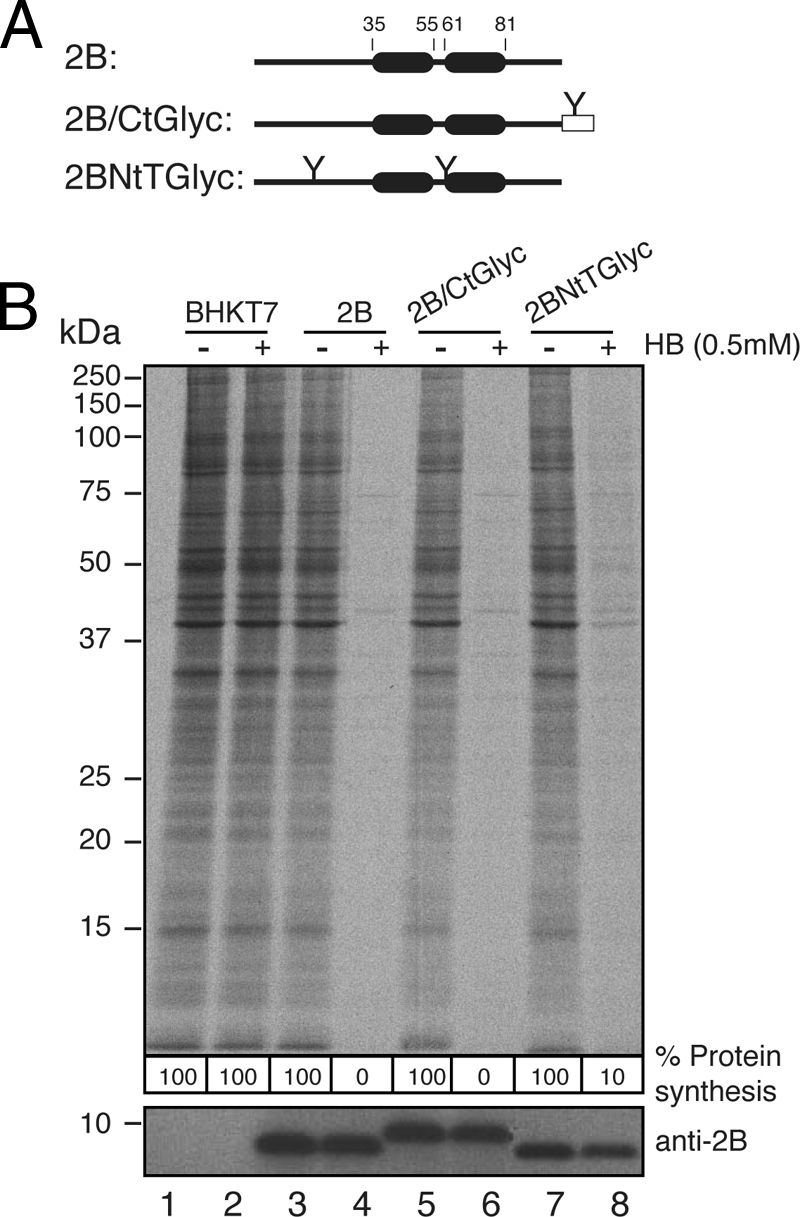
Permeabilization activity of 2B-derived constructs in BHKT7 cells. (A) Structural organization of the transfected 2B variants. (B) BHKT7 cells were transfected with pTM1-2Bwt or with pTM1-2B variants. At 3 h posttransfection, cells were pretreated with hygromycin B (HB) at 0.5 mM for 15 min and then labeled with [35S]Met/Cys for 45 min in the presence of the inhibitor. After labeling, the proteins were analyzed by SDS-PAGE (17.5%) followed by fluorography and autoradiography (top). The synthesis of 2B protein was detected by Western blotting using specific rabbit polyclonal antibodies (bottom). The numbers below each lane represent the percentage of protein synthesis in the presence of hygromycin B as calculated by densitometric scanning.
Fig. 9.
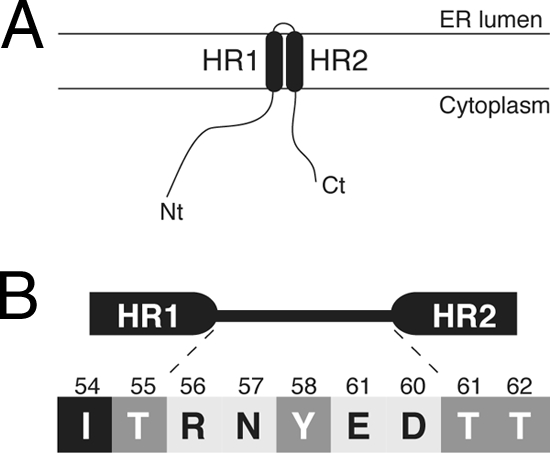
(A) Topological model for 2B association with membranes. (B) Turn-inducing propensity at the interhelical region of the viroporin 2B helical hairpin according to the scale of Monné and coworkers (39). The five amino acid residues interconnecting HR1 and HR2 are shown flanked by the putative last two residues from the HR1 helix and the putative first two residues from HR2. All residues connecting HR1 and HR2 are turn inducers (normalized turn potential, >1). The residues are highlighted according to their turn potential: black for a potential lower than 1 (Ile54), dark gray for a potential between 1 and 2 (Thr55, Tyr58, and Thr61), and light gray for a potential above 2 (Arg56, Asn57, Glu59, and Asp60).
DISCUSSION
Viroporins are a group of proteins responsible for alterations in the permeability of cellular membranes during virus infection, favoring the release of viral particles from infected cells (reviewed in reference 22). The molecular mechanisms by which viroporins insert into cell membranes remain largely unknown. In this study, we demonstrate that poliovirus viroporin 2B is a double-spanning integral membrane protein that can be inserted into the ER membrane through the translocon machinery.
Computer-assisted membrane protein topology prediction is a useful starting point for experimental studies of membrane proteins. We have used 10 popular prediction methods and found large discrepancies between their predictions. Two of the algorithms failed to predict 2B as an integral membrane protein, and two of them assigned two TM segments for the protein. It should be noted that the reliability of a topology prediction can be estimated by the number of prediction methods that agree. Since six of the algorithms predicted 2B as a membrane protein with only one TM segment, these results clearly highlight that the presence of helical hairpin structures, which was not detected even by the methods predicting reentrant loops (MEMSAT-SVM and SPOCTOPUS), may be missed by current predictive methods, as previously suggested for a different TM helical hairpin (40).
The membrane association of 2B/P2 fusion protein was resistant to alkaline extraction. Since this treatment disrupts microsomal membranes and releases any soluble luminal protein, this result indicates that the fusion protein is not translocated to the lumen of the microsomes. Urea treatments solubilized our fusion protein (Fig. 1), indicating that secondary and tertiary structures in 2B play an important role in 2B insertion. The latter results contrast with previous work that showed that coxsackievirus 2B/enhanced green fluorescent protein fusions were resistant to urea extraction (18). This discrepancy could be derived either from the differences found in the amino acid sequence of both 2B proteins or from the use of different fusion proteins in both cases. In fact, a significant influence of the P2 domain can be observed in our Triton X-114 partition experiments. Fusions containing the full P2 domain partition significantly into the aqueous phase, whereas the addition of the 50 N-terminal residues from this domain promoted the partitioning of the shorter chimera into the organic phase. These results clearly demonstrated integral membrane protein behavior (compare lanes 5 and 6 with lanes 7 and 8 in Fig. 2B), as corroborated by partition experiments using full-length 2B (Fig. 2C, lanes 1 and 2).
By challenging the hydrophobic regions of 2B in a model protein construct (Lep), we demonstrate first that these regions do not integrate as TM segments in the presence of ER-derived membranes when expressed separately (Fig. 3). It should be mentioned that, in these Lep-derived constructs, the HR1 segment is forced to insert into the membrane with an Nlum/Ccyt topology, and this topological effect can prevent the proper TM disposition of this region. In fact, using a Lep-derived variant (Lep′), we demonstrated the TM disposition of HR1 when expressed with an Ncyt/Clum orientation (Fig. 4). The glycosylation data obtained in the context of the parental 2B sequence using engineered and truncated proteins provided compelling evidence that HR1-HR2 also may integrate cotranslationally into the membrane in the absence of fused domains. Hence, a truncated 2B protein containing HR1 inserted efficiently into ER microsomal membranes adopting an Ncyt/Clum topology (Fig. 5), and the addition of HR2 residues to this construct resulted in the cytoplasmic reorientation of the C terminus (Fig. 6). Furthermore, the lack of glycosylation at N-glycosylation acceptor sites engineered at different positions, both in an in vitro microsomal system (Fig. 7) and in cultured cells (Fig. 8), suggests that both the N and C termini of viroporin 2B protein reside on the cytosolic side of the membrane. In the context of the P2 polyprotein, the membrane topology found in the present work leaves the protease cleavage sites of P2 facing the cytosol, which is suitable for polypeptide processing by viral proteases.
Taken together, these data support the capacity of the HR domains to act as interacting TM segments in their natural contexts. In this sense, we have described previously the integration into the ER membrane of two closely spaced membrane-spanning segments of viral origin, where both TM segments of the nascent protein bind to one or more translocon proteins and are held until the termination of translation, whereupon they are released laterally as a helical hairpin into the lipid phase (50, 51). More recently, this mechanism of partition into the membrane as a pair of helices has been observed by others using a nonviral membrane protein (13). Thus, the retention of a first cationic amphipathic segment (HR1) at the ER translocon to generate a helical hairpin might facilitate partitioning into the lipid phase by shielding the polar amino acids that could compromise effective membrane integration (36).
These findings provide important new insights into the molecular architecture and the molecular mechanism of 2B integration into the membrane. The synergic effect found for pore formation between HR1 and HR2 in a previous peptide-based analysis, which indicated that both HRs cooperate in membrane permeabilization (48), suggests that HR1 and HR2 interact with each other to form a helix-turn-helix (helical hairpin) motif that traverses the lipid bilayer. An additional source of stability of this motif can be the turn between the two helices. It has been shown that charged and polar residues (plus prolines and glycines) display turn induction in a TM polyleucine stretch (38); in our case, we do not know exactly which amino acid residues form the turn in the membrane-bound viroporin, although in all likelihood the turn occurs between the highly hydrophilic residues 55 and 61. Figure 9B shows this region of viroporin 2B, where the residues are highlighted according to their turn-inducing propensities (38). All residues present in this region are strongly turn inducing (normalized turn potential, >1). Among them, four (Arg56, Asn57, Glu59, and Asp60) have a high turn potential (>2). In essence, a great concentration of turn-promoting residues is found in the region connecting HR1 and HR2. Thus, in the membrane-bound form, we can expect the turn of viroporin 2B to be centered on 56RNYED60. Interestingly, previous mutagenesis studies using the CBV 2B protein showed that the negatively charged residues found in this short hydrophilic turn between HR1 and HR2 are indeed important for the membrane-active character of 2B protein (16). Moreover, recent molecular dynamic simulations of the poliovirus 2B channel/pore-forming regions suggested that Glu59 and Asp60 are involved in the helical hairpin formation (45). In any case, it seems clear that the turn may play a significant role in the stability and integration of the membrane-bound 2B protein. In addition, the topology observed in the present work agrees with previous data obtained with different fusion proteins (18). Furthermore, the localization of the C terminus at the cytosolic side of the membrane is consistent with the need for the proteolytic liberation of the 2B protein from the precursor 2BC polyprotein by a cytosolic viral protease cleavage, which is accomplished by 3Cpro (54). This could occur after the membrane insertion of 2BC or even the entire P2 precursor (2ABC).
Our findings further suggest a physiological role for translocon-mediated 2B integration into the ER membrane. Our combined analysis predicts a marginal propensity for 2B polypeptide to insert into membranes (Table 1 and Fig. 3D). On the other hand, upon viral entry, initially synthesized 2B or 2BC proteins will remain diluted in the cytosol of the infected cell. Marginal hydrophobicity together with low concentration are predicted to reduce the probability of the spontaneous insertion of 2B and 2BC into their primary target organelle: the Golgi complex (18, 49). Thus, we speculate that cotranslational insertion into the ER membrane is a particularly relevant phenomenon at the initial stages of the infectious cycle, during which both the viral mRNA levels and the concentration of the translated viral proteins are predictably low. Under those conditions, the cellular protein biosynthesis and vesicular transport machineries remain functional, and 2B and its 2BC precursor likely are synthesized as additional cell membrane proteins. From the ER, these proteins may reach the Golgi compartment membrane to fulfill regulatory and/or signaling functions that result in the disruption of Ca2+ homeostasis (5, 15) and vesicular transport inhibition (7, 15). At later stages, the canonical ER-Golgi protein-trafficking pathway no longer is functional and/or required for viral replication. The massive proliferation of cell endomembranes (viroplasm) and the high levels of viral protein synthesis result in higher effective concentrations of these components inside the infected cell system. Under those conditions, it is predicted that the spontaneous insertion into the membrane of a significant amount of synthesized 2B and 2BC will ensue.
In summary, viroporin 2B may use common structural arrangements to integrate into the ER membrane through the translocon, at least during the initial stages of the viral replicative cycle. The development of in vitro assays designed to dissect the membrane integration process will lead to a better understanding of the membrane permeabilization mechanisms that act during enterovirus infection.
ACKNOWLEDGMENTS
We thank Arne Elofsson (Stockholm University) for helpful comments on the manuscript.
This work was supported by grants BFU2009-08401 (to I.M.), BFU2009-07352 (to L.C.), and BIO2008-00772 (to J.L.N.) from the Spanish Ministry of Science and Innovation (MICINN, ERDF; supported by the European Union), as well as by ACOMP/2011/025 from the Generalitat Valenciana to I.M. M.B.-P. was the recipient of an FPU fellowship from the Spanish Ministry of Education.
Footnotes
Published ahead of print on 10 August 2011.
REFERENCES
- 1. Agirre A., Barco A., Carrasco L., Nieva J. L. 2002. Viroporin-mediated membrane permeabilization. Pore formation by nonstructural poliovirus 2B protein. J. Biol. Chem. 277:40434–40441 [DOI] [PubMed] [Google Scholar]
- 2. Agol V. 2002. Picornavirus genome: an overview, p. 127–148. In Semler B. L., Wimmer E. (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC [Google Scholar]
- 3. Aldabe R., Barco A., Carrasco L. 1996. Membrane permeabilization by poliovirus proteins 2B and 2BC. J. Biol. Chem. 271:23134–23137 [DOI] [PubMed] [Google Scholar]
- 4. Aldabe R., Carrasco L. 1995. Induction of membrane proliferation by poliovirus proteins 2C and 2BC. Biochem. Biophys. Res. Commun. 206:64–76 [DOI] [PubMed] [Google Scholar]
- 5. Aldabe R., Irurzun A., Carrasco L. 1997. Poliovirus protein 2BC increases cytosolic free calcium concentrations. J. Virol. 71:6214–6217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bañó-Polo M., Baldin F., Tamborero S., Marti-Renom M. A., Mingarro I. 2011. N-glycosylation efficiency is determined by the distance to the C-terminus and the amino acid preceding an Asn-Ser-Thr sequon. Protein Sci. 20:179–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barco A., Carrasco L. 1995. A human virus protein, poliovirus protein 2BC, induces membrane proliferation and blocks the exocytic pathway in the yeast Saccharomyces cerevisiae. EMBO J. 14:3349–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barco A., Carrasco L. 1998. Identification of regions of poliovirus 2BC protein that are involved in cytotoxicity. J. Virol. 72:3560–3570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baumgärtner A. 1996. Insertion and hairpin formation of membrane proteins: a Monte Carlo study. Biophys. J. 71:1248–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bordier C. 1981. Phase separation of integral membrane proteins in Triton X-114 solution. J. Biol. Chem. 256:1604–1607 [PubMed] [Google Scholar]
- 11. Buchholz U. J., Finke S., Conzelmann K. K. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73:251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Claros M. G., von Heijne G. 1994. TopPred II: an improved software for membrane protein structure prediction. Comput. Appl. Biosci. (Cambridge) 10:685–686 [DOI] [PubMed] [Google Scholar]
- 13. Cross B. C., High S. 2009. Dissecting the physiological role of selective transmembrane-segment retention at the ER translocon. J. Cell Sci. 122:1768–1777 [DOI] [PubMed] [Google Scholar]
- 14. Cserzö M., Wallin E., Simon I., von Heijne G., Elofsson A. 1997. Prediction of transmembrane a-helices in prokaryotic membrane proteins: the dense alignment surface method. Protein Eng. 10:673–676 [DOI] [PubMed] [Google Scholar]
- 15. de Jong A. S., et al. 2008. Functional analysis of picornavirus 2B proteins: effects on calcium homeostasis and intracellular protein trafficking. J. Virol. 82:3782–3790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Jong A. S., Melchers W. J., Glaudemans D. H., Willems P. H., van Kuppeveld F. J. 2004. Mutational analysis of different regions in the coxsackievirus 2B protein: requirements for homo-multimerization, membrane permeabilization, subcellular localization, and virus replication. J. Biol. Chem. 279:19924–19935 [DOI] [PubMed] [Google Scholar]
- 17. de Jong A. S., et al. 2002. Multimerization reactions of coxsackievirus proteins 2B, 2C and 2BC: a mammalian two-hybrid analysis. J. Gen. Virol. 83:783–793 [DOI] [PubMed] [Google Scholar]
- 18. de Jong A. S., et al. 2003. Determinants for membrane association and permeabilization of the coxsackievirus 2B protein and the identification of the Golgi complex as the target organelle. J. Biol. Chem. 278:1012–1021 [DOI] [PubMed] [Google Scholar]
- 19. Engelman D. M., Steitz T. A. 1981. The spontaneous insertion of proteins into and across membranes: the helical hairpin hypothesis. Cell 23:411–422 [DOI] [PubMed] [Google Scholar]
- 20. Enquist K., et al. 2009. Membrane-integration characteristics of two ABC transporters, CFTR and P-glycoprotein. J. Mol. Biol. 387:1153–1164 [DOI] [PubMed] [Google Scholar]
- 21. García-Sáez A. J., Mingarro I., Perez-Paya E., Salgado J. 2004. Membrane-insertion fragments of Bcl-xL, Bax, and Bid. Biochemistry 43:10930–10943 [DOI] [PubMed] [Google Scholar]
- 22. Gonzalez M. E., Carrasco L. 2003. Viroporins. FEBS Lett. 552:28–34 [DOI] [PubMed] [Google Scholar]
- 23. Hedin L. E., et al. 2010. Membrane insertion of marginally hydrophobic transmembrane helices depends on sequence context. J. Mol. Biol. 396:221–229 [DOI] [PubMed] [Google Scholar]
- 24. Hessa T., et al. 2005. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433:377–381 [DOI] [PubMed] [Google Scholar]
- 25. Hessa T., et al. 2007. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450:1026–1030 [DOI] [PubMed] [Google Scholar]
- 26. Hirokawa T., Boon-Chieng S., Mitaku S. 1998. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14:378–379 [DOI] [PubMed] [Google Scholar]
- 27. Johnson A. E., van Waes M. A. 1999. The translocon: a dynamic gateway at the ER membrane. Annu. Rev. Cell Dev. Biol. 15:799–842 [DOI] [PubMed] [Google Scholar]
- 28. Jones D. T. 2007. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 23:538–544 [DOI] [PubMed] [Google Scholar]
- 29. Krogh A., Larsson B., von Heijne G., Sonnhammer E. L. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567–580 [DOI] [PubMed] [Google Scholar]
- 30. Luik P., et al. 2009. The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 106:12712–12716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Madan V., Castello A., Carrasco L. 2008. Viroporins from RNA viruses induce caspase-dependent apoptosis. Cell Microbiol. 10:437–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Madan V., et al. 2007. Plasma membrane-porating domain in poliovirus 2B protein. A short peptide mimics viroporin activity. J. Mol. Biol. 374:951–964 [DOI] [PubMed] [Google Scholar]
- 33. Martínez-Gil L., Johnson A. E., Mingarro I. 2010. Membrane insertion and biogenesis of the Turnip crinkle virus p9 movement protein. J. Virol. 84:5520–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martínez-Gil L., Perez-Gil J., Mingarro I. 2008. The surfactant peptide KL4 sequence is inserted with a transmembrane orientation into the endoplasmic reticulum membrane. Biophys. J. 95:L36–L38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Martínez-Gil L., et al. 2009. Plant virus cell-to-cell movement is not dependent on the transmembrane disposition of its movement protein. J. Virol. 83:5535–5543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martínez-Gil L., Sauri A., Marti-Renom M. A., Mingarro I. 2011. Membrane protein integration into the ER. FEBS J. [Epub ahead of print.] doi:10.1111/j.1742-4658.2011.08185.x [DOI] [PubMed] [Google Scholar]
- 37. Martínez-Gil L., Sauri A., Vilar M., Pallas V., Mingarro I. 2007. Membrane insertion and topology of the p7B movement protein of melon necrotic spot virus (MNSV). Virology 367:348–357 [DOI] [PubMed] [Google Scholar]
- 38. Monné M., Hermansson M., von Heijne G. 1999. A turn propensity scale for transmembrane helices. J. Mol. Biol. 288:141–145 [DOI] [PubMed] [Google Scholar]
- 39. Monné M., Nilsson I., Elofsson A., von Heijne G. 1999. Turns in transmembrane helices: determination of the minimal length of a “helical hairpin” and derivation of a fine-grained turn propensity scale. J. Mol. Biol. 293:807–814 [DOI] [PubMed] [Google Scholar]
- 40. Nagy A., Turner R. J. 2007. The membrane integration of a naturally occurring alpha-helical hairpin. Biochem. Biophys. Res. Commun. 356:392–397 [DOI] [PubMed] [Google Scholar]
- 41. Navarro J. A., et al. 2006. RNA-binding properties and membrane insertion of Melon necrotic spot virus (MNSV) double gene block movement proteins. Virology 356:57–67 [DOI] [PubMed] [Google Scholar]
- 42. Nilsson I., von Heijne G. 1993. Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J. Biol. Chem. 268:5798–5801 [PubMed] [Google Scholar]
- 43. Nugent T., Jones D. T. 2009. Transmembrane protein topology prediction using support vector machines. BMC Bioinformatics 10:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Orzáez M., Salgado J., Gimenez-Giner A., Perez-Paya E., Mingarro I. 2004. Influence of proline residues in transmembrane helix packing. J. Mol. Biol. 335:631–640 [DOI] [PubMed] [Google Scholar]
- 45. Patargias G., Barke T., Watts A., Fischer W. B. 2009. Model generation of viral channel forming 2B protein bundles from polio and coxsackie viruses. Mol. Membr. Biol. 26:309–320 [DOI] [PubMed] [Google Scholar]
- 46. Rapoport T. A., Goder V., Heinrich S. U., Matlack K. E. 2004. Membrane-protein integration and the role of the translocation channel. Trends Cell Biol. 14:568–575 [DOI] [PubMed] [Google Scholar]
- 47. Rost B., Fariselli P., Casadio R. 1996. Topology prediction for helical transmembrane proteins at 86% accuracy. Protein Sci. 5:1704–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sánchez-Martínez S., et al. 2008. Functional and structural characterization of 2B viroporin membranolytic domains. Biochemistry 47:10731–10739 [DOI] [PubMed] [Google Scholar]
- 49. Sandoval I. V., Carrasco L. 1997. Poliovirus infection and expression of the poliovirus protein 2B provoke the disassembly of the Golgi complex, the organelle target for the antipoliovirus drug Ro-090179. J. Virol. 71:4679–4693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saurí A., McCormick P. J., Johnson A. E., Mingarro I. 2007. Sec61alpha and TRAM are sequentially adjacent to a nascent viral membrane protein during its ER integration. J. Mol. Biol. 366:366–374 [DOI] [PubMed] [Google Scholar]
- 51. Saurí A., Saksena S., Salgado J., Johnson A. E., Mingarro I. 2005. Double-spanning plant viral movement protein integration into the endoplasmic reticulum membrane is signal recognition particle-dependent, translocon-mediated, and concerted. J. Biol. Chem. 280:25907–25912 [DOI] [PubMed] [Google Scholar]
- 52. Shakin-Eshleman S. H., Spitalnik S. L., Kasturi L. 1996. The amino acid at the X position of an Asn-X-ser sequon is an important determinant of N-linked core-glycosylation efficiency. J. Biol. Chem. 271:6363–6366 [DOI] [PubMed] [Google Scholar]
- 53. Silberstein S., Gilmore R. 1996. Biochemistry, molecular biology, and genetics of the oligosaccharyltransferase. FASEB J. 10:849–858 [PubMed] [Google Scholar]
- 54. Skern T., et al. 2002. Structure and function of picornavirus proteinases, p. 199–212. In Semler B. L., Wimmer E. (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC [Google Scholar]
- 55. Tamborero S., Vilar M., Martinez-Gil L., Johnson A. E., Mingarro I. 2011. Membrane insertion and topology of the translocating chain-associating membrane protein (TRAM). J. Mol. Biol. 406:571–582 [DOI] [PubMed] [Google Scholar]
- 56. van der Werf S., Bradley J., Wimmer E., Studier F. W., Dunn J. J. 1986. Synthesis of infectious poliovirus RNA by purified T7 RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 83:2330–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. van Kuppeveld F. J., Galama J. M., Zoll J., van den Hurk P. J., Melchers W. J. 1996. Coxsackie B3 virus protein 2B contains cationic amphipathic helix that is required for viral RNA replication. J. Virol. 70:3876–3886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. van Kuppeveld F. J., et al. 1997. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J. 16:3519–3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. van Kuppeveld F. J., Melchers W. J., Willems P. H., Gadella T. W., Jr 2002. Homomultimerization of the coxsackievirus 2B protein in living cells visualized by fluorescence resonance energy transfer microscopy. J. Virol. 76:9446–9456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Viklund H., Bernsel A., Skwark M., Elofsson A. 2008. SPOCTOPUS: a combined predictor of signal peptides and membrane protein topology. Bioinformatics 24:2928–2929 [DOI] [PubMed] [Google Scholar]
- 61. Vilar M., et al. 2002. Insertion and topology of a plant viral movement protein in the endoplasmic reticulum membrane. J. Biol. Chem. 277:23447–23452 [DOI] [PubMed] [Google Scholar]
- 62. Wimmer E., Hellen C. U., Cao X. 1993. Genetics of poliovirus. Annu. Rev. Genet. 27:353–436 [DOI] [PubMed] [Google Scholar]