

Abstract
An ordered silencing of Epstein-Barr virus (EBV) latency gene transcription is critical for establishment of persistent infection within B lymphocytes, yet the mechanisms responsible and the role that the virus itself may play are unclear. Here we describe two B-cell superinfection models with which to address these problems. In the first, Burkitt lymphoma (BL) cells that maintain latency I, when superinfected, initially supported transcription from the common EBNA promoters Wp and Cp (latency III) but ultimately transitioned to latency I (Cp/Wp silent), an essential requirement for establishment of EBV latency in vivo. We used this model to test whether the early lytic-cycle gene BHLF1, implicated in silencing of the Cp/Wp locus, is required to establish latency I. Upon superinfection with EBV deleted for the BHLF1 locus, however, we have demonstrated that BHLF1 is not essential for this aspect of EBV latency. In the second model, BL cells that maintain Wp-restricted latency, a variant program in which Cp is silent but Wp remains active, sustained the latency III program of transcription from the superinfecting-virus genomes, failing to transition to latency I. Importantly, there was substantial reduction in Wp-mediated protein expression from endogenous EBV genomes, in the absence of Cp reactivation, that could occur independent of a parallel decrease in mRNA. Thus, our data provide evidence of a novel, potentially posttranscriptional mechanism for trans-repression of Wp-dependent gene expression. We suggest that this may ensure against overexpression of the EBV nuclear antigens (EBNAs) prior to the transcriptional repression of Wp in cis that occurs upon activation of Cp.
INTRODUCTION
Persistent infection by Epstein-Barr virus (EBV) is dependent on an ordered silencing of genes encoding the viral latency-associated proteins as the virus establishes latency within memory B cells, the principal reservoir of EBV (reviewed in reference 67). This enables infected B cells to evade the host's anti-EBV immune surveillance and limits the oncogenic potential of the virus. The latency proteins of EBV are composed of the six EBV nuclear antigens (EBNAs) and the three so-called latent membrane proteins (LMPs), one of which, LMP1, has potent transforming activity (73). Upon primary infection of B lymphocytes, expression of the EBV latency-associated proteins begins with the EBNAs, whose mRNAs initiate from a common and B-cell-specific promoter, Wp (2, 5, 27, 68, 69, 76). Subsequently, Wp is downregulated (though not completely) upon EBNA2-mediated activation of a second common EBNA gene promoter, Cp, ∼3 kbp upstream of Wp (19, 49, 63, 75, 76). With the exception of the genome maintenance functions provided by EBNA1, the principal contributions of the EBNAs to EBV latency are as transcription factors that regulate their own expression and that of the LMPs and of cellular gene expression (reviewed in reference 26).
Expression of the full complement of EBV latency proteins, designated the latency III or growth program, is sustained within EBV-immortalized B lymphoblastoid cell lines (LCLs)—generated by infection of primary B cells in vitro—that require EBV gene expression for continued growth. Latency III is also supported within a subset of B-cell lines derived from Burkitt lymphomas (BLs) (13). In vivo, however, all EBV protein expression is believed to be silenced (latency 0) as infected B cells enter the pool of resting memory B cells (4, 67). This critical event in the EBV life cycle is thought to initiate with silencing of EBNA gene transcription from Wp and Cp. Prior to entering the resting state and subsequently during periods of limited proliferation, and also in many BL cell lines that retain the EBV protein expression profile of their parent tumors (EBNA1 only; latency I), latently infected B cells express EBNA1 (necessary for viral genome maintenance in dividing cells) through activation of an EBNA1-exclusive promoter, Qp, ∼39 kbp downstream of Cp (14, 38, 59). Little or no mRNA expression from Qp is detectable during latency III, probably as a consequence of negative autoregulation by EBNA1 expressed at higher levels from Wp or Cp and bound to Qp immediately downstream of its transcription start site (53, 58, 64, 81). Within resting B cells, Qp is believed to be transcriptionally repressed through the action of the retinoblastoma protein, targeted to Qp via its interaction with E2F transcription factors bound near the transcription start site (8, 51). Unlike Cp and Wp, Qp appears to escape epigenetic silencing through the insulator function of CCCTC-binding factor (CTCF), which by binding upstream of Qp prevents the spread of repressive histone and DNA methylation across the Qp locus (66). Thus, while Cp and Wp become stably silenced, Qp resists silencing to serve as a default promoter to ensure expression of EBNA1 when needed, i.e., during cell proliferation.
EBNA1 alone has little if any acute oncogenic potential (21, 22) and possesses properties that interfere with its detection by the host's anti-EBNA1 immune surveillance (32–34, 65, 78). Consequently, its expression appears to be tolerated for limited durations as it performs its genome maintenance functions. This is not true of other latency-associated proteins that directly or indirectly rely on Wp or Cp for their expression, and thus silencing of these EBNA promoters is critical to long-term persistence of EBV within an immunocompetent host. While much has been learned about the factors that regulate transcription (primarily activation) from Cp and Wp during latency III, the mechanisms through which these promoters are silenced during the establishment of latency in vivo remain poorly defined. Methylation of the EBV DNA genome, and Cp and Wp specifically, has long been known to suppress EBV latency gene expression (17, 37, 40, 45–47, 58, 70), though more recent work has indicated that promoter methylation (at least of Wp) lags behind reduction in transcription (16); thus, DNA methylation may be more critical to maintenance as opposed to initiation of EBV gene silencing. An enhancer-blocking function of CTCF, furthermore, has been implicated in the initiation and maintenance of Cp silencing through CTCF binding between the transcription start site and the EBNA1-dependent enhancer for Cp within the upstream latent-infection origin of DNA replication, oriP (7). However, quantitative assessment of CTCF occupancy at this locus in the EBV genomes within a broad collection of B-cell lines that maintain either latency III or I failed to note a consistent correlation between CTCF binding and Cp inactivity (52).
An equally important gap in our understanding of this process is the question of whether EBV itself plays an active role in the epigenetic silencing of its gene expression. One finding suggesting that it might comes from an analysis by Kelly and colleagues of a subset of BL-derived cell lines that maintain a variant program of EBV latency gene expression known as Wp-restricted latency, during which Wp remains transcriptionally active and Cp is silent (23). The EBV genomes in these BL lines contain a deletion that ranges between 6,754 and 8,540 bp (depending on the cell line) that removes the coding information for the C-terminal portion of EBNA-LP (a transcriptional coactivator with EBNA2), the entire open reading frame (ORF) for EBNA2, and the majority of the ORF to the complete ORF and 5′ promoter region of the adjacent gene BHLF1, an early lytic-cycle gene (23, 25). Consistent with findings of earlier studies, the apparent consequence of deleting the EBNA2 ORF in these cells is the failure to support EBNA expression from Cp (25), interference in cis from which is believed to be the primary mechanism of Wp repression upon the switch to Cp usage (42, 43, 79, 80). Thus, sustained use of Wp in these BL cells would appear to be a default response in B cells unable to silence EBNA expression. However, a subpopulation of cells within some Wp-restricted BL lines contain wild-type (wt) EBV genomes that interestingly are transcriptionally silent (25), suggesting that these cells do have the potential to silence the Cp/Wp locus. An attractive interpretation of this phenomenon is that the locus deleted from the transcriptionally active EBV genomes normally acts in the context of a wt genome to silence Cp/Wp in cis. Thus, EBV may have adopted a mechanism to direct silencing of its own gene expression that perhaps is analogous to some forms of gene silencing employed by mammalian cells, e.g., X-linked inactivation. The initial objective of the current work was to address the possible contribution of the BHLF1 locus (deleted from the transcriptionally active EBV genomes within in Wp-restricted BLs) to Cp/Wp silencing.
Although BHLF1 encodes an abundant early lytic-cycle transcript (18, 29), in our early studies it appeared to be actively transcribed during latent infection (albeit in an LCL maintaining latency III) in the absence of appreciable cytoplasmic mRNA (55). A subsequent analysis of the gene, furthermore, revealed that it may possess a latency-specific promoter(s) in addition to its previously identified lytic-cycle promoter (77). While protein encoded by BHLF1 has been tentatively identified and reported by one group to localize within the nucleolus (35, 39), BHLF1's role as a protein-encoding gene has been questioned (9). Indeed, the ORF is composed mostly of a 125-bp direct repeat and has an unusually high G/C content (82%), and glycine and proline make up 16% and 22% of the predicted protein, respectively. In light of the above, we considered the hypothesis that BHLF1, possibly as a long noncoding RNA, performs the latency-specific function of promoting silencing of the Cp/Wp locus in cis implicated in Wp-restricted BL cells.
To test our hypothesis, we first developed two new in vitro B-cell infection models that should prove useful for elucidating the mechanisms responsible for the establishment of EBV latency. In the first, we demonstrated that upon superinfection with a wt recombinant EBV (rEBV), BL cells that maintain their endogenous EBV genomes in latency I support a transition from latency III to I, thus recapitulating an important event in the establishment of restricted latency in vivo. We then used this system to test the possible role of BHLF1 in this process. Using mutant rEBV deleted for the BHLF1 locus, however, we found that BHLF1 is not required for establishment of latency I. Our second model employed Wp-restricted BL cells that, by contrast, do not support the transition to latency I upon superinfection with wt rEBV but instead sustain the latency III program of expression from the superinfecting-EBV genomes, yet Cp within the endogenous genomes remains silent despite the presence of EBNA2. This suggests that BL cells that maintain Wp-restricted latency are programmed to support latency III and that transcriptionally silent wt EBV genomes originally present within these cells (25) likely represented those stably silenced during establishment of latency within the precursor B cells of the tumors from which these cell lines were derived. Our most significant observation was that Wp-mediated protein expression from the endogenous (deleted) genomes in these BL cells is repressed following superinfection, possibly through a posttranscriptional mechanism. Since Cp within the endogenous genomes remains silent, the mechanism at play must be acting in trans. Thus, our data reveal the existence of a novel mechanism for the regulation of Wp-dependent gene expression, possibly one that acts to ensure proper levels of the EBNAs prior to the switch to Cp.
MATERIALS AND METHODS
Cell lines.
Akata (Ak-BL), Kem I, and Mutu I are EBV-positive BL cell lines that maintain a latency I program of EBV gene expression. Kem III and Mutu III are BL lines derived from the same tumor as their counterparts Kem I and Mutu I but which maintain a latency III program. Mutu− is an EBV-negative line derived from Mutu I. Sal and Oku are BL lines that maintain a Wp-restricted program of EBV latency gene expression and harbor only EBV genomes that contain an 8.5-kbp (Sal) or 6.7-kbp (Oku) deletion that encompasses the contiguous BYRF1 (EBNA2) and BHLF1 ORFs and all or a portion of the leftmost lytic cycle origin of DNA replication, oriLytL (23). Akata-LCL (Ak-LCL) is a human LCL generated by infection of primary B lymphocytes in vitro with the strain of EBV produced from Ak-BL cells. All lymphoid cell lines were maintained in RPMI 1640 medium supplemented with 2 mM l-glutamine and 10% fetal bovine serum (HyClone). The human embryonic kidney-derived cell line HEK293 was maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 2 mM l-glutamine and 10% fetal bovine serum, except as noted below.
Immunoblot analysis.
EBV and cellular proteins within whole-cell extracts (5 × 105 or 1 × 106 cell equivalents per sample, depending on the primary antibody used) were detected by standard immunoblotting techniques using the following antibodies: EBNA1, rabbit antiserum (gift of J. Herring); EBNA3 proteins, sheep antiserum to EBNA3A, -3B, and -3C (Exalpha Biologicals, Inc.); EBNA2, monoclonal antibody (MAb) PE2 (82); LMP1, MAb S12 (36); EBNA-LP, MAb JF186 (11); BHRF1, MAb EA-R-p17 (Millipore); Bcl-6, rabbit polyclonal antibody (Cell Signaling); β-tubulin, MAb H-234 (Santa Cruz); and β-actin, MAb JLA20 (Calbiochem).
RT-PCR.
Total cellular RNA was extracted using RNA-BEE (Tel-Test) according to the manufacturer's instructions and treated with RQ1-DNase (Promega) to remove any residual DNA. cDNA was generated from 2 μg total RNA in 20-μl reaction mixtures with 200 U SuperScript III reverse transcriptase (RT) (Invitrogen) according to the manufacturer's instructions, using either 0.1 pmol gene-specific primer (GSP) or 5 ng oligo(dT)12–18 (Invitrogen). Nucleotide sequences of all GSPs and forward and reverse PCR primers are provided in Table 1. Corresponding negative-control reactions lacked reverse transcriptase (−RT). Two microliters of the RT or −RT reaction was subjected to PCR in 25-μl reaction mixtures containing 0.5 μM (each) primer, 0.25 mM (each) deoxynucleoside triphosphate (dNTP), 2 mM MgSO4, 1× HiFi PCR buffer (Invitrogen), and 1 U Platinum Taq High Fidelity DNA polymerase (Invitrogen). PCR parameters were as follows: 3 min of denaturation at 95°C, followed by 35 cycles of 60 s at 95°C, 60 s at annealing temperature (Table 1), and 60 s at 68°C, followed by a final extension for 10 min at 68°C. For semiquantitative analysis of BHRF1 mRNAs, cDNA was diluted 5-fold and then subjected to 5 3-fold serial dilutions; a 2-μl aliquot from each of the 6 dilutions was then subjected to 30 cycles of amplification as described above. GAPDH cDNA generated with oligo(dT)12–18 was amplified in parallel to ensure that equivalent amounts of RNA from each sample had been subjected to analysis.
Table 1.
Oligodeoxynucleotide primers used for PCR and RT-PCR
| Primer | Annealing site | Sequence (5′-3′) | EBV genome coordinatesa | Notes [PCR annealing temp, °C] |
|---|---|---|---|---|
| 3′-W2 | W2 exon | CCCTGAAGGTGAACCGCTTA | 14832–14813 | GSPb and reverse PCR primer to detect Cp and Wp usage |
| 5′-C1/C2 | C1/C2 exon junction | CATCTAAACCGACTGAAGAA | 11470–11476/11626–11635 | Forward primer to detect Cp usage [55] |
| 5′-W0/W01 | W0/W01 exon junction | GTCCACACAAATGGGAG | 14399–14410/14559–14563 | Forward primer to detect Wp usage [52] |
| 5′-C1C2 | C1 exon | AGATCAGATGGCATAGAGAC | 11336–11355 | Forward primer to amplify Cp-specific cDNAs for MscI restriction analysis [55] |
| 3′-C1C2 | C2 exon | ATGCTCACGTGCAGGAGG | 11657–11640 | Reverse primer to amplify Cp-specific cDNAs for MscI restriction analysis [55] |
| K-GSP | EBNA1 coding exon (K) | CTTTGCAAGCCAATGCAA | 95911–95895 | GSP to detect Qp-derived EBNA1 mRNA (Q/U/K exon structure) |
| 5′-Q | EBNA1 5′ (Q) exon | AAGGCGCGGGATAGC | 50137–50151 | Forward primer to detect Qp-derived EBNA1 mRNA [58] |
| 3′-K | EBNA1 3′ coding exon (K) | CTCTATGTCTTGGCCCT | 95863–95847 | Reverse primer to detect Qp-derived EBNA1 mRNA [58] |
| 3′-BERF4 | EBNA3C BERF4 exon | GTGGTGCATTCCACGGGTAA | 87123–87104 | GSP and reverse PCR primer to amplify EBNA3C cDNAs [58] |
| 5′-BERF3 | EBNA3C BERF3 exon | AGACACGGAAGACAATGTGCC | 86346–86366 | Forward primer to amplify EBNA3C cDNAs [58] |
| BHRF1-GSP | BHRF1 HF exon | TTCTCTTGCTGCTAGCT | 42192–42176 | GSP to detect BHRF1 mRNAs |
| 5′-W2 | W2 exon | TGGTAAGCGGTTCACCTTCAG | 14810–14830 | Forward primer to detect BHRF1 mRNAs [60] |
| 3′-BHRF1 | BHRF1 HF exon | TCCCGTATACACAGGGCTAACAGT | 42134–42111 | Reverse primer to detect BHRF1 mRNAs [60] |
| 5′-GAPDH | Exon 2 | GATATTGTTGCCATCAATGAC | NAc | Forward primer to detect GAPDH mRNA [58] |
| 3′-GAPDH | Exon 4 | TTGATTTTGGAGGGATCTCG | NA | Reverse primer to detect GAPDH mRNA [58] |
Based on complete EBV genome sequence (type 1 strain), accession number NC_007605.
GSP, gene-specific primer; used for synthesis of cDNA.
NA, not applicable.
Viral source of EBNA mRNAs.
To identify the source(s) of Cp-specific transcripts within superinfected cells, we took advantage of a single-nucleotide polymorphism in the C1 exon of EBV that creates an MscI restriction site in Akata EBV DNA, thus allowing the differentiation of Akata EBV Cp transcripts from other genotypes of EBV (10). cDNA produced using oligo(dT) was amplified by PCR as described above with primers (5′/3′ C1C2; Table 1) that encompass the C1 and C2 exons of Cp-derived transcripts. Approximately 50 pmol of amplified cDNA was end labeled in 25-μl reaction mixtures using 10 μCi [γ-32P]ATP and 10 U T4 polynucleotide kinase (Invitrogen) in forward reaction buffer for 10 min at 37°C, after which EDTA was added to 5 mM and the samples were heated at 65°C for 10 min. Unincorporated [γ-32P]ATP was removed by spin column chromatography, and equivalent cpm of labeled cDNA were precipitated in ethanol in the presence of 10 μg yeast tRNA (Invitrogen) and then digested with MscI. Digested DNA was electrophoresed through a 6% polyacrylamide gel that was then dried and processed by autoradiography. To investigate the source(s) of Wp and EBNA3C transcription in superinfected cells, RT-PCR products generated with the appropriate primers (Table 1) were cloned into pCR2.1-TOPO (Invitrogen), cDNA inserts were subjected to DNA sequence analysis, and the source of cDNA (transcription) was determined based on nucleotide polymorphisms that differentiated endogenous from superinfecting EBV genomes as illustrated in Fig. 9. These experiments were performed with two independent cDNA synthesis reactions for each RNA sample to account for potential bias during PCR amplification.
Fig. 9.

Source of Wp-derived and EBNA3C transcripts within superinfected Sal BL cells. (A) The source of Wp-derived transcripts early (∼1 month) and late (12 to 17 months) postsuperinfection (SI) was determined by cDNA sequence analysis of cloned RT-PCR products to detect nucleotide polymorphisms P1 and P2 within the W01 exon (left). For each experiment (Exp), amplified cDNA was generated from a separate cDNA synthesis reaction of the same RNA sample; the numerator indicates the number of cDNA clones originating from RNA encoded by the endogenous (Sal) EBV genome out of the total number of cDNA clones (endogenous plus superinfecting virus) sequenced per experiment. (B) The source of EBNA3C transcripts was determined by cDNA sequence analysis to detect nucleotide polymorphisms P1, P2, and P3 (left) within the large coding exon of EBNA3C cDNAs (left), amplified from RNA isolated ∼1 month (early) or 12 to 17 months (late) postsuperinfection. Numbers refer to nucleotide coordinates of the EBV genome (accession number NC_007605). Asterisks, nucleotide coordinates given in panel A refer to those within the 5′-most copy of the BamHI-W/IR1 repeats, each of which encodes a copy of the W01/W1 exon.
Generation of rEBV.
All rEBV used in this study was derived from Ak-GFP-BAC (clone 12-15) (20) and produced from HEK293 cells stably transfected with Ak-GFP-BAC or its derivative. Ak-GFP-BAC DNAs, which contain a chloramphenicol resistance (Cmr) gene, were maintained in Escherichia coli strain DH10B under chloramphenicol selection. To generate ΔB-S rEBV lacking the BHLF1 locus, a 3.3-kbp deletion was introduced into Ak-GFP-BAC by recombination-mediated genetic engineering (recombineering) in E. coli strain SW105 (obtained from the Mouse Cancer Genetics Program, NCI-Frederick). The ΔB-S deletion encompasses nucleotide coordinates 38287 to 41550 of the EBV genome (NCBI accession number NC_007605) and extends rightward from the stop codon of the BHLF1 ORF that is 566 bp downstream of the EBNA2 mRNA poly(A) addition site (54) to the right boundary of the 8.5-kbp deletion present in EBV genomes within the Sal BL cell line (23) (see Fig. 4). Thus, ΔB-S removes the entire BHLF1 ORF and approximately 1 kbp of the 5′ promoter elements, which also includes oriLytL (40301 to 41293), which abuts the 5′ end of the BHLF1 ORF. To generate the DNA targeting construct, a tetracycline resistance gene cassette was flanked by 250-bp EBV homology arms that were generated by PCR from Ak-GFP-BAC and which represented DNA immediately upstream and downstream of the locus to be deleted in Ak-GFP-BAC. Briefly, the upstream arm (nucleotide coordinates 38038 to 38287) was amplified with the forward primer 5′-GCctcgagGGCTGCTTTTAGCCTAATTGTG-3′ (lowercase, XhoI site) and reverse primer 5′-GCaagcttgcgatcgcTGCAGTGTCCCTGCTGCC-3′ (lowercase, HindIII site; underlined, AsiSI site) and following digestion was ligated between the XhoI and HindIII restriction sites of the multiple cloning site in pBluescript II KS(+) (Stratagene), making pBS-US. The downstream arm (coordinates 41550 to 41799) was similarly generated using the forward primer 5′-GCgcgatcgcTTTCGTCTGTGTGTTGAAGGG-3′ (lowercase, AsiSI) and reverse primer 5′-GCgaattcACACAGACCTGAAACACAACTC-3′ (lowercase, EcoRI), digested, and ligated into pBS-US between the AsiSI restriction site present within the upstream arm fragment and the EcoRI site in pBluescript II KS(+), yielding pBS-USDS, which was verified by DNA sequence analysis. Next, an AsiSI restriction fragment containing a tetracycline resistance gene flanked by flippase (Flp) recombinase recognition target (FRT) sites was purified from the plasmid pFRT-rpsL-Tet-FRT (rpsL is a counterselection gene irrelevant to these studies) and ligated into the AsiSI site of pBS-USDS. From this, the targeting DNA fragment consisting of US-FRT-rpsL-Tet-FRT-DS was amplified by PCR with the forward primer 5′-GGCTGCTTTTAGCCTAATTGTG-3′ and reverse primer 5′-ACACAGACCTGAAACACAACTC-3′, digested with DpnI to destroy the plasmid template, and purified by agarose gel electrophoresis. PCRs (25 μl) contained 20 to 50 ng template DNA, 0.5 μM (each) primer, 0.25 mM (each) dNTP, 2 mM MgSO4, 1× HiFi PCR buffer (Invitrogen), and 1 U Platinum Taq High Fidelity DNA polymerase (Invitrogen). Cycling parameters were as follows: 10 min of denaturation at 95°C, followed by 25 cycles of 45 s at 95°C, 30 s at 60°C, and 45 s at 72°C and a final 10-min extension at 68°C.
Fig. 4.

Generation of ΔB-S rEBV. Shown is the recombineering strategy used to generate rEBV mutant ΔB-S, which is deleted for the entire BHLF1 ORF and 5′ promoter region up to the right (3′) boundary of the deletion in the EBV genomes within the Sal BL cell line (23); the deletion also removed the leftmost lytic-cycle origin of DNA replication (oriLyt). Left, agarose gel analysis of BamHI-digested and corresponding Southern blot of the parental Ak-BAC-GFP (lane 1), intermediate BAC clone containing the targeting cassette FRT-rpsL-tet-FRT in place of deleted EBV DNA (lane 2), and the final BAC clone of ΔB-S (lane 3) after Flp-mediated removal of the targeting cassette, resulting in a 3.3-kbp deletion and insertion of one Flp target sequence (FRT). Right, configuration of the domain targeted within the EBV genome. Shown are the BamHI restriction sites (vertical arrows) defining the BamHI-Y (1.8-kbp) and BamHI-H (6.0-kbp) restriction fragments of the wt EBV genome (top) and within the intermediate (middle) and final (bottom) mutated EBV BAC clones. In addition to BamHI-H and BamHI-Y, diagnostic 2.8- and 2.9-kbp BamHI restriction fragments expected to hybridize to the probe are indicated below each DNA construct (right) and by white dots on the ethidium bromide-stained agarose gel (left). Open arrows depict the ORFs present in the targeted domain. The DNA diagram is not to scale.
To generate ΔB-S-Ak-GFP-BAC, SW105 cells containing Ak-GFP-BAC were grown at 32°C to an optical density at 600 nm (OD600) of 0.5 and then induced to express recombination proteins by shifting to 42°C for 15 min, followed by rapid cooling and washing. These cells were then transformed (by electroporation) with 300 ng targeting fragment and selected for tetracycline resistance (Tetr). To remove DNA flanked by the 250-bp EBV DNA targeting arms, Tetr clones were grown at 32°C to an OD600 of 0.5; l-arabinose was then added to 10% (wt/vol) (to induce expression of the Flp recombinase), and incubation was continued for 1 h. Bacteria were then plated on chloramphenicol-containing LB agar plates, and Cmr colonies were replica plated on tetracycline-containing agar to identify those that had lost the tetracycline resistance cassette. To verify proper recombination, bacterial artificial chromosome (BAC) DNA from Cmr Tets colonies was amplified by PCR and subjected to sequence analysis using primers with annealing sites within the EBV genome outside of the 250-bp homology arms present in the targeting fragment. To ensure against illegitimate recombination elsewhere in the EBV genome, BAC DNA that had been purified using the NucleoBond BAC100 kit (Clonetech) was examined by agarose gel electrophoresis and Southern blot analysis following digestion with BamHI (see Fig. 4).
Production of rEBV and superinfection of BL cells.
To generate wt or ΔB-S rEBV, HEK293 cells were transfected with 2 μg Ak-GFP-BAC or ΔB-S-Ak-GFP-BAC DNA, respectively, using TransIT-293 transfection reagent (Mirus), and individual clones were selected based on green fluorescent protein (GFP) expression and resistance to Geneticin (500 μg/ml). To induce EBV replication, HEK293 clones were transiently transfected with 1 μg each of expression plasmids encoding the EBV proteins BZLF1 and BALF4, and at 24 h posttransfection, 12-O-tetradecanoylphorbol 13-acetate (TPA) and sodium butyrate were added to the culture medium to 20 ng/ml and 1.47 mM, respectively. After 3 h, cell monolayers were rinsed and then incubated in fresh RPMI growth medium (instead of DMEM) for 3 days, after which the culture medium was clarified by centrifugation and passed through a 0.45-μm filter. Successful virus production was determined by infecting Raji BL cells using a “spin infection” protocol in which 5 × 105 cells were mixed with 1 ml virus-containing HEK293 supernatant per well in 6-well plates and centrifuged at 200 × g for 1 h at 4°C. Afterwards, cells were incubated at 37°C for 24 h, followed by addition of 2 ml fresh growth medium. At 3 days postinfection, Raji cells were microscopically scored for GFP expression to identify the HEK293 clones that most efficiently produce rEBV. Virus produced in this manner was used to superinfect EBV-positive Kem I and Sal BL cells by the same method; at 5 to 7 days postsuperinfection, cells were placed under selection with Geneticin (Kem I, 400 μg/ml; Sal, 700 μg/ml) in the 6-well plates and subsequently expanded for further analysis.
RESULTS
Establishment of restricted latency within superinfected BL cells.
To address the role of the BHLF1 locus and other potential mechanisms in the establishment of restricted latency (e.g., latency I), we first sought to establish a B-cell infection model in which EBV latency-associated gene expression would consistently proceed from a latency III program to a restricted program of latency in which the EBNA promoters Wp and Cp are silent. While we and others have shown that infection of some EBV-negative BL cell lines, such as those derived from the EBV-positive BL lines Akata and Mutu, can result in the establishment of a latency I program (28, 50, 71), we found these infection systems less than adequate. Specifically, the time necessary to obtain complete silencing of latency III-associated gene expression (e.g., of EBNA2) often varied by several months between experiments. Further, we not infrequently obtained cell lines that continued to express EBNA2 but which did not express the EBNA3 proteins and/or LMP1. This was a particularly common but not exclusive complication when we introduced the viral genome (as a BAC clone) directly by transfection instead of by infection with rEBV. Quite possibly this was a consequence of a high rate of EBV genome integration, which we noted based on a lack of the episomal (latency-associated) form of the EBV genome as determined by Gardella gel analysis (12) or by a Southern blot analysis designed to detect integration through or near the terminal repeats of the EBV genome (15). Our findings were also consistent with the previous observation that infection of EBV-negative B-cell lines frequently results in integration of the EBV genome (15).
To circumvent these issues, we next explored whether superinfection of BL cells that maintain their endogenous EBV genome in a state of restricted latency could serve as an appropriate model. It had been previously demonstrated that superinfection of EBV-positive Akata BL cells (latency I) results in a latency III pattern of EBNA expression but only from the superinfecting-virus genomes (10). However, since latency gene expression was not evaluated beyond 72 h postsuperinfection, it was unclear whether these Akata cells ultimately supported the transition to a latency I program from the superinfecting-virus genome. Because efficient infection of Akata cells required stable exogenous expression of the cellular receptor for EBV attachment (10), we asked whether Kem I BL cells, which also strictly maintain a latency I program, could substitute for Akata cells and support a latency III to I transition in latency gene expression.
Following superinfection of Kem I cells with wt rEBV generated from Ak-BAC-GFP and encoding resistance to neomycin, cells were expanded under selection (Geneticin) and then harvested for analysis of EBV latency-associated gene expression beginning approximately 1 month postsuperinfection. As shown in Fig. 1A, we initially observed a latency III pattern of gene expression in the superinfected Kem I cells, as indicated by the detection of EBNA2, -3A, -3B (very low), and -3C and of LMP1. Because the respective EBNA3C proteins encoded by the superinfecting rEBV and endogenous viral genomes are distinctly different in size (unlike for EBNA1, -2, -3A, and -3B), the larger EBNA3C (relative to EBNA3C in Kem III BL cells) detected at the early time point postsuperinfection (Fig. 1A) indicated that the latency III pattern of EBNA expression originated from the superinfecting-virus genome, as previously noted for superinfecetd Akata BL cells (10). Importantly, these cells ultimately restricted expression to EBNA1, consistent with latency I (Fig. 1B). Further, we have found no evidence of significant integration by the superinfecting-virus genomes (data not shown), which could potentially influence EBV gene expression.
Fig. 1.
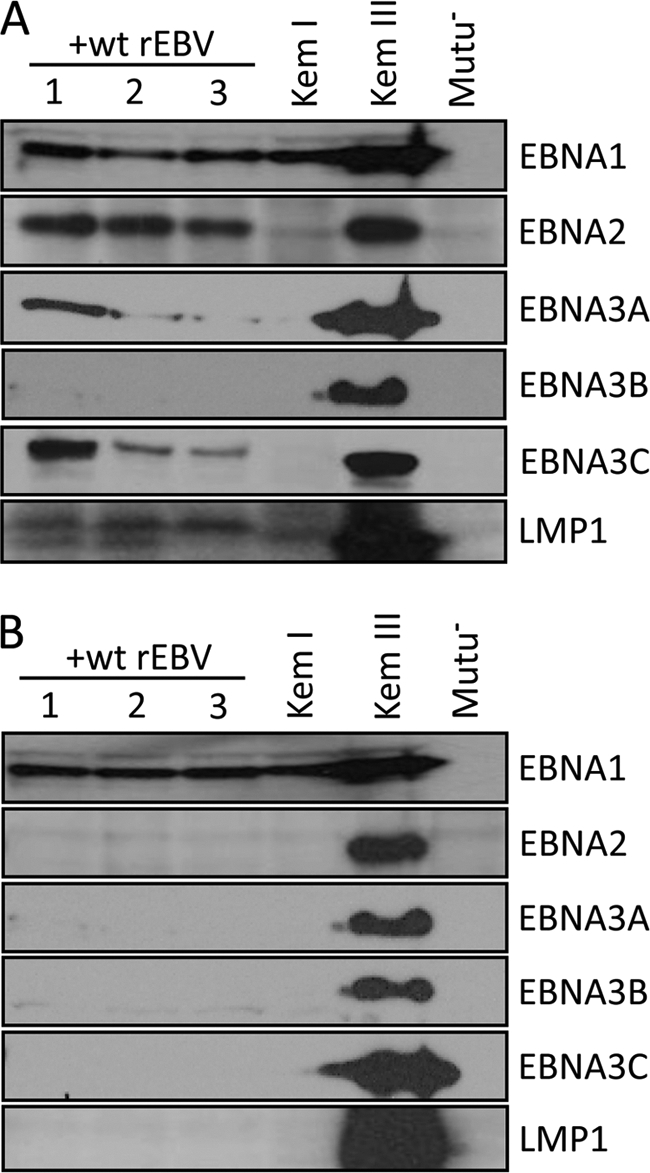
Restricted latency is established upon superinfection of latently infected BL cells. Kem I BL cells were superinfected with wt rEBV, and three resulting cell lines, each derived from an independent infection, were monitored by immunoblotting for the expression of EBV EBNA and LMP1 proteins at approximately 1 month (A) and 12 months (B) postsuperinfection. Detection of EBNA and LMP1 expression in Kem I and Kem III BL cells serves as a positive control for latency I and III, respectively; Mutu− is an EBV-negative BL cell line derived from Mutu I cells. Loading of cell lysates was equivalent within panels A and B as determined by blotting for EBNA1 or β-tubulin (not shown).
We next assessed EBNA promoter usage within superinfected Kem I cells by RT-PCR. In agreement with the results of our protein expression analysis, Cp- and Wp-specific transcripts, indicative of latency III, were detected at the early time postsuperinfection (Fig. 2A) but not at later times (Fig. 2B). Although the results shown in Fig. 1B and 2B were obtained with cells harvested approximately 12 months postsuperinfection, we consistently observe a complete conversion to latency I by 2 months, if not earlier. Because the endogenous and rEBV-encoded EBNA1s are difficult to distinguish by immunoblotting (Fig. 1B), we examined whether superinfected cells continued to express GFP at late times postsuperinfection to ensure that loss of the latency III pattern of gene expression was due to silencing of EBV gene expression and was not a consequence of a loss of the superinfecting-virus genomes. As shown in Fig. 3, a high percentage of cells of two of the three superinfected Kem I BL lines examined remained GFP positive. Moreover, since superinfected cells remained under G418 selection, loss or lower levels of GFP expression may actually reflect epigenetic silencing of the GFP gene instead of loss of the rEBV genome itself. We concluded, therefore, that superinfection of Kem I BL cells is an appropriate model with which to address the establishment of restricted EBV latency.
Fig. 2.
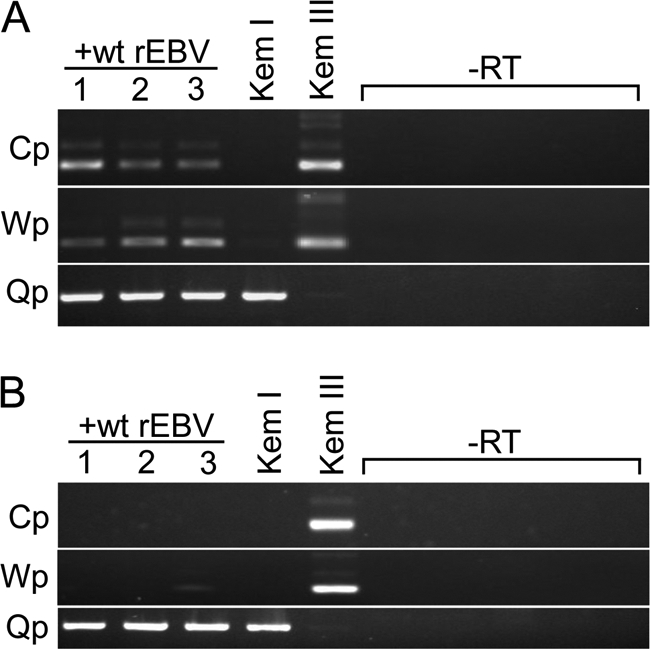
EBNA promoter silencing upon superinfection of Kem I BL cells. Total RNA from the superinfected Kem I cell lines analyzed in Fig. 1 was subjected to RT-PCR to determine EBNA promoter usage. (A) ∼1 month postsuperinfection, Cp and Wp usage (indicative of latency III) was detected by amplification of the common 5′ ends of the EBNA mRNAs containing either the C1-C2-W1-W2 or W0-W01/W1-W2 exon structure (48), respectively. The faint band immediately above the major products for Cp and Wp in the lanes for the superinfected and Kem III lines (latency III control) are cDNAs representing transcripts in which the 81-nucleotide intron between the W01/W1 and W2 exons had been retained, as determined by DNA sequence analysis. Detection of Qp-specific transcripts is consistent with expression of EBNA1 from the endogenous EBV genome. (B) Approximately 12 months postsuperinfection, Cp and Wp usage is undetectable, consistent with full establishment of latency I. −RT, amplification of parallel cDNA synthesis reactions (for all RNA samples) that did not contain reverse transcriptase.
Fig. 3.

The superinfecting EBV genome is retained within Kem I BL cells. Fluorescence microscopy was used to detect GFP expression within Kem I lines at 17 (cell line 1) or 16 (cell lines 2 and 3) months postsuperinfection with wt rEBV. Phase-contrast microscopy of the same field indicates that a majority of cells within lines 2 and 3 remained GFP positive, whereas much fewer cells of line 1 were GFP positive, though all cells were maintained under G418 selection to select for retention of the superinfecting-virus genome. Magnification, ×20.
Does the BHLF1 locus contribute to restricted latency?
We next tested the hypothesis that the BHLF1 locus is required for the establishment of restricted latency. To do this, we generated a mutant rEBV, ΔB-S, from whose genome the entire BHLF1 ORF and 5′ promoter region were deleted as illustrated in Fig. 4. As a consequence of removing the 5′ promoter region, this 3,264-bp deletion also removed oriLytL, which abuts the BHLF1 ORF and is one of two origins of EBV DNA replication, separated by ∼102 kbp, that are active during virus replication (one oriLyt is sufficient for virus replication). The right boundary of the introduced deletion matches that of the deletion within the EBV genomes within the BL cell line Sal, which is the largest known deletion of this locus among the BL cell lines that support the Wp-restricted latency program (23). Thus, with the ΔB-S rEBV, we would be able to assess the potential role in latency of all elements downstream of the EBNA2 ORF normally targeted in this deletion.
Upon superinfection of Kem I cells with ΔB-S rEBV, at approximately 1 month postsuperinfection, we observed a latency III pattern of EBNA and LMP1 expression, as we had following superinfection with wt rEBV (data not shown). However, by 4 months, an analysis of EBNA and LMP1 expression within superinfected Kem I cells revealed that a latency I pattern of gene expression had been fully established (Fig. 5). Further, as we had found for superinfection with wt rEBV, continued expression of GFP indicated that the ΔB-S rEBV genome had not been lost over time (data not shown). Thus, within this infection model it appeared that neither BHLF1 nor other elements within the introduced deletion are critical for the establishment of a restricted latency in B cells.
Fig. 5.
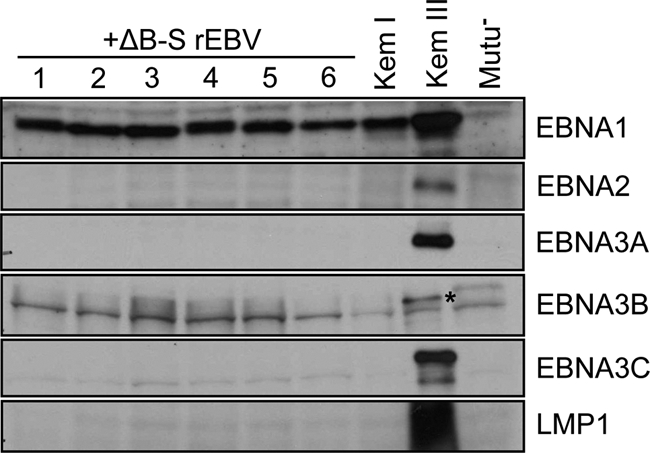
The BHLF1 locus is not required to establish restricted latency. Comparable to superinfection with wt rEBV (Fig. 1), EBNA and LMP1 expression indicative of latency III could be detected at 1 month postsuperinfection with ΔB-S rEBV (data not shown) but as demonstrated here was no longer evident at later times (>4 months); the blot shown is of protein samples harvested at 8 months postsuperinfection. The authentic EBNA3B band is marked with an asterisk in the Kem III lane; others are common background bands, since they were also detected in the EBV-negative Mutu− cells.
Wp-restricted BL cells support latency III.
Coexistence of silent full-length EBV genomes and transcriptionally active counterparts that contain deletions spanning the EBNA2 and BHLF1 loci has thus far been observed only in BL cell lines and their parental tumors that support Wp-restricted latency (25). Consequently, we considered that a contribution of the BHLF1 locus to the establishment of restricted latency might be dependent on the state of B-cell differentiation or other factors that may distinguish classic BL cell lines (such as Kem I and Akata) from those that support Wp-restricted latency, such as the Sal BL line. Therefore, we repeated the above analysis following superinfection of Sal BL cells. Unlike the case in Kem I cells, however, superinfection of Sal cells with wt rEBV resulted in a sustained latency III pattern of gene expression, as shown by immunoblot analysis of EBNA and LMP1 expression at 12 months postsuperinfection (Fig. 6A). Results of EBNA promoter usage analysis at early (1 month) and late (out to 17 months) times postsuperinfection was consistent with latency III (Cp and Wp positive) (Fig. 6B). We did detect some Qp usage for EBNA1 expression predominantly at early times postsuperinfection. However, because Akata BL cells support some early lytic-cycle gene expression shortly after superinfection (10), the Qp signal we detected in superinfected Sal cells may actually reflect lytic-cycle-specific EBNA1 transcripts originating with the upstream (∼200 bp) promoter Fp (30, 38, 57). Regardless, at late times postsuperinfection, Fp/Qp activity was much reduced and was comparable to that in Sal cells alone, which primarily use Wp for EBNA1 expression (Fig. 6B, right panel) (23). Further, it is not uncommon to detect a low level of Qp usage even within established LCLs that support latency III.
Fig. 6.
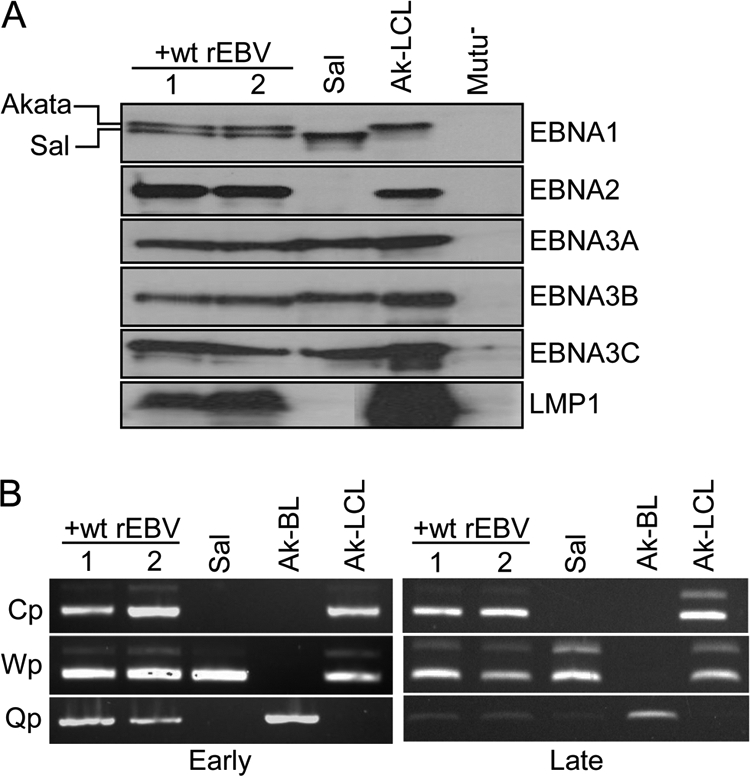
BL cells maintaining Wp-restricted latency support latency III upon superinfection. Two lines derived by the wt rEBV superinfection of cells from the Sal BL cell line that maintains a Wp-restricted latency program were subjected to analysis. (A) Immunoblot analysis of EBNA and LMP1 expression at ∼12 months postsuperinfection. Note that EBNA1 encoded by the BAC-derived EBV (Akata isolate) and that encoded by the endogenous (Sal) EBV genome can be easily distinguished by their different sizes; this is evident to a lesser extent for the Sal EBV EBNA3A and EBNA3B proteins, which are larger than their Akata EBV counterparts. Due to the deletion in the Sal EBV genome and the lack of any wt EBV genomes within the Sal cell line, EBNA2 can only be expressed from the superinfecting-EBV genome. Ak-LCL is an LCL generated by infection of primary B cells in vitro with the Akata isolate of EBV. (B) EBNA promoter usage in Sal cells early (∼1 month) and late (∼12 months) postsuperinfection, in comparison to parental Sal BL (Wp restricted), Ak-BL (latency I; Qp only), and Ak-LCL (latency III; Cp and Wp). As indicated in the legend to Fig. 2, the larger minor-band products in the Cp and Wp amplifications are cDNAs representing transcripts in which the 81-nucleotide intron between exons W01/W1 and W2 is present.
Thus, despite the fact that Wp-restricted BL cells originally maintained a full-length EBV genome in a transcriptionally silent state (our Wp-restricted BL lines do not contain wt genomes as concluded by our inability to amplify DNA within the EBNA2 ORF by PCR) (25), our results suggest that these cells are otherwise programmed to support the latency III program. This is also supported by our analysis of the expression of Bcl-6, a germinal center B-cell marker tightly associated with the latency I program. Specifically, we found that while Bcl-6 was abundantly expressed in BL lines Mutu I, Kem I, and Ak-BL (all latency I) as expected, it was not detectable within the two Wp-restricted BL lines analyzed, Oku and Sal (Fig. 7).
Fig. 7.
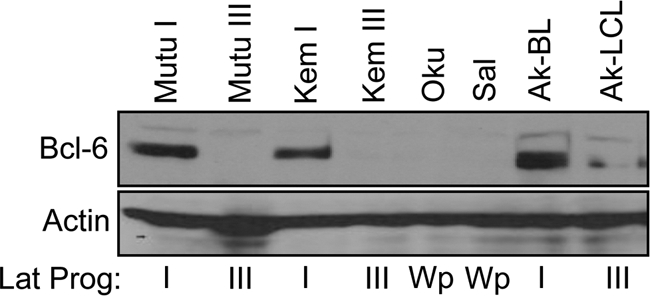
Bcl-6 expression correlates with EBV latency program. The indicated cell lines were subjected to immunoblotting to detect expression of Bcl-6, a marker for germinal center B cells that is highly expressed in BL cells that maintain latency I but is very low or undetectable in BL cells and LCLs that support latency III. The lack of Bcl-6 expression in the BL lines Oku and Sal, which maintain a Wp-restricted latency, suggests that these cells are otherwise programmed to support latency III, consistent with the EBV latency-associated protein expression pattern established within superinfected Sal cells (see Fig. 6A). The faint bands in the Ak-LCL lane are due to spillover from Ak-BL in the adjacent lane.
Repression of endogenous-genome protein expression in Wp-restricted BL cells.
Our superinfection of Sal BL cells also yielded an unexpected and particularly noteworthy result. Strain-specific differences in the apparent sizes of EBNA1, EBNA3A, and EBNA3B expressed in superinfected Sal cells suggested that while EBNA1 expression occurred from both genomes, detectable expression of the remaining EBNAs favored the superinfecting EBV genomes (Fig. 6A; note that Sal EBNA3A and EBNA3B are slightly larger than their counterparts expressed from the Akata EBV genome in Ak-LCL). To investigate this further, we took advantage of nucleotide polymorphisms to determine the genomic source of Cp- and Wp-derived transcripts, as well as that of the EBNA3C mRNA, since EBNA3C encoded by the superinfecting (Akata) EBV and that encoded by endogenous (Sal) EBV are indistinguishable by immunoblotting. For Cp-derived transcripts, we screened for a previously described single-nucleotide polymorphism that results in an MscI restriction site in the Akata EBV C1 exon (10) that we determined is not present in the Sal EBV genome. Because Cp is silent in the parental Sal cells, we used Kem III RNA to generate Cp-specific cDNA as a positive control, since the Kem and Sal EBV genomes are identical at this locus. As shown in Fig. 8, MscI digestion of Cp-specific cDNAs revealed that even at 1 month postsuperinfection, all detectable Cp transcripts originated from the superinfecting virus, indicating that silencing of endogenous Cp in these cells is maintained, as it is in superinfected Akata BL cells (10), even in the presence of EBNA2 (its primary EBV transactivator) expressed from the superinfecting-virus genome.
Fig. 8.
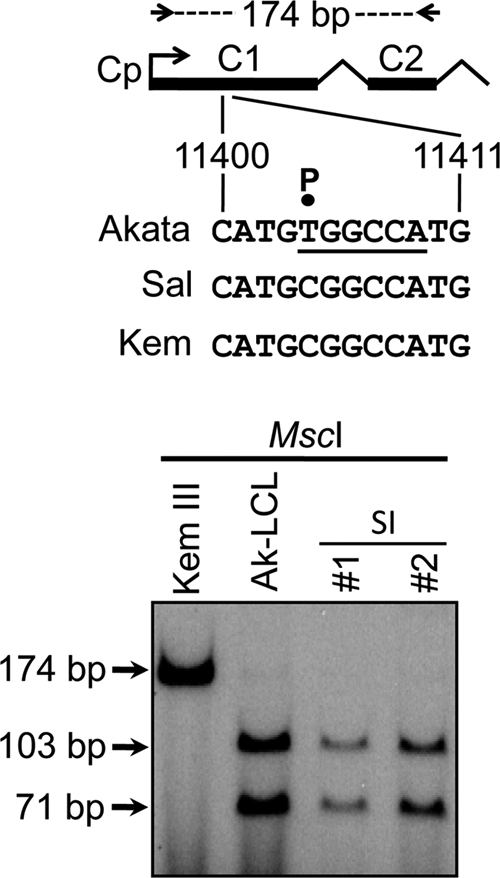
Cp within the endogenous EBV genome remains silent in superinfected Sal BL cells. The source of Cp-derived transcripts was determined based on a single-nucleotide polymorphism (P) resulting in an MscI restriction site (underlined) in the C1 exon of the superinfecting (Akata) strain of EBV (10). A 174-bp cDNA spanning the C1 and C2 exons (top) was amplified from RNA isolated from Sal cells ∼1 month postsuperinfection, end labeled with 32P, digested with MscI, and then subjected to polyacrylamide gel electrophoresis and autoradiography; cDNAs generated from Kem III BL and Ak-LCL cells served as controls for detection of the absence and presence of the MscI site, respectively. The lack of detectable cDNA resistant to MscI digestion in the superinfection (SI) lanes indicates that Cp-derived transcripts originated from the superinfecting virus.
To determine the source of Wp-derived transcripts, we identified and then screened by cDNA sequence analysis for two closely spaced nucleotide differences between the Akata and Sal EBV genomes within the second exon of mRNAs originating from Wp. As shown in Fig. 9A, at the earlier time postsuperinfection the majority of Wp-initiated transcripts originated from the endogenous EBV genomes, consistent with the exclusive usage of Wp for EBNA expression in parental Sal cells and predominately Cp usage within the superinfecting genome. When we determined the relative contributions of the endogenous and superinfecting-virus genomes to Wp transcripts at late times, the endogenous EBV genomes were clearly the predominate source of Wp transcripts within one cell line (SI Sal #1), though in the second line assessed (SI Sal #2) they accounted for 14 to 47% of Wp transcripts (Fig. 9A). Note that even under nonquantitative conditions for RT-PCR, one can clearly see that while at late times the levels of Wp transcripts detected in the superinfected Sal cells were less than those in parental Sal cells, they did not appear substantially lower (Fig. 6B, right panel, compare lanes 1 and 2 to lane 3). This was especially evident for line SI Sal #1, in which virtually all Wp transcripts originated from the endogenous-virus genomes at late times (Fig. 9A). Thus, even though there was an apparent shift to protein expression predominantly from the superinfecting-virus genomes (at least for EBNA3A and -3B) (Fig. 6A), there did not appear to be a substantial corresponding decrease in Wp transcripts expressed from the endogenous genomes.
Taking a similar tack for EBNA3C, we screened for three nucleotide polymorphisms that we had identified in the 5′ end of the large coding exon of the mRNA. Our results indicated that even by 1 month postsuperinfection, the majority of EBNA3C mRNAs were being expressed from the superinfecting-virus genome (Fig. 9B). At late times this had been sustained within one line (SI Sal #2), while the endogenous genome accounted for approximately 50% of EBNA3C transcripts in the other (SI Sal #1). In general, the proportion of EBNA3C transcripts encoded by the endogenous genomes in both lines at the late time corresponded to the relative amount of endogenous Wp transcript levels, i.e., a higher proportion of endogenous viral transcripts in SI Sal #1 than in SI Sal #2 (compare Fig. 9A and B). However, because the EBNA3Cs encoded by endogenous and superinfecting viruses are indistinguishable by immunoblotting, it was unclear whether levels of endogenous Wp-derived EBNA3C mRNAs were sufficient to express detectable EBNA3C protein.
Repression of endogenous EBNA-LP and BHRF1 expression.
Although the endogenous Sal EBV EBNA3A and EBNA3B proteins and their respective counterparts encoded by the superinfecting virus are distinguishable by size, because their respective molecular masses are so similar (unlike the case for EBNA1) it was difficult to conclude by immunoblotting what proportion of the total detectable EBNA3A and EBNA3B, if any, was contributed by the endogenous virus in superinfected Sal cells (Fig. 6A). Therefore, to better assess the degree of repression of Wp-dependent protein expression and thus its potential for biological significance, we determined the effect of superinfection on the expression of EBNA-LP and BHRF1, both of which are expressed solely from Wp within BL lines that support Wp-restricted latency (23, 24). As shown in Fig. 10A, superinfection of Sal BL cells with wt rEBV resulted in a virtually complete loss of detectable EBNA-LP expression. Note that because the monoclonal antibody used does not detect EBNA-LP encoded by the superinfecting virus strain, e.g., as in Ak-LCL (Fig. 10A), the observed effect solely reflects repression of Wp within the endogenous EBV genome. Similarly, we observed a complete loss of detectable expression of BHRF1, which is not normally evident in established LCLs that support latency III, as in Ak-LCL (Fig. 10B), even though latency-associated BHRF1 transcripts (presumably from Wp) may be detectable by RT-PCR (24). Thus, the level of trans-repression observed is clearly able to produce a substantial effect on non-EBNA1 protein expression and consequently is likely to be of biological significance.
Fig. 10.
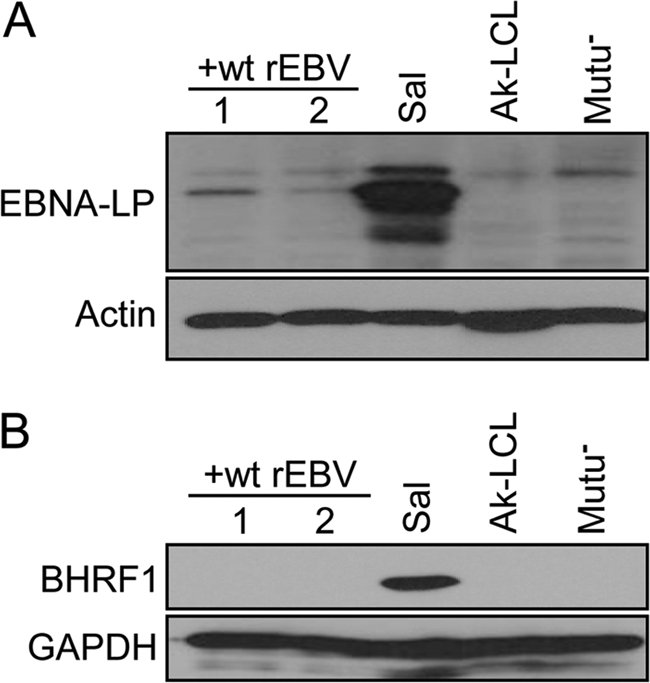
Wp-dependent protein expression is repressed in superinfected Sal BL cells. Immunoblot detection of the EBV protein EBNALP (A) or BHRF1 (B), believed to arise predominately if not exclusively from transcription originating from Wp in Sal and other Wp-restricted BL lines, is shown. Note that the antibody used to detect EBNA-LP does not detect EBNA-LP encoded by the Akata strain of EBV (as in Ak-LCL and rEBV used to superinfect Sal cells). Lysates from superinfected Sal cells were harvested ∼12 months postsuperinfection. Detection of β-actin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading controls.
Finally, to better determine whether the effect we observed is posttranscriptional in nature as suggested by data presented in Fig. 6B and 9A, we took advantage of the distinct differences in structures of the BHRF1 mRNAs expressed in Wp-restricted BL lines and Cp/Wp-using LCLs containing wt EBV (24). As illustrated in Fig. 11, due to the deletion in the Sal EBV genome that removes several of the exons normally present within latency-associated BHRF1 mRNAs from Wp (or Cp), the genomic origin of these transcripts and their relative abundance can be determined more directly by RT-PCR, which we were unable to do for the EBNA3C mRNAs and the 5′ region of Wp-derived transcripts. Due to the presence of several cDNA products that represent the different BHRF1 mRNAs, however, we had to employ a semiquantitative approach instead of a standard real-time RT-PCR assay to assess relative contributions of the superinfecting-virus and endogenous EBV genomes in our superinfected Sal BL cells.
Fig. 11.
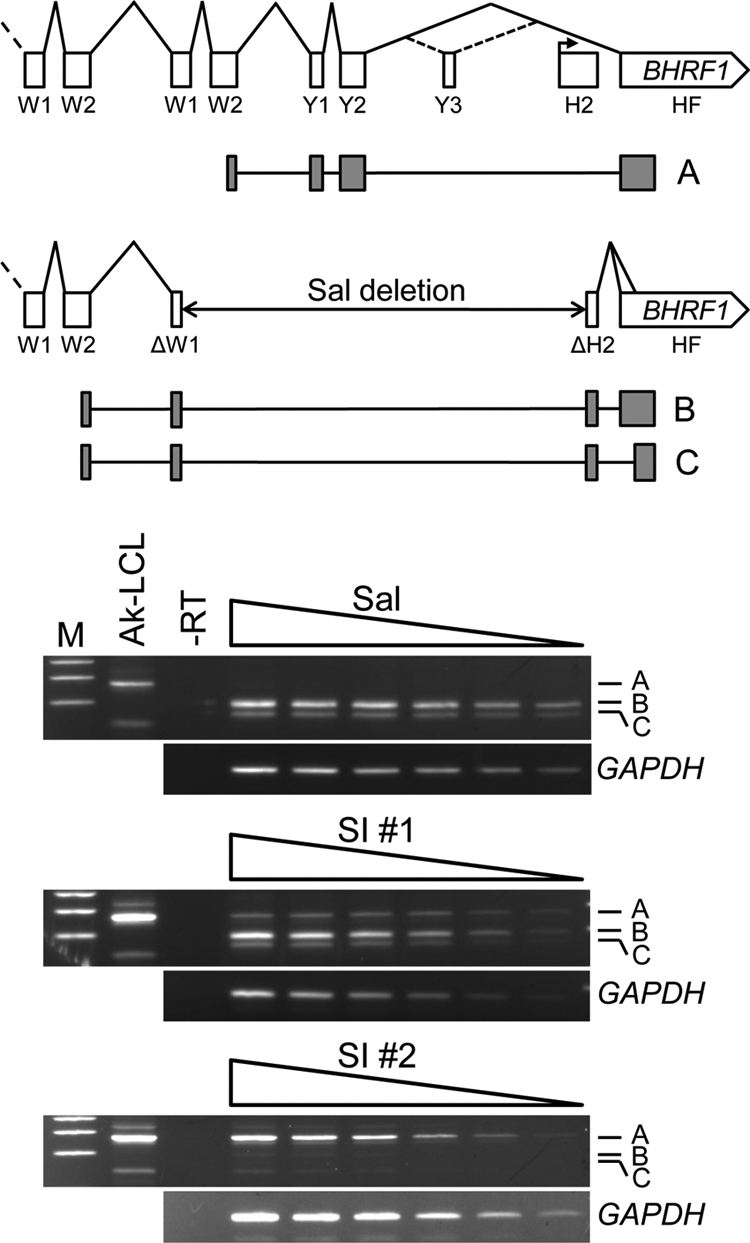
Repression of Wp-dependent BHRF1 protein expression can occur independent of a parallel decrease in mRNA. Top, structures of the 3′ portion of latency-associated BHRF1 mRNAs expressed from the wt EBV genome during latency III (upper) (41) or from the deletion-containing EBV genome within Sal Wp-restricted BL cells (lower) (24). W1 and W2 are exons derived from each copy of the BamHI-W (IR1) repeat that comprise the 5′ leader of BHRF1 and EBNA mRNAs from Wp or Cp. The BHRF1 ORF is entirely contained within the 3′ exon, HF. H2 is a noncoding exon present in spliced BHRF1 mRNAs that arise from a promoter (bent arrow) active early in the lytic cycle (41). The deletion within the Sal EBV genome fuses the 5′ end of the last W1 exon to the 3′ end of the H2 exon, resulting in a novel exon within mRNAs expressed in Sal cells (24). Note that the EBNA2 and BHLF1 ORFs within the deletion are not shown (see Fig. 1). The major RT-PCR products (A, B, and C) generated with forward and reverse primers within W2 and HF, respectively, are composed of the exon-derived cDNA represented by the shaded boxes shown below their positions within the respective mRNA structure. The diagram is not precisely to scale. Bottom, analysis of BHRF1 mRNA expression in parental and two superinfected Sal BL cell lines by semiquantitative RT-PCR. Equal aliquots of serially diluted (left to right) BHRF1 and GAPDH cDNAs were amplified and assessed by agarose gel electrophoresis. Amplification of BHRF1 mRNAs encoded by wt genomes in Ak-LCL is shown for comparison; the unlabeled minor band above band A represents an alternatively spliced mRNA that includes the Y3 exon, whereas as the structure of the cDNA that is the smaller unlabeled band is unknown. M, DNA size markers (400, 300, and 200 bp).
Within Sal BL cells, we detected two cDNA products (Fig. 11, bands B and C) that represent the two alternatively spliced BHRF1 mRNAs previously shown to be expressed from the endogenous, deletion-containing EBV genomes in this Wp-restricted line (24). Consistent with a posttranscriptional mechanism of repression, within the (superinfected) line SI #1, we detected the endogenous-genome-specific transcripts at approximately the same levels as in the parental Sal cells and in greater amounts overall than the cDNA representing the major BHRF1 mRNA originating from the wt superinfecting rEBV (Fig. 11, compare the relative amounts of band A to those of bands B and C in the top and middle gel panels). In contrast, in SI #2, the predominant cDNA originated from the wt EBV genomes of the superinfecting virus, suggesting that Wp within the endogenous EBV genomes had been largely silenced transcriptionally at this point, possibly by DNA methylation (see below). Nonetheless, the fact that in SI #1 cells we observed a loss of protein expression with little if any corresponding decrease in mRNA from the endogenous genomes supports the existence of a novel posttranscriptional mechanism involved in the regulation of Wp-dependent gene expression. Moreover, the fact that in the absence of the BHRF1 protein we observed a level of “wt” BHRF1 mRNA within SI #2 cells comparable to that of endogenous-virus-encoded mRNA in parental Sal BL cells (BHRF1 protein positive) would appear to also support a posttranscriptional mechanism of regulation in this superinfected-cell line. We note also that the respective results obtained from the two superinfected Sal lines in Fig. 11 corresponded well with those we obtained by sequence analysis of Wp transcript cDNAs also generated at late times postsuperinfection (Fig. 9A).
DISCUSSION
Here we have described two new in vitro B-cell infection models that should prove useful for exploring the mechanisms that regulate EBV latency gene expression, notably through the EBNA promoters Cp and Wp, which are critical to the success of EBV as a human pathogen. In the first, BL cells that normally maintain latency I, upon superinfection, consistently appear to support a transition from the latency III to latency I program of transcription from the superinfecting-virus genomes. While we have not excluded the possibility that over an extended period in culture there may be a selection against cells that sustain latency III in this model, the fact that in a high percentage of cells the superinfecting-virus genome adopts a restricted latency program relatively early after superinfection supports the use of this model for delineating the mechanisms responsible for the initiation and maintenance of restricted latency. Unfortunately, this cannot be done in primary B cells that depend upon maintenance of the latency III program of EBV for continued growth and survival in vitro and which regardless are unlikely to be able to autonomously support a transition to restricted latency. Employing this model, we demonstrated that the BHLF1 locus of the EBV genome is not essential for the establishment of restricted latency, as we had hypothesized. Using the second model, the superinfection of BL cells maintaining Wp-restricted latency that are unable to transition to latency I, we identified a novel trans-repression of gene expression originating from the EBNA and BHRF1 promoter Wp.
The only previously defined negative regulation of Wp during a latency III program occurs during the switch from Wp to Cp in the initial stages after EBV infection of B lymphocytes (76). Based on a number of observations, the downregulation of Wp in this process is believed to be mediated by transcriptional interference in cis upon activation of Cp by EBNA2 (initially produced from Wp). For example, when primary B cells are infected with rEBV lacking either a functional EBNA2 response element within Cp or a functional EBNA2 gene, there is a failure to switch to Cp usage and EBNA expression continues to predominate through Wp (19, 49, 60, 75, 79, 80). Further, conditional loss of functional EBNA2 in an established LCL results in an increase in Wp activity that is concomitant with decreased Cp activity (79, 80). The best direct evidence for the repression of Wp in cis by a transcriptionally active Cp are the observations that Wp-specific transcription within reporter plasmids in transient-transfection assays is elevated upon either deletion of the upstream Cp or the reversal of Cp orientation relative to Wp (42, 43). The observation here that Wp-dependent gene expression can be downregulated from EBV genomes lacking transcriptionally active Cp indicates that this effect is mediated in trans and therefore is distinct mechanistically from the event currently understood to be responsible for Wp-to-Cp switching.
While it is possible that the trans-repression we describe contributes to the switch to EBNA expression from Cp, we believe that this is unlikely to have a significant influence on this process. Notably, at our earliest times of analysis post-superinfection of Wp-restricted Sal BL cells, which occurred ∼3 weeks later than is required to obtain maximal Cp usage following infection of primary B cells (76), the vast majority of Wp-specific transcripts originated with the endogenous EBV genomes (Fig. 9A). Assuming that the much lower proportion of Wp-specific transcripts expressed from the superinfecting-virus genomes reflects the additional influence of transcriptional interference from Cp, trans-repression of Wp-mediated gene expression would appear relatively insignificant compared to repression in cis in the context of an active Cp. Further, whereas cis-mediated repression of Wp is transcriptional in nature, our data suggest that there is a significant posttranscriptional contribution to the trans-repression of Wp-dependent gene expression reported here. The apparent lack of significant inhibition of Wp transcription associated with repression of protein expression would also seem inconsistent with involvement of this regulatory mechanism in the ultimate silencing of Wp during the transition to restricted programs of latency gene transcription. Interestingly, while our manuscript was in revision, Leonard and colleagues reported direct methylation of Wp following infection of germinal center B cells, which appears to be mediated by the de novo DNA methyltransferase DNMT3A, whose expression is induced by EBV infection (31). This supports the existence of a second mechanism of trans-repression distinct from that revealed here and may account for the largely silent Wp in one of the superinfected Sal lines that we analyzed (SI #2).
If trans-repression of Wp-dependent gene expression described here is not a critical component of Wp-to-Cp promoter switching or of the ultimate transcriptional silencing of Wp, then what is its contribution to EBV latency? We propose instead that this may reflect a mechanism that ensures the appropriate level of latency gene expression during the exclusive use of Wp for EBNA and EBNA-dependent gene expression. This would be important, for example, to prevent cytotoxic overexpression of LMP1 (56), whose expression is activated transcriptionally by EBNA2 and EBNA3C (1, 3, 72, 74, 83). Because the respective EBNA mRNAs derived from Cp and Wp differ only in their small 5′ exons—C1 and C2 for Cp and W0 for Wp (see Fig. 8 and 9A) (6, 54, 61)—if this trans-repression is primarily posttranscriptional, then one would expect that Cp may be similarly regulated, though our data suggest that the more prominent effect is on Wp, since the majority of EBNA expression, with the exception of EBNA1 (see below), would appear to be derived from Cp-mediated transcription (i.e., from the superinfecting-virus genomes in Sal BL cells). Further, whereas previous studies have identified EBNA2 and EBNA3C as positive and negative regulators of Cp, respectively (44, 49, 63, 75), Wp is believed to be principally a constitutively active promoter under no such virus-dependent control (prior to activation of Cp). Thus, unlike the case for Cp, this may be the primary mechanism to negatively regulate latency gene expression dependent on Wp.
Additional support for such a role for the trans-repression of Wp-dependent expression may come from our observations with respect to EBNA1. In all of the Sal lines that we have examined, either superinfected with wt rEBV (as in Fig. 6A) or ΔB-S rEBV (data not shown), the relative contributions of endogenous and superinfecting-virus genomes to EBNA1 expression are approximately 1:1, with the total EBNA1 level equivalent to that observed in parental Sal cells. The fact that expression of EBNA1 from the Wp-utilizing endogenous genomes in superinfected Sal BL cells is easily detectable whereas that of the other proteins is not—this is most evident for EBNA-LP and BHRF1 (Fig. 10)—suggests that there are indeed mechanisms at play that monitor and regulate levels of EBV proteins. Moreover, since the EBNA1 and EBNA3 mRNAs are derived from the same transcription unit, in which the EBNA1 coding exon is downstream of those for the EBNA3s, the presence of Sal EBV EBNA1 in the apparent absence of detectable EBNA3A and EBNA3B from the same genomes (EBNA3Cs encoded by the Sal and Akata EBV isolates are indistinguishable by immunoblotting) may be further evidence of a posttranscriptional mechanism of action, possibly one that regulates alternative splicing. Interestingly, this apparent maintenance of specific EBNA1 levels is consistent with an earlier report that the number of EBNA1 molecules per cell is relatively constant (a less than 2-fold variance) within B-cell lines supporting latency III, despite the fact that the EBV genome copy number among these lines varied widely and up to 40-fold (62).
Perhaps the most important questions moving forward are whether this trans-repression is regulated by EBV itself and, if so, what the EBV gene product(s) directly or indirectly responsible is. Our preliminary findings suggest that the BHLF1 locus is not required for the observed downregulation of Wp activity (data not shown), just as it does not appear to be required for the establishment of latency I (Fig. 5). Although EBNA2 and/or full-length EBNA-LP would be obvious candidates, since they are not encoded by the endogenous EBV genomes of Wp-restricted BL lines such as Sal, our current understanding of the functions of these two EBNAs, which are generally known to activate transcription, does not provide an obvious explanation for them in the trans-repression of Wp-dependent protein expression, particularly through a posttranscriptional mode of action. Should this prove to be a posttranscriptional mechanism, certainly one or more of the EBV microRNAs (miRNAs) would be obvious candidates, although it could as likely be mediated by a cellular miRNA(s).
ACKNOWLEDGMENTS
We thank Rebekah Templin and Kristen Yetming for excellent technical assistance, Teru Kanda and Kenzo Takada for their kind gift of Ak-GFP-BAC, John Sixbey for the EBV-negative Mutu cell line, Lindsey Hutt-Fletcher, Shannon Kenney, and Sankar Swaminathan for helpful advice on generation of rEBV, and the Molecular Genetics & DNA Sequencing Core Facility of the Penn State Hershey Cancer Institute.
This work was supported by U.S. Public Health Service grants CA056645 and CA117827 to C.E.S. and CA073544 and AI073215 to J.T.S., by the Penn State Hershey Cancer Institute, and in part under a grant with the Pennsylvania Department of Health using Tobacco Settlement Funds.
C.E.S. receives royalties from the sale of EBNA3 antibodies that were used in this work.
Footnotes
Published ahead of print on 24 August 2011.
REFERENCES
- 1. Abbot S. D., et al. 1990. Epstein-Barr virus nuclear antigen 2 induces expression of the virus-encoded latent membrane protein. J. Virol. 64:2126–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alfieri C., Birkenbach M., Kieff E. 1991. Early events in Epstein-Barr virus infection of human B lymphocytes. Virology 181:595–608 [DOI] [PubMed] [Google Scholar]
- 3. Allday M. J., Crawford D. H., Thomas J. A. 1993. Epstein-Barr virus (EBV) nuclear antigen 6 induces expression of the EBV latent membrane protein and an activated phenotype in Raji cells. J. Gen. Virol. 74:361–369 [DOI] [PubMed] [Google Scholar]
- 4. Babcock G. J., Hochberg D., Thorley-Lawson A. D. 2000. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 13:497–506 [DOI] [PubMed] [Google Scholar]
- 5. Bell A., Skinner J., Kirby H., Rickinson A. 1998. Characterisation of regulatory sequences at the Epstein-Barr virus BamHI W promoter. Virology 252:149–161 [DOI] [PubMed] [Google Scholar]
- 6. Bodescot M., Perricaudet M., Farrell P. J. 1987. A promoter for the highly spliced EBNA family of RNAs of Epstein-Barr virus. J. Virol. 61:3424–3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chau C. M., Zhang X. Y., McMahon S. B., Lieberman P. M. 2006. Regulation of Epstein-Barr virus latency type by the chromatin boundary factor CTCF. J. Virol. 80:5723–5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davenport M. G., Pagano J. S. 1999. Expression of EBNA-1 mRNA is regulated by cell cycle during Epstein-Barr virus type I latency. J. Virol. 73:3154–3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dolan A., Addison C., Gatherer D., Davison A. J., McGeoch D. J. 2006. The genome of Epstein-Barr virus type 2 strain AG876. Virology 350:164–170 [DOI] [PubMed] [Google Scholar]
- 10. Evans T. J., Jacquemin M. G., Farrell P. J. 1995. Efficient EBV superinfection of group I Burkitt's lymphoma cells distinguishes requirements for expression of the Cp viral promoter and can activate the EBV productive cycle. Virology 206:866–877 [DOI] [PubMed] [Google Scholar]
- 11. Finke J., et al. 1987. Monoclonal and polyclonal antibodies against Epstein-Barr virus nuclear antigen 5 (EBNA-5) detect multiple protein species in Burkitt's lymphoma and lymphoblastoid cell lines. J. Virol. 61:3870–3878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gardella T., Medveczky P., Sairenji T., Mulder C. 1984. Detection of circular and linear herpesvirus DNA molecules in mammalian cells by gel electrophoresis. J. Virol. 50:248–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gregory C. D., Rowe M., Rickinson A. B. 1990. Different Epstein-Barr virus-B cell interactions in phenotypically distinct clones of a Burkitt's lymphoma cell line. J. Gen. Virol. 71:1481–1495 [DOI] [PubMed] [Google Scholar]
- 14. Hochberg D., et al. 2004. Demonstration of the Burkitt's lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc. Natl. Acad. Sci. U. S. A. 101:239–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hurley E. A., et al. 1991. When Epstein-Barr virus persistently infects B-cell lines, it frequently integrates. J. Virol. 65:1245–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hutchings I. A., et al. 2006. Methylation status of the Epstein-Barr virus (EBV) BamHI W latent cycle promoter and promoter activity: analysis with novel EBV-positive Burkitt and lymphoblastoid cell lines. J. Virol. 80:10700–10711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jansson A., Masucci M., Rymo L. 1992. Methylation of discrete sites within the enhancer region regulates the activity of the Epstein-Barr virus BamHI W promoter in Burkitt lymphoma lines. J. Virol. 66:62–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jeang K. T., Hayward S. D. 1983. Organization of the Epstein-Barr virus DNA molecule. III. Location of the P3HR-1 deletion junction and characterization of the NotI repeat units that form part of the template for an abundant 12-O-tetradecanoylphorbol-13-acetate-induced mRNA transcript. J. Virol. 48:135–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jin X. W., Speck S. H. 1992. Identification of critical cis elements involved in mediating Epstein-Barr virus nuclear antigen 2-dependent activity of an enhancer located upstream of the viral BamHI C promoter. J. Virol. 66:2846–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kanda T., Yajima M., Ahsan N., Tanaka M., Takada K. 2004. Production of high-titer Epstein-Barr virus recombinants derived from Akata cells by using a bacterial artificial chromosome system. J. Virol. 78:7004–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kang M. S., et al. 2005. Epstein-Barr virus nuclear antigen 1 does not induce lymphoma in transgenic FVB mice. Proc. Natl. Acad. Sci. U. S. A. 102:820–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kang M. S., Soni V., Bronson R., Kieff E. 2008. Epstein-Barr virus nuclear antigen 1 does not cause lymphoma in C57BL/6J mice. J. Virol. 82:4180–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kelly G., Bell A., Rickinson A. 2002. Epstein-Barr virus-associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat. Med. 8:1098–1104 [DOI] [PubMed] [Google Scholar]
- 24. Kelly G. L., et al. 2009. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in Burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS Pathog. 5(3):e1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kelly G. L., et al. 2005. Epstein-Barr virus nuclear antigen 2 (EBNA2) gene deletion is consistently linked with EBNA3A, -3B, and -3C expression in Burkitt's lymphoma cells and with increased resistance to apoptosis. J. Virol. 79:10709–10717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kieff E. D., Rickinson A. B. 2006. Epstein-Barr virus and its replication, p. 2603–2654 In Knipe D. M., Howley P. M., Griffin D. E., Lamb R. A., Martin M. A., Roizman B., Strauss S. E.(ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 27. Kirby H., Rickinson A., Bell A. 2000. The activity of the Epstein-Barr virus BamHI W promoter in B cells is dependent on the binding of CREB/ATF factors. J. Gen. Virol. 81:1057–1066 [DOI] [PubMed] [Google Scholar]
- 28. Komano J., Sugiura M., Takada K. 1998. Epstein-Barr virus contributes to the malignant phenotype and to apoptosis resistance in Burkitt's lymphoma cell line Akata. J. Virol. 72:9150–9156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Laux G., Freese U. K., Bornkamm G. W. 1985. Structure and evolution of two related transcription units of Epstein-Barr virus carrying small tandem repeats. J. Virol. 56:987–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lear A. L., et al. 1992. The Epstein-Barr virus (EBV) nuclear antigen 1 BamHI F promoter is activated on entry of EBV-transformed B cells into the lytic cycle. J. Virol. 66:7461–7468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leonard S., et al. 13 July 2011. Epigenetic and transcriptional changes which follow Epstein-Barr virus infection of germinal centre B cells and their relevance to the pathogenesis of Hodgkin's lymphoma. J. Virol. doi:10.1128/JVI.00468-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leung C. S., Haigh T. A., Mackay L. K., Rickinson A. B., Taylor G. S. 2010. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc. Natl. Acad. Sci. U. S. A. 107:2165–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Levitskaya J., et al. 1995. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature 375:685–688 [DOI] [PubMed] [Google Scholar]
- 34. Levitskaya J., Sharipo A., Leonchiks A., Ciechanover A., Masucci M. G. 1997. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. U. S. A. 94:12616–12621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lieberman P. M., Hardwick J. M., Hayward S. D. 1989. Responsiveness of the Epstein-Barr virus NotI repeat promoter to the Z transactivator is mediated in a cell-type-specific manner by two independent signal regions. J. Virol. 63:3040–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mann K. P., Staunton D., Thorley-Lawson D. A. 1985. Epstein-Barr virus-encoded protein found in plasma membranes of transformed cells. J. Virol. 55:710–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Masucci M. G., et al. 1989. 5-Azacytidine up regulates the expression of Epstein-Barr virus nuclear antigen 2 (EBNA-2) through EBNA-6 and latent membrane protein in the Burkitt's lymphoma line Rael. J. Virol. 63:3135–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nonkwelo C., Skinner J., Bell A., Rickinson A., Sample J. 1996. Transcription start sites downstream of the Epstein-Barr virus (EBV) Fp promoter in early-passage Burkitt lymphoma cells define a fourth promoter for expression of the EBV EBNA-1 protein. J. Virol. 70:623–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nuebling C. M., Mueller-Lantzsch N. 1989. Identification and characterization of an Epstein-Barr virus early antigen that is encoded by the NotI repeats. J. Virol. 63:4609–4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Paulson E. J., Speck S. H. 1999. Differential methylation of Epstein-Barr virus latency promoters facilitates viral persistence in healthy seropositive individuals. J. Virol. 73:9959–9968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pearson G. R., et al. 1987. Identification of an Epstein-Barr virus early gene encoding a second component of the restricted early antigen complex. Virology 160:151–161 [DOI] [PubMed] [Google Scholar]
- 42. Puglielli M. T., Desai N., Speck S. H. 1997. Regulation of EBNA gene transcription in lymphoblastoid cell lines: characterization of sequences downstream of BCR2 (Cp). J. Virol. 71:120–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Puglielli M. T., Woisetschlaeger M., Speck S. H. 1996. oriP is essential for EBNA gene promoter activity in Epstein-Barr virus-immortalized lymphoblastoid cell lines. J. Virol. 70:5758–5768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Radkov S. A., et al. 1997. Epstein-Barr virus EBNA3C represses Cp, the major promoter for EBNA expression, but has no effect on the promoter of the cell gene CD21. J. Virol. 71:8552–8562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Robertson K. D., Ambinder R. F. 1997. Mapping promoter regions that are hypersensitive to methylation-mediated inhibition of transcription: application of the methylation cassette assay to the Epstein-Barr virus major latency promoter. J. Virol. 71:6445–6454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Robertson K. D., Ambinder R. F. 1997. Methylation of the Epstein-Barr virus genome in normal lymphocytes. Blood 90:4480–4484 [PubMed] [Google Scholar]
- 47. Robertson K. D., Hayward S. D., Ling P. D., Samid D., Ambinder R. F. 1995. Transcriptional activation of the Epstein-Barr virus latency C promoter after 5-azacytidine treatment: evidence that demethylation at a single CpG site is crucial. Mol. Cell. Biol. 15:6150–6159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rogers R. P., Woisetschlaeger M., Speck S. H. 1990. Alternative splicing dictates translational start in Epstein-Barr virus transcripts. EMBO J. 9:2273–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rooney C. M., et al. 1992. Host cell and EBNA-2 regulation of Epstein-Barr virus latent-cycle promoter activity in B lymphocytes. J. Virol. 66:496–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ruf I. K., et al. 1999. Epstein-Barr virus regulates c-MYC, apoptosis, and tumorigenicity in Burkitt lymphoma. Mol. Cell. Biol. 19:1651–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ruf I. K., Sample J. 1999. Repression of Epstein-Barr virus EBNA-1 gene transcription by pRb during restricted latency. J. Virol. 73:7943–7951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Salamon D., et al. 2009. Binding of CCCTC-binding factor in vivo to the region located between Rep* and the C promoter of Epstein-Barr virus is unaffected by CpG methylation and does not correlate with Cp activity. J. Gen. Virol. 90:1183–1189 [DOI] [PubMed] [Google Scholar]
- 53. Sample J., Henson E. B., Sample C. 1992. The Epstein-Barr virus nuclear protein 1 promoter active in type I latency is autoregulated. J. Virol. 66:4654–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sample J., Hummel M., Braun D., Birkenbach M., Kieff E. 1986. Nucleotide sequences of mRNAs encoding Epstein-Barr virus nuclear proteins: a probable transcriptional initiation site. Proc. Natl. Acad. Sci. U. S. A. 83:5096–5100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sample J., Kieff E. 1990. Transcription of the Epstein-Barr virus genome during latency in growth-transformed lymphocytes. J. Virol. 64:1667–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sandberg M. L., Kaykas A., Sugden B. 2000. Latent membrane protein 1 of Epstein-Barr virus inhibits as well as stimulates gene expression. J. Virol. 74:9755–9761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schaefer B. C., Strominger J. L., Speck S. H. 1995. The Epstein-Barr virus BamHI F promoter is an early lytic promoter: lack of correlation with EBNA 1 gene transcription in group 1 Burkitt's lymphoma cell lines. J. Virol. 69:5039–5047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schaefer B. C., Strominger J. L., Speck S. H. 1997. Host-cell-determined methylation of specific Epstein-Barr virus promoters regulates the choice between distinct viral latency programs. Mol. Cell. Biol. 17:364–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schaefer B. C., Strominger J. L., Speck S. H. 1995. Redefining the Epstein-Barr virus-encoded nuclear antigen EBNA-1 gene promoter and transcription initiation site in group I Burkitt lymphoma cell lines. Proc. Natl. Acad. Sci. U. S. A. 92:10565–10569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schlager S., Speck S. H., Woisetschlager M. 1996. Transcription of the Epstein-Barr virus nuclear antigen 1 (EBNA1) gene occurs before induction of the BCR2 (Cp) EBNA gene promoter during the initial stages of infection in B cells. J. Virol. 70:3561–3570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Speck S. H., Pfitzner A., Strominger J. L. 1986. An Epstein-Barr virus transcript from a latently infected, growth-transformed B-cell line encodes a highly repetitive polypeptide. Proc. Natl. Acad. Sci. U. S. A. 83:9298–9302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sternas L., Middleton T., Sugden B. 1990. The average number of molecules of Epstein-Barr nuclear antigen 1 per cell does not correlate with the average number of Epstein-Barr virus (EBV) DNA molecules per cell among different clones of EBV-immortalized cells. J. Virol. 64:2407–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sung N. S., Kenney S., Gutsch D., Pagano J. S. 1991. EBNA-2 transactivates a lymphoid-specific enhancer in the BamHI C promoter of Epstein-Barr virus. J. Virol. 65:2164–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sung N. S., Wilson J., Davenport M., Sista N. D., Pagano J. S. 1994. Reciprocal regulation of the Epstein-Barr virus BamHI-F promoter by EBNA-1 and an E2F transcription factor. Mol. Cell. Biol. 14:7144–7152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tellam J., et al. 2007. Translation efficiency of EBNA1 encoded by lymphocryptoviruses influences endogenous presentation of CD8+ T cell epitopes. Eur. J. Immunol. 37:328–337 [DOI] [PubMed] [Google Scholar]
- 66. Tempera I., Wiedmer A., Dheekollu J., Lieberman P. M. 2010. CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog. 6(8):e1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Thorley-Lawson D. A. 2001. Epstein-Barr virus: exploiting the immune system. Nat. Rev. Immunol. 1:75–82 [DOI] [PubMed] [Google Scholar]
- 68. Tierney R., Kirby H., Nagra J., Rickinson A., Bell A. 2000. The Epstein-Barr virus promoter initiating B-cell transformation is activated by RFX proteins and the B-cell-specific activator protein BSAP/Pax5. J. Virol. 74:10458–10467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tierney R., et al. 2007. Epstein-Barr virus exploits BSAP/Pax5 to achieve the B-cell specificity of its growth-transforming program. J. Virol. 81:10092–10100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tierney R. J., et al. 2000. Methylation of transcription factor binding sites in the Epstein-Barr virus latent cycle promoter Wp coincides with promoter down-regulation during virus-induced B-cell transformation. J. Virol. 74:10468–10479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Trivedi P., et al. 2001. Differential regulation of Epstein-Barr virus (EBV) latent gene expression in Burkitt lymphoma cells infected with a recombinant EBV strain. J. Virol. 75:4929–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tsang S. F., Wang F., Izumi K. M., Kieff E. 1991. Delineation of the cis-acting element mediating EBNA-2 transactivation of latent infection membrane protein expression. J. Virol. 65:6765–6771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang D., Liebowitz D., Kieff E. 1985. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell 43:831–840 [DOI] [PubMed] [Google Scholar]
- 74. Wang F., Tsang S. F., Kurilla M. G., Cohen J. I., Kieff E. 1990. Epstein-Barr virus nuclear antigen 2 transactivates latent membrane protein LMP1. J. Virol. 64:3407–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Woisetschlaeger M., et al. 1991. Role for the Epstein-Barr virus nuclear antigen 2 in viral promoter switching during initial stages of infection. Proc. Natl. Acad. Sci. U. S. A. 88:3942–3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Woisetschlaeger M., Yandava C. N., Furmanski L. A., Strominger J. L., Speck S. H. 1990. Promoter switching in Epstein-Barr virus during the initial stages of infection of B lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 87:1725–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Xue S. A., Griffin B. E. 2007. Complexities associated with expression of Epstein-Barr virus (EBV) lytic origins of DNA replication. Nucleic Acids Res. 35:3391–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yin Y., Manoury B., Fahraeus R. 2003. Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus-encoded EBNA1. Science 301:1371–1374 [DOI] [PubMed] [Google Scholar]
- 79. Yoo L., Speck S. H. 2000. Determining the role of the Epstein-Barr virus Cp EBNA2-dependent enhancer during the establishment of latency by using mutant and wild-type viruses recovered from cottontop marmoset lymphoblastoid cell lines. J. Virol. 74:11115–11120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yoo L. I., Mooney M., Puglielli M. T., Speck S. H. 1997. B-cell lines immortalized with an Epstein-Barr virus mutant lacking the Cp EBNA2 enhancer are biased toward utilization of the oriP-proximal EBNA gene promoter Wp1. J. Virol. 71:9134–9142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yoshioka M., Crum M. M., Sample J. T. 2008. Autorepression of Epstein-Barr virus nuclear antigen 1 expression by inhibition of pre-mRNA processing. J. Virol. 82:1679–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Young L., et al. 1989. Expression of Epstein-Barr virus transformation-associated genes in tissues of patients with EBV lymphoproliferative disease. N. Engl. J. Med. 321:1080–1085 [DOI] [PubMed] [Google Scholar]
- 83. Zhao B., Sample C. E. 2000. Epstein-Barr virus nuclear antigen 3C activates the latent membrane protein 1 promoter in the presence of Epstein-Barr virus nuclear antigen 2 through sequences encompassing an spi-1/spi-B binding site. J. Virol. 74:5151–5160 [DOI] [PMC free article] [PubMed] [Google Scholar]