

Abstract
Hepatitis C virus (HCV)-infected patients undergoing liver transplantation universally experience rapid reinfection of their new liver graft. Current treatment protocols do not prevent graft reinfection and, in addition, an accelerated disease progression is observed. In the present study, we have evaluated a novel strategy to prevent HCV infection using a lectin, griffithsin (GRFT) that specifically binds N-linked high-mannose oligosaccharides that are present on the viral envelope. The antiviral effect of GRFT was evaluated in vitro using the HCV pseudoparticle (HCVpp) and HCV cell culture (HCVcc) systems. We show here that preincubation of HCVpp and HCVcc with GRFT prevents infection of Huh-7 hepatoma cells. Furthermore, GRFT interferes with direct cell-to-cell transmission of HCV. GRFT acts at an early phase of the viral life cycle by interfering in a genotype-independent fashion with the interaction between the viral envelope proteins and the viral receptor CD81. The capacity of GRFT to prevent infection in vivo was evaluated using uPA+/+-SCID mice (uPA stands for urokinase-type plasminogen activator) that harbor human primary hepatocytes in their liver (chimeric mice). In this proof-of-concept trial, we demonstrated that GRFT can mitigate HCV infection of chimeric mice. Treated animals that did become infected demonstrated a considerable delay in the kinetics of the viral infection. Our data demonstrate that GRFT can prevent HCV infection in vitro and mitigate HCV infection in vivo. GRFT treatment of chronically infected HCV patients undergoing liver transplantation may be a suitable strategy to prevent infection of the liver allograft.
INTRODUCTION
Worldwide, about 170 million people are estimated to be chronically infected with the hepatitis C virus (HCV), and about one third of them are at risk of developing liver fibrosis, cirrhosis, and hepatocellular carcinoma in the coming decades (7). These long-term sequelae of chronic HCV infection represent today's leading indication for liver transplantation in the Western world. Following transplantation, the donor liver immediately becomes reinfected by the circulating virus, causing an infection that rapidly progresses toward fibrosis and cirrhosis (50). The current standard therapy, a combination of pegylated interferon and ribavirin, is successful in only 50 to 75% of patients, depending on the genotype of the infecting virus. The efficacy of this combination therapy in a liver transplantation setting is however much lower and can induce considerable complications (4, 51). Although more-potent combination therapies have recently been approved for treatment of patients chronically infected with HCV (40), their use in liver transplant patients will probably be complicated by possible drug-drug interactions (12). Therefore, new strategies to prevent infection are urgently needed, not least because a protective vaccine has not yet been developed (25).
Different strategies for the prevention of HCV infection have already been evaluated. First, HCV entry can be inhibited in vitro and in vivo by targeting the virus with monoclonal or polyclonal neutralizing antibodies (2, 24, 48). Although neutralizing antibodies appear to be very potent in neutralizing viral strains of different genotypes in vitro (15, 33, 43), their in vivo efficacy turns out to be much lower (29, 44). The discrepancy between in vitro and in vivo studies may be related to the different characteristics of naturally produced viral particles that are highly associated with lipoproteins, unlike HCV pseudoparticles (HCVpp) and cell culture-produced HCV (HCVcc) (27, 42). In addition, HCV can spread efficiently from one infected cell to a neighboring one (47, 52). Importantly, this alternative transmission route is resistant to neutralizing antibodies (47).
To overcome the high variability of the viral envelope proteins, the well-conserved (co)receptors may be a more promising target for inhibition of HCV entry. Blockade of CD81, scavenger receptor B1 (SR-B1), and claudin 1 with monoclonal antibodies or small molecules has been shown to be a very efficacious way to prevent HCV infection in a genotype-independent manner (11, 30, 32a, 45). However, the interactions of blocking agents with viral receptors could interfere with the natural function of these host proteins and induce unwanted side effects. Another way of preventing HCV infection is to target the HCV envelope proteins with small compounds, as has recently been described (1). However, such molecules seem to be genotype specific. An alternative way to block the interaction between the virus and its receptors takes advantage of the highly glycosylated nature of the viral envelope proteins E1 and E2. These glycosylations, up to 5 in E1 and 11 in E2, are located at specific sites that are conserved in the different genotypes and are involved in protein folding, HCV entry, and protection of the virus from antibody-dependent neutralization (5, 9, 13, 18, 19). We have previously shown that the lectin cyanovirin N binds to the glycans present on the viral particle and thereby inhibits HCV entry by blocking the interaction between E2 and CD81 (20). We have now evaluated the anti-HCV effect of another lectin, griffithsin (GRFT), using the HCVpp and HCVcc systems, and a small animal model for the study of HCV (28, 31). GRFT is a homodimeric protein composed of two 121-amino-acid (12.5-kDa) monomers, each containing three identical carbohydrate-binding sites that tightly interact with the terminal mannose residues present on N-linked high-mannose oligosaccharides (53, 54). GRFT was originally isolated from the red alga Griffithsia sp. and displays picomolar and nanomolar range activity against HIV-1 and severe acute respiratory syndrome (SARS) coronavirus, respectively (34, 37). We show here that not only can GRFT efficiently prevent HCV infection in cell culture in a genotype-independent manner but that it also interferes with the direct cell-to-cell transmission of HCV. GRFT interacts with the glycans present on the viral envelope proteins, thereby preventing the attachment of the virus to its receptor CD81. Our data were successfully validated in vivo in chimeric mice with a humanized liver, indicating that GRFT may be a novel molecule for the prevention of graft reinfection in HCV-infected liver transplant patients.
MATERIALS AND METHODS
Reagents and cell culture.
Recombinant griffithsin (GRFT) was produced in Nicotiana benthamiana plants as described previously (38). A synthetic cDNA encoding a lectin activity-deficient mutant of GRFT, termed GRFTMUT (MUT stands for mutant), was designed with a conservative amino acid substitution of aspartic acid to asparagine in each of the 3 carbohydrate-binding pockets identified in the primary amino acid sequence and crystal structures of GRFT (23, 34, 53). The nonmutated GRFT with full lectin activity is termed GRFTWT (WT stands for wild type) to distinguish it from the lectin activity-defective GRFTMUT. GRFTMUT was expressed in N. benthamiana and purified exactly as previously described for GRFTWT (38). Proteins were purified to >99% purity and formulated in phosphate-buffered saline (PBS) (pH 7.4) at 10-mg/ml protein concentration. Endotoxin was removed from protein samples using Detoxi-Gel endotoxin-removing gel gravity flow columns (Thermo Scientific). Endotoxin levels were measured using the ToxinSensor chromogenic Limulus amebocyte lysate (LAL) endotoxin assay kit from GenScript (Piscataway, NJ). A polyclonal rabbit anti-GRFTWT antiserum was produced in New Zealand White rabbits by a commercial vendor (Antibodies Inc., Davis, CA). Animals were immunized with antigen emulsified in RIBI adjuvant system.
HEK293T human embryonic kidney cells and Huh-7 hepatoma cells (36) were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum (FCS), 100 U/ml penicillin, and 100 μg/ml streptomycin. Monoclonal antibodies against E1 (clone A4) (8) and E2 (clone 3/11; kindly provided by J. A. McKeating, University of Birmingham, United Kingdom) (10) were produced in vitro in a MiniPerm bioreactor (Heraeus) according to the manufacturer's recommendations. The oligosaccharide mannonanose-di-(N-acetyl-d-glucosamine) (Man-9) was purchased from Sigma-Aldrich (St. Louis, MO). Possible in vitro toxicity of GRFT on Huh-7 cells was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (Promega, Charbonieres, France).
Production of HCVpp and viral entry assay.
HCVpp were produced essentially as described before (39). Briefly, 293T cells were cotransfected with the plasmids MLV Gag-Pol (MLV stands for murine leukemia virus), MLV-FLuc (FLuc stands for firefly luciferase), and pcDNA3.1 containing the E1E2 region of one of the following strains: H77 (gt1a; GenBank accession number AAB67037), UKN1B-5.23 (gt1b; GenBank accession number AY734976), JFH1 (gt2a; GenBank accession number AB047639), UKN2B-1.1 (gt2b; GenBank accession number AY734982), UKN3A-1.28 (gt3a; GenBank accession number AY734984), UKN4-11.1 (gt4; GenBank accession number AY734986), and UKN6-5.340 (gt6; GenBank accession number AY736194) kindly provided by J. K. Ball (Nottingham University, United Kingdom) and R. Bartenschlager (University of Heidelberg, Germany). Culture supernatants containing pseudoparticles were harvested 48 h later and filtered through 0.45-μm-pore-size membranes. To evaluate HCVpp infectivity, Huh-7 cells were seeded 1 day before infection at a density of 3 × 104 cells/well in a 24-well plate. HCVpp solutions were preincubated with or without GRFT for 1 h at 37°C and then transferred to the Huh-7 culture plate. Four hours later, the medium was replaced. Two days after infection, firefly luciferase reporter gene activity was evaluated with a commercial kit according to the protocol provided by the manufacturer (Promega). All experiments were performed three times with all conditions run in duplicate.
Production of HCVcc and infection assay.
Huh-7 cells were electroporated with in vitro-transcribed RNA of JFH1 engineered to express the A4 epitope with or without Renilla luciferase (RLuc), or with RNA of H77/JFH1-RLuc (unpublished data). Three versions of JFH1 were used: one that contained two mutations in the capsid (JFH1-CS-A4 [JFH1]), a second one with the same mutations but also expressing RLuc (JFH1-CS-A4-RLuc), and a third variant containing an additional mutation in E2 resulting in the loss of glycosylation at position N6 (JFH1-CS-A4-N6-RLuc [JFH1ΔN6]). These mutations result in a more robust expansion and higher infectious titers of the virus in cell culture (6, 16). HCVcc was collected over a 1-week period, filtered (0.22-μm-pore-size filter), divided into single-use aliquots, and stored at −80°C until further use. One day after seeding (3 × 104 cells/24-well plate), Huh-7 cells were incubated with an HCVcc solution that was preincubated for 1 h at 37°C with different concentrations of GRFT. After 4 h, virus was washed away, and fresh medium was added. In some cases, Huh-7 cells were pretreated with GRFT (1 μg/ml), or GRFT (1 μg/ml) was added 4 h after infection when the medium was replaced. Two days after infection, intracellular Renilla luciferase activity was measured with a commercial kit according to the instructions provided by the manufacturer (Promega). JFH1 infections were performed at a multiplicity of infection (MOI) of 0.0033 to avoid saturation of the RLuc signal. To evaluate the impact of GRFT treatment on the extracellular infectivity, supernatants from Huh-7 cell cultures treated with different concentrations of GRFTWT or GRFTMUT were collected 2 days after infection, serially diluted, and added to naïve Huh-7 cells that were grown in 96-well plates seeded with 7 × 103 cells/well. The HCVcc infectivity of the supernatants was expressed as 50% tissue culture infectious units per milliliter (TCID50/ml) and was calculated as described before (26).
Direct cell-to-cell transmission assay.
One day before infection, Huh-7 cells were seeded at 5 × 104 cells in a 24-well plate containing microscopy coverslips. The next day, the cultures were infected with JFH1-CS-A4 HCVcc (see above) at an MOI of 0.025. After 2 h of incubation, the virus was washed away, and new medium containing 10% FCS, 1% agarose, and GRFTMUT or GRFTWT was added. The effect of GRFTWT and GRFTMUT was evaluated at two concentrations, 2 and 10 μg/ml (final concentrations). After 72 h, the cells were fixed with 8% paraformaldehyde (PFA), and infected cells were visualized via immunofluorescence microscopy using a monoclonal anti-E1 antibody (epitope A4). For each condition, the mean number of HCV-positive cells/HCV-positive focus was determined in 17 separate foci. All conditions were tested in duplicate. A similar experiment was conducted without agarose overlay.
In a second experimental setup, we seeded 1.6 × 104 Huh-7.5 cells in a 96-well imaging plate (BD Biosciences, Erembodegem, Belgium). One day later, 2.50 × 102 Jc1-infected Huh-7.5-RFP-NLS-IPS cells were added per well (22). Cultures were treated with 150 μg/ml of a monoclonal neutralizing antibody (nAb) to prevent cell-free infection, with 10 μg/ml of GRFTWT, or with a combination of GRFTWT with the neutralizing antibody. All conditions were performed in duplicate. Two days after coculture, the cells were fixed with PFA, and HCV-infected acceptor cells were visualized with the anti-NS5A monoclonal antibody 9E10 in combination with an Alexa Fluor 647-conjugated goat anti-mouse antibody (Invitrogen). HCV-infected acceptor cells (Alexa Fluor 647 positive, red fluorescent protein [RFP] negative) were counted in at least 100 separate clusters (at least 50 foci/well). Images were acquired using a BD Pathway 435 high-content bioimager (BD Biosciences) with a 10× objective.
Coimmunoprecipitation assay.
After preincubation with different concentrations of GRFTWT or GRFTMUT, genotype 1a HCVpp (strain H77) were lysed with Triton X-100 (final concentration of 1%), and rabbit polyclonal antibodies against GRFT were added. HCVpp-GRFT complexes were then precipitated with protein G-Sepharose beads (Pharmacia). To avoid nonspecific binding of HCVpp, protein G-Sepharose beads were preincubated with pseudoviral particles containing the envelope proteins of vesicular stomatitis virus (VSVpp). After the proteins were separated by 10% SDS-PAGE, they were transferred to nitrocellulose membranes (Hybond-ECL; Amersham), and HCV E1 and E2 were visualized using monoclonal anti-E1 (A4) and anti-E2 (3/11) antibodies.
CD81 pulldown assay.
The interaction between HCV E1E2 and CD81 was investigated with a CD81 pulldown assay. Briefly, HCVpp (strain H77 and JFH1) were incubated with different concentrations of GRFTWT (0, 0.1, 1, and 10 μg/ml) or GRFTMUT (10 μg/ml). After 2 h of incubation at 4°C, Triton X-100 was added to achieve a final concentration of 1%. This solution was added to a suspension of glutathione Sepharose beads (Pharmacia) that had been preincubated with a fusion protein consisting of the large extracellular loop of CD81 (CD81-LEL) and glutathione S-transferase. After overnight incubation at 4°C, complexes were isolated by centrifugation and separated on a 10% SDS-polyacrylamide gel. E1 and E2 proteins were visualized by Western blotting using antibodies A4 and 3/11, respectively, as described above.
Chimeric mice.
Chimeric uPA+/+-SCID mice (uPA stands for urokinase-type plasminogen activator, and SCID stands for severe combined immune deficient) were produced as described before (49). Briefly, homozygous uPA-SCID mice (32) were anesthetized with isoflurane and approximately one million cryopreserved human primary hepatocytes were transplanted into their spleens (donor HH223; BD Biosciences, Erembodegem, Belgium). To evaluate successful engraftment in the mouse liver, the amount of human albumin present in mouse plasma was quantified by enzyme-linked immunosorbent assay (ELISA) (Bethyl Laboratories, Montgomery, TX). All animals used in this study had human albumin plasma levels of at least 2.1 mg/ml. GRFTWT or GRFTMUT was administered daily via subcutaneous injection at a dose of 5 mg/kg of body weight. The GRFT treatment was started 3 days before the mice were challenged with 105 IU of H77 virus (3) and was continued until day 2 (2 mice), day 7 (2 mice), or day 14 (2 mice) postinfection. The animals were bled at weekly intervals, and HCV RNA was quantified using the COBAS Ampliprep/COBAS TaqMan HCV test (Roche Diagnostics, Vilvoorde, Belgium). All animal experiments were approved by the Animal Ethics Committee of Ghent University.
Graphs and statistics.
All graphs were prepared using Prism v5.0c (GraphPad Software Inc., La Jolla, CA). Fifty percent inhibitory concentration (IC50) and 90% inhibitory concentration (IC90) values were calculated via nonlinear regression analysis using a variable-slope log (dose)-versus-response curve with a least-squares (ordinary) fit.
To analyze whether the difference between treatment groups was statistically significant, the data obtained from in vitro and in vivo experiments were analyzed using the unpaired nonparametric two-tailed Mann-Whitney test. Data were analyzed using GraphPad InStat v3.0b (GraphPad Software Inc.).
RESULTS
GRFTWT inhibits HCVcc infection.
To investigate the capacity of GRFTWT to inhibit the infection of hepatocytes by HCV, cell culture-derived JFH1 luciferase reporter virus (HCVcc) was mixed with increasing concentrations of griffithsin (GRFT), and 1 h later, this HCVcc-GRFT mixture was transferred to Huh-7 target cells. After 2 days, the intracellular Renilla luciferase signal was measured, and a clear, dose-dependent reduction in HCVcc infectivity was observed (Fig. 1A). GRFTWT efficiently inhibits JFH1 HCVcc infection with a 50% inhibitory concentration (IC50) of 181 ng/ml (13.9 nM) and a 90% inhibitory concentration (IC90) of 644 ng/ml (49.5 nM) (Fig. 1D). Comparable antiviral activity was observed against the JFH1ΔN6 reporter virus (IC50 of 89.9 ng/ml or 6.9 nM and IC90 of 1.45 μg/ml or 112 nM) lacking glycosylation at position N-6 (Fig. 1B and D). No antiviral effect was seen when GRFT with lectin activity eliminated by mutating all six carbohydrate-binding sites (GRFTMUT) was used. We did not observe any in vitro toxicity at concentrations up to 450 μg/ml (34.6 μM), leading to a therapeutic index exceeding 2,486 (data not shown).
Fig. 1.

Prevention of Huh-7 cell infection. JFH1 (A), JFH1ΔN6 (B), and H77/JFH1 (C) hepatitis C virus cell culture (HCVcc) were preincubated with different concentrations (μg/ml) of GRFTWT or GRFTMUT and then transferred to Huh-7 cells. Two days later, intracellular Renilla luciferase signal was quantified and normalized to the GRFTMUT-treated group. (D) Fifty percent inhibitory concentration (IC50) and 90% inhibitory concentration (IC90) values were calculated via nonlinear regression analysis using a variable-slope log dose-versus-response curve with a least-squares (ordinary) fit. All conditions were tested in duplicate, and error bars represent the standard errors of the means. The 10 μg/ml concentration of griffithsin (GRFT) corresponds to 769 nM.
To evaluate whether GRFT treatment reduces extracellular infectivity, Huh-7 cells were exposed for 4 h to mixtures of JFH1 HCVcc that were pretreated with different concentrations of GRFT. After 2 days of culture, the amount of secreted infectious particles present in the supernatant was determined by limiting dilution assay as described before (26). As shown in Table 1, a dose-dependent reduction in extracellular infectivity could be observed, with a 2-log10-fold reduction in 50% tissue culture infectious dose (TCID50) at 10 μg/ml (770 nM) GRFTWT.
Table 1.
Extracellular infectivity of Huh-7 cultures 2 days after infection with JFH1 HCVcc
| Treatmenta | Extracellular infectivity (TCID50/ml) |
|---|---|
| GRFTMUT | |
| 10 μg/ml | 1.41 × 104 |
| GRFTWT | |
| 0.01 μg/ml | 7.70 × 103 |
| 0.1 μg/ml | 1.33 × 103 |
| 1.0 μg/ml | 1.05 × 102 |
| 10 μg/ml | 1.41 × 102 |
The 10 μg/ml concentration of GRFT corresponds to 769 nM.
GRFTWT interferes with direct cell-to-cell transmission.
To investigate whether GRFT can block direct cell-to-cell transmission of HCV, we investigated the spread of the virus in an agarose overlay cell culture experiment. Huh-7 hepatoma cells were infected with JFH1 HCVcc at a low multiplicity of infection (0.025), and the culture was overlaid with agarose-containing medium to prevent cell-free infection. GRFTWT or GRFTMUT was added to the medium at a final concentration of 2 or 10 μg/ml. Three days later, infected cells were visualized via immunofluorescence microscopy, and the number of infected cells in 17 HCV-positive foci was counted. As shown in Fig. 2, a clear reduction of the average number of infected cells per focus was observed when GRFTWT was added to the medium, indicating that GRFTWT inhibited direct cell-to-cell transmission of the virus. The average number of infected cells per focus in GRFTMUT-treated cultures was comparable to that in nontreated control cultures, indicating that GRFTMUT had no impact on cell-to-cell transmission. The difference between GRFTWT- and GRFTMUT-treated cultures was highly significant (P < 0.0001). It has to be noted that we obtained similar results in the absence of an agar overlay (data not shown).
Fig. 2.

GRFT interferes with direct cell-to-cell spread of HCV in vitro. (A) The effect of GRFT on direct cell-to-cell transmission of HCV was investigated by incubating HCV-infected Huh-7 cells with different concentrations (in micrograms per milliliter) of GRFTWT or GRFTMUT in agarose-containing medium. At 72 h postinfection, the cultures were immunostained with an anti-E1 antibody, and the number of HCV-positive cells per focus was enumerated in 17 foci. The experiments were performed two times. Each symbol represents the value for an individual focus, and the short horizontal lines indicate the average number of cells per infected focus. (B) Representative pictures of HCV-infected agarose overlay cultures treated with 10 μg/ml GRFTMUT (top panel) or GRFTWT (bottom panel), indicating that GRFTWT interferes with direct cell-to-cell transmission of HCV. HCV-infected cells are visualized via immunofluorescence microscopy using an E1-specific antibody. (C) Cocultures of Huh-7.5 target cells with Jc1-infected Huh-7.5-RFP-NLS-IPS were treated with an anti-HCV neutralizing antibody (nAb; 150 μg/ml) or with 10 μg/ml GRFTWT alone or in combination with the neutralizing antibodies. More than 100 foci were analyzed per condition. (D) Representative images taken from a nAb-treated coculture (top panel) showing a focus of infected target cells (green cytoplasm and blue nucleus) that surround an infected donor cell (green cytoplasm and pink nucleus). In cocultures treated with GRFTWT alone (bottom panel), the infection remains restricted to the donor cells (green cytoplasm and pink nucleus). The 10 μg/ml concentration of GRFT corresponds to 769 nM.
In an alternative approach, we cocultured naïve Huh-7.5 cells with Jc1-infected Huh-7.5-RFP-NLS-IPS cells. The latter cells are stably transduced to express a red fluorescent dye in the cytoplasm that translocates to the nucleus upon HCV infection (22). This approach allows us to easily discriminate between donor and acceptor cells and rules out the possibility that clusters of infected cells are derived from dividing donor cells rather than from spread of the virus. In cocultures that were treated with 150 μg/ml of a neutralizing antibody, to prevent cell-free infection, we identified HCV-infected clusters consisting of up to 50 acceptor cells (Fig. 2C and D). On average, 7.8 infected acceptor cells were present per focus. Cultures that were treated with GRFT alone or GRFT combined with neutralizing antibodies had on average 0.2 infected acceptor cell per cluster with a maximal amount of 4 cells/cluster (Fig. 2C and D). The difference between GRFT-treated and antibody-treated cultures was highly significant (P < 0.0001) and indicates that GRFT can prevent direct cell-to-cell transmission of HCV, while neutralizing antibodies that target the virus do not.
Mode of action of the inhibitory effect of GRFT.
GRFTWT inhibits HCVcc infection by affecting an early step of the viral life cycle. Indeed, while a 1-h preincubation of the viral particles with GRFT efficiently inhibited viral infection in cell culture, the addition of GRFT 4 h postinfection, when the virus is washed away, had no effect (Fig. 3A). This indicates that GRFT is acting exclusively on the viral entry phase. Furthermore, GRFT pretreatment of the Huh-7 cells had no effect on HCV infectivity (Fig. 3A). Although we have evidence that GRFT, like cyanovirin N (CV-N), binds glycan structures on the surfaces of epithelial cells and peripheral blood mononuclear cells (PBMC) (23), these results suggest that the anti-HCV activity of GRFT is via a direct interaction with the viral particles and not with the target cells.
Fig. 3.

GRFT inhibits HCV infection at an early step in the viral life cycle. (A) Huh-7 cells were infected with untreated HCVcc (JFH1ΔN6) (a) or with HCVcc that was preincubated with 1 μg/ml of GRFTWT (b). Treatment of the Huh-7 cells with GRFTWT (1 μg/ml) before infection (c) or 4 h after infection (d) did not have any effect. (B) The antiviral effect of GRFTWT could be circumvented by coincubation of the lectin mannonanose-di-(N-acetyl-d-glucosamine) (Man-9). GRFTWT (1 μg/ml) was preincubated for 1 h with Man-9 (0, 1, or 10 μg/ml) before JFH1ΔN6 HCVcc was added. After 1 h, the GRFT-Man9-HCVcc mixture was added to Huh-7 cells. Four hours later, the medium was replaced, and intracellular Renilla luciferase (RLuc) signal was measured 2 days later. While Man-9 pretreatment reversed the inhibitory action of GRFT, Man-9 pretreatment of the Huh-7 cells did not have any effect on viral infection. All conditions were tested in duplicate, and error bars represent the standard errors of the means. Intracellular RLuc signal was normalized to the values for non-GRFT-treated cultures.
Since GRFT is known to interact with the terminal mannose residues present on N-linked high-mannose oligosaccharides (53, 54), we tested whether N-linked high-mannose oligosaccharide can interfere with the inhibitory effect of GRFT. Interestingly, the antiviral effect of GRFT could be significantly reduced when GRFT was preincubated for 1 h with 10 μg/ml of the N-linked high-mannose oligosaccharide mannonanose-di-(N-acetyl-d-glucosamine) (Man-9) (Fig. 3B). The fact that Man-9 pretreatment could not fully revert the inhibitory effect of GRFT might be due to a higher affinity of GRFT to clusters of oligomannose glycans present on a protein backbone (35). Similar results were observed with cyanovirin N lectin (20). Our data suggest that GRFT is interacting with glycans present on the viral envelope glycoproteins. Furthermore, pretreatment of the virus with Man-9 had no effect on viral infectivity (Fig. 3B).
Additional proof of the interaction between GRFTWT and E1E2 heterodimer was generated by preincubation of HCV pseudoparticles (HCVpp) of genotype 1a (strain H77) with different concentrations of either GRFTWT or GRFTMUT. After addition of Triton X-100, the potential GRFT-E1E2 complexes were coimmunoprecipitated with a polyclonal anti-GRFT antibody conjugated to protein G-Sepharose beads. As shown in Fig. 4, Western blot analysis indicates that the HCV envelope proteins could be coimmunoprecipitated only in the presence of wild-type GRFT. Neither E1 nor E2 could be detected in the absence of GRFT or when GRFT containing a nonfunctional carbohydrate-binding site was utilized (GRFTMUT).
Fig. 4.
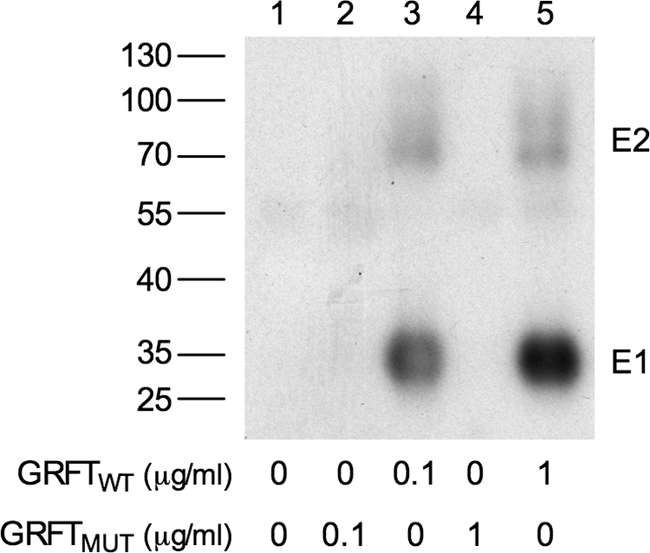
GRFT interacts with the viral envelope proteins. E1E2 heterodimers were coimmunoprecipitated with different concentrations of GRFTMUT (lanes 2 and 4) or GRFTWT (lanes 3 and 5). Coimmunoprecipitation without GRFT served as a negative control (lane 1). GRFTWT and GRFTMUT were added at a final concentration of 0.1 μg/ml (lanes 2 and 3) or 1 μg/ml (lanes 4 and 5). After Western blotting, E1 and E2 proteins were visualized using specific monoclonal antibodies. The positions of molecular mass markers (in kilodaltons) (PagerRuler; Fermentas) are shown to the left of the gel as a reference.
Since we have shown that GRFT interferes with HCV entry and that this antiviral effect is most likely caused by the binding of GRFT to the viral envelope proteins, we can assume that this interaction prevents the attachment of the viral particle to one of its cellular receptors. To verify this hypothesis, genotype 1a H77 HCVpp and genotype 2a JFH1 HCVpp were preincubated with increasing concentrations of GRFTWT (0, 0.1, 1, and 10 μg/ml) or with GRFTMUT (10 μg/ml) as a control. This mixture was then incubated with a fusion protein containing the large extracellular loop of the human CD81 (CD81-LEL) and glutathione S-transferase. Complexes were pulled down with glutathione-Sepharose beads. As shown in Fig. 5, increasing concentrations of GRFTWT reduced the amount of H77 E1E2 heterodimer that interacted with CD81-LEL, suggesting that GRFT inhibits HCV entry by blocking E2-CD81 interaction. Likewise, increasing concentrations of GRFTWT interfered with the interaction of JFH1 E1E2 with CD81-LEL (data not shown). Addition of GRFTMUT had no effect on the interaction between the viral envelope proteins and CD81.
Fig. 5.
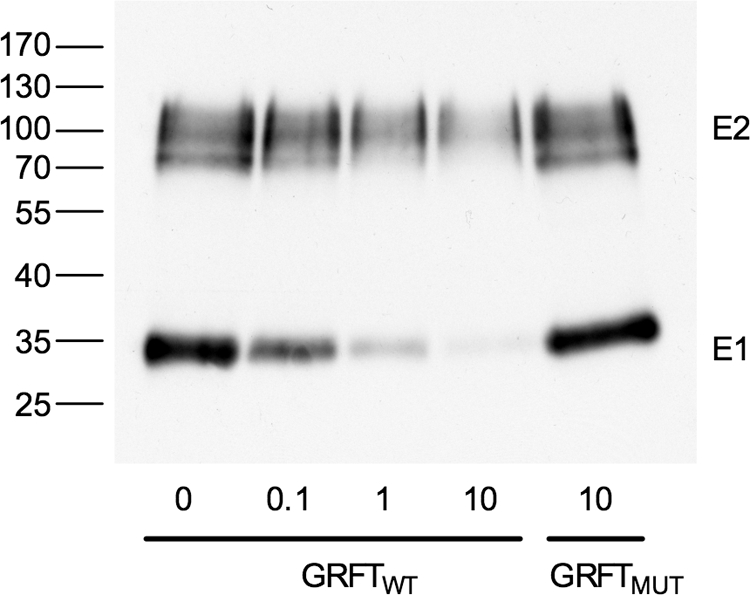
CD81 pulldown assay. HCV pseudoparticles (HCVpp) were preincubated with different concentrations (in micrograms per milliliter) of GRFTWT or GRFTMUT and large extracellular loop of CD81 (CD81-LEL) fused to glutathione S-transferase. Complexes were pulled down with glutathione-Sepharose beads. After Western blotting, the viral E1 and E2 proteins were visualized using specific monoclonal antibodies. The positions of molecular mass markers (in kilodaltons) (PagerRuler; Fermentas) are shown to the left of the gel as a reference.
GRFT prevents HCV infection in a genotype-independent manner.
To confirm that GRFT exerts its inhibitory effect on the entry phase of the viral life cycle, we also utilized the HCVpp system. HCV pseudoparticles containing genotype 1a HCV envelope proteins were preincubated with GRFT and then transferred to Huh-7 cells. As shown in Fig. 6, a clear inhibition of HCV entry could be observed while GRFT treatment had no effect on particles pseudotyped with the glycoproteins of vesicular stomatitis virus (VSV) or the feline retrovirus RD114. HCVpp generated with envelope glycoproteins from HCV strains of genotypes 1b, 2a, 2b, 3a, 4a, and 6 showed susceptibility to GRFT treatment (Fig. 6). The slight differences observed in the efficacy of treatment for some genotypes could potentially be due to variations in the glycosylation of HCV envelope proteins in the context of the HCVpp system. The cross-genotype antiviral effect of GRFT was corroborated in the HCV cell culture system using chimeric H77/JFH1 virus (IC50 of 87.2 ng/ml or 6.7 nM and IC90 of 223 ng/ml or 17.1 nM) (Fig. 1C and D).
Fig. 6.

Inhibition of HCV infection by GRFTWT is genotype independent. HCVpp of different genotypes (genotypes 1a to 6) were preincubated with GRFTWT (1 μg/ml) or without GRFTWT and then transferred to Huh-7 cells. Intracellular firefly luciferase signal was measured and normalized to untreated cultures. GRFTWT had no effect on the infectivity of particle pseudotypes with VSV and RD114 envelope proteins. Experiments were performed three times, and all conditions were tested in duplicate. Error bars represent the standard errors of the means.
GRFT partially protects chimeric mice from HCV infection.
To study the efficacy of GRFT in preventing in vivo HCV infections, we used the chimeric uPA-SCID mouse model (28, 31). The livers of these chimeric mice are largely engrafted with functional primary human hepatocytes and can be infected with serum-derived HCV of all genotypes (3). Six chimeric uPA-SCID mice received daily subcutaneous injections with 5 mg/kg GRFTWT. In addition, five control animals were dosed daily with GRFTMUT. The GRFT treatment was initiated 3 days before the animals were challenged with serum-derived HCV of genotype 1a (strain H77) and was continued for up to 2 weeks after injection of the virus. GRFT treatment was stopped on day 4 (2 mice) and day 7 (2 mice) after viral challenge because of physical deterioration of the treated mice. As shown in Fig. 7, 1 week after infection, HCV RNA could easily be detected in all five control animals, while HCV RNA was detected in only two of six GRFTWT-treated animals. In the remaining four treated animals, the viral load was below the limit of detection (750 IU/ml). One week later, viral RNA was detected in five out of six treated animals, albeit at much lower concentrations than in the control animals, which experienced full-blown viremia. From week 2 to week 3, one control animal and two treated animals died spontaneously, but one treated chimeric mouse remained completely HCV negative (<750 IU/ml) throughout the 4-week observation period. At weeks 1 and 2, the median viral loads of the GRFTWT- and GRFTMUT-treated chimeric mice were statistically significantly different (P = 0.0173 and P = 0.0667, respectively). When we compared the viral load of only the animals that were HCV positive at week two, thereby excluding the one protected mouse, there still was a 1-log10-unit difference in viremia between GRFTWT- and GRFTMUT-treated groups, indicating a delay in the kinetics of the infection in GRFTWT-treated mice.
Fig. 7.
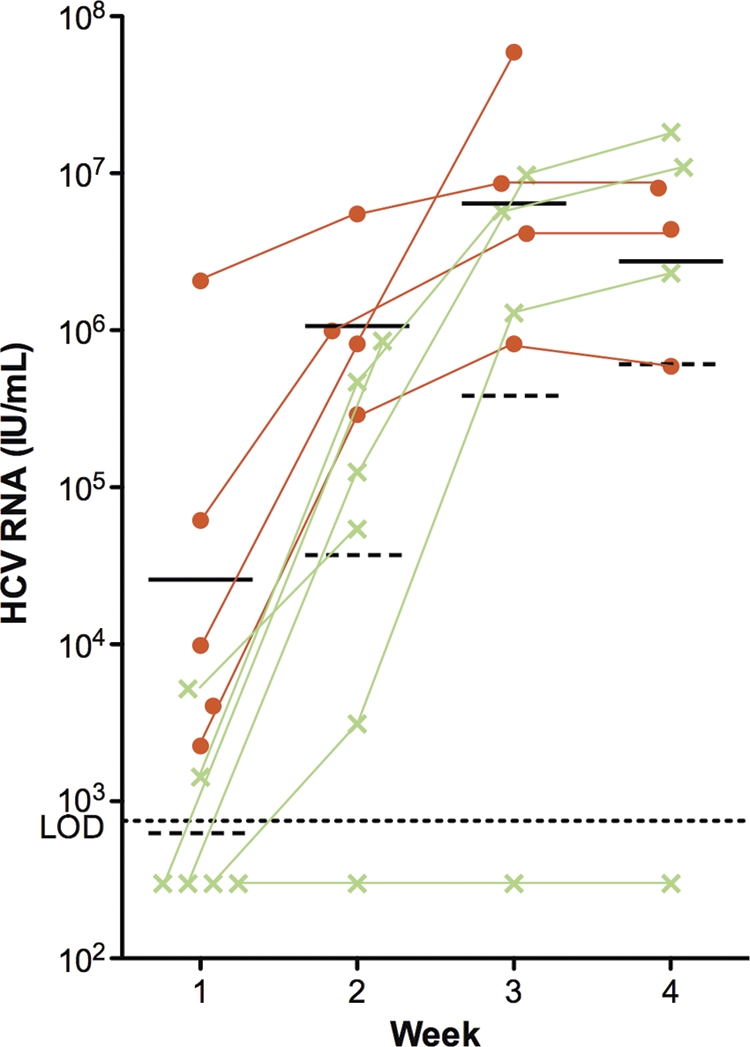
In vivo evaluation of the anti-HCV effect of GRFT. Chimeric mice were treated daily with GRFTWT (green) or GRFTMUT (orange) at a dose of 5 mg/kg of body weight. Three days after the first dose, the mice were challenged with serum-derived HCV of strain H77 (genotype 1a). Each symbol represents the value for an individual mouse, and the short horizontal solid line and broken line represent the geometric mean viral load of GRFTMUT- and GRFTWT-treated mice, respectively. LOD, limit of detection (750 IU/ml).
DISCUSSION
New strategies to prevent HCV infection, especially in the context of liver transplantation in chronically infected HCV patients, are urgently needed. Current standard therapy before transplantation is poorly tolerated and hardly effective, especially in patients with decompensated liver disease (17). A recent randomized controlled trial of prophylactic peginterferon plus ribavirin therapy after liver transplantation resulted in a sustained virological response of only 22% (4). In addition, only a minority of liver transplant recipients are eligible for combination therapy in the early postoperative period, and most of those who commence treatment are not able to tolerate it. A protective vaccine is not yet available, and the development of such a vaccine is severely hampered by the high variability of the virus and of its envelope proteins in particular. More-conserved features of the virus may be attractive targets for antiviral therapy. Across the different genotypes and strains, the envelope proteins are highly glycosylated at specific, well-defined locations (14). The fact that these glycans play a pivotal role in protein folding, HCV entry, and immune evasion (5, 13, 18) combined with their conserved nature makes them attractive targets for novel broad-spectrum antiviral therapy.
Here we investigated the anti-HCV activity of the oligomannose-specific lectin griffithsin (GRFT). This 13-kDa protein contains three almost identical carbohydrate-binding sites, each capable of interacting separately with monosaccharides through multiple contact points. The active homodimeric form of GRFT therefore possesses 6 carbohydrate-binding sites. Atomic-resolution crystal structures of an engineered monomeric GRFT showed that each monomer can bind to two different nonamannoside molecules through all three carbohydrate-binding sites (35, 53), which explains its tight interaction with high-mannose oligosaccharides that are commonly found on the surfaces of viral glycoproteins. In fact, GRFT exhibits strong anti-HIV and anti-SARS coronavirus activity at picomolar- and nanomolar-range concentrations, respectively (34, 37). In line with these findings, we show here that GRFT can efficiently prevent HCV infection at nanomolar-range levels, provided that the mannose-binding site is completely functional. A mutated GRFT variant in which all three binding sites are rendered nonfunctional no longer displays antiviral activity against HCV. Although we cannot exclude the possibility that GRFT may also affect the interaction between the viral particle and other receptors, our data clearly show that GRFT shields the viral glycoproteins, thereby at least preventing the interaction with CD81. Using both the HCV pseudoparticle (HCVpp) and HCV cell culture (HCVcc) systems, we could clearly demonstrate that GRFT can inhibit HCV infection of different genotypes, which is probably due to the high degree of glycosylation of the envelope proteins of all HCV genotypes. Interestingly, GRFT did not have any inhibitory effect on the entry of pseudoparticles containing the envelope glycoproteins of VSV or RD114. Since the glycoprotein G of VSV contains only two N-linked glycans of a complex type, it is unlikely to be recognized by GRFT (21). RD114 envelope protein contains 11 potential N-linked glycosylation sites. However, based on endoglycosidase H digestion, the RD114 envelope protein associated with the viral particle contains complex glycans instead of high-mannose glycans (D. Lavilette, Ecold Normale Supérieure [ENS], Lyon, France, personal communication).
Since the glycans present on the viral envelope proteins are known to play a pivotal role in the viral life cycle, this approach may be less prone to viral escape mechanisms. In fact, we showed that a mutated virus that has lost its glycosylation at position N-6 in E2 is as sensitive to GRFT treatment as the wild-type virus is. In the unlikely situation that the virus eliminates its glycans, this variant would also become more susceptible to the activity of neutralizing antibodies that are ubiquitously present in the serum of chronically infected patients (18, 33). The present data are also in line with a previous publication by our group, which showed the anti-HCV activity of nanomolar concentrations of another lectin, cyanovirin N (20).
HCV can infect naïve hepatocytes in two different ways. In the first classical way, circulating viral particles bind the different coreceptors that are expressed on the membrane of the hepatocyte, after which the particle is internalized (41). However, recently, an alternative transmission route has been described wherein viral particles directly disseminate from one infected hepatocyte to a neighboring one (47, 52). This direct cell-to-cell transmission route is resistant to the activity of neutralizing antibodies, which hampers the use of these antibodies in a clinical setting. Our in vitro experiments unambiguously show that GRFT can inhibit this alternative transmission route.
Finally, in a proof-of-concept in vivo study, we demonstrated that GRFT treatment could mitigate HCV infections in chimeric mice that harbor primary human hepatocytes in their livers.
However, soon after cessation of GRFTWT therapy, 2 out of 6 animals died spontaneously. In addition, one GRFTMUT-treated chimeric mouse died within 2 weeks. Spontaneous death of chimeric mice is not an uncommon event, and the precise cause of death can rarely be determined (46, 49). These animals are very small and fragile (average weight, 9.3 g) even though a large proportion of their diseased liver is replaced by human hepatocytes. In fact, animals with a high level of chimerism seem to be more fragile probably because of nephrotoxicity of human complement factors produced by the resident human hepatocytes (46). We have evaluated toxicity of GRFT in healthy mouse and guinea pig models and found that treatments up to 50 mg/kg are well tolerated, with only mild toxicities (unpublished data). During our experiments in chimeric mice, we noticed a decrease in body weight associated with GRFT treatment, but given the already small size of these animals, this may have had a more pronounced impact than in healthy mice. Previous published data indicate that GRFT treatment does not induce significant alterations in the levels of an extensive panel of cytokines and chemokines in human cervical explants and it does not have a mitogenic effect on human peripheral blood mononuclear cells (23, 38). Using the MTS assay, we did not observe any toxic effect of GRFT on human hepatoma cells (data not shown). Nevertheless, a more comprehensive toxicological study in different animal species should be performed to assess the safety profile of GRFT before this compound can be evaluated in the clinic.
Because of the negative effect of GRFT on the physical condition of the chimeric mice, we were unable to increase the dose or prolong the treatment. With the current dose (5 mg/kg subcutaneously), we achieved plasma GRFT levels of around 90 ng/ml (6.9 nM) at the time of viral challenge (data not shown), which is more or less in the range of the IC50 in cell culture (6.7 to 13.9 nM). Higher plasma levels, which are easily achievable in nonchimeric mice and guinea pigs, might have improved the outcome of our in vivo study.
Although this study examined only a limited number of chimeric animals, the results obtained nonetheless were statistically significant and support the idea that targeting the glycans present on the viral particle could be an alternative and original strategy to prevent infection of the donor liver in HCV-infected patients undergoing liver transplantation. A protocol could be devised whereby just before liver transplantation the chronic HCV patient is infused with GRFT to saturate and sterilize the circulating viral particles. A major advantage of GRFT is that it also inhibits direct cell-to-cell transmission of HCV. Therefore, continuous GRFT therapy could also prevent dissemination of the virus in case not all viral particles were initially neutralized and the new donor liver nevertheless became infected. Ultimately, GRFT treatment could also be combined with standard treatment of chronic HCV patients to prevent rebound after cessation of therapy or to inhibit the spread of resistant mutants that appeared during treatment with (a combination of) direct antiviral compounds.
ACKNOWLEDGMENTS
This work was supported by the French “Agence Nationale de Recherche sur le Sida et les Hépatites Virales” (ANRS), National Institutes of Health (grant AI076169), and Ghent University by a Concerted Research Action (grant 01G00507). A.A. was supported first by a Marie Curie Research Training Network grant (MRTN-CT-2006-035599) and then by a fellowship from the ANRS. J.D. is an international scholar of the Howard Hughes Medical Institute. P.M. was supported by a postdoctoral fellowship and an individual research grant (grant 31500910) of The Research Foundation–Flanders (FWO-Vlaanderen). K.E.P. was supported by NIH grant AI076169.
We thank J. K. Ball, R. Bartenschlager, F. L. Cosset, J. McKeating, T. Pietschmann, C. Rice, and T. Wakita for providing essential reagents.
Footnotes
Published ahead of print on 6 September 2011.
REFERENCES
- 1. Baldick C. J., et al. 2010. A novel small molecule inhibitor of hepatitis C virus entry. PLoS Pathog. 6:e1001086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bartosch B., Dubuisson J., Cosset F. L. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197:633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bukh J., et al. 2010. Challenge pools of hepatitis C virus genotypes 1-6 prototype strains: replication fitness and pathogenicity in chimpanzees and human liver-chimeric mouse models. J. Infect. Dis. 201:1381–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bzowej N., et al. 2011. PHOENIX: a randomized controlled trial of peginterferon alfa-2a plus ribavirin as a prophylactic treatment after liver transplantation for hepatitis C virus. Liver Transpl. 17:528–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choukhi A., Ung S., Wychowski C., Dubuisson J. 1998. Involvement of endoplasmic reticulum chaperones in the folding of hepatitis C virus glycoproteins. J. Virol. 72:3851–3858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Delgrange D., et al. 2007. Robust production of infectious viral particles in Huh-7 cells by introducing mutations in hepatitis C virus structural proteins. J. Gen. Virol. 88:2495–2503 [DOI] [PubMed] [Google Scholar]
- 7. Dienstag J. L., McHutchison J. G. 2006. American Gastroenterological Association technical review on the management of hepatitis C. Gastroenterology 130:231–264 [DOI] [PubMed] [Google Scholar]
- 8. Dubuisson J., et al. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 68:6147–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Falkowska E., Kajumo F., Garcia E., Reinus J., Dragic T. 2007. Hepatitis C virus envelope glycoprotein E2 glycans modulate entry, CD81 binding, and neutralization. J. Virol. 81:8072–8079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Flint M., et al. 1999. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol. 73:6235–6244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fofana I., et al. 2010. Monoclonal anti-claudin 1 antibodies prevent hepatitis C virus infection of primary human hepatocytes. Gastroenterology 139:953–964 [DOI] [PubMed] [Google Scholar]
- 12. Garg V., et al. 2011. Effect of telaprevir on the pharmacokinetics of cyclosporine and tacrolimus. Hepatology 54:20–27 [DOI] [PubMed] [Google Scholar]
- 13. Goffard A., et al. 2005. Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J. Virol. 79:8400–8409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goffard A., Dubuisson J. 2003. Glycosylation of hepatitis C virus envelope proteins. Biochimie 85:295–301 [DOI] [PubMed] [Google Scholar]
- 15. Gottwein J. M., et al. 2009. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49:364–377 [DOI] [PubMed] [Google Scholar]
- 16. Goueslain L., et al. 2010. Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J. Virol. 84:773–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guillouche P., Feray C. 2011. Systematic review: anti-viral therapy of recurrent hepatitis C after liver transplantation. Aliment. Pharmacol. Ther. 33:163–174 [DOI] [PubMed] [Google Scholar]
- 18. Helle F., et al. 2007. The neutralizing activity of anti-hepatitis C virus antibodies is modulated by specific glycans on the E2 envelope protein. J. Virol. 81:8101–8111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Helle F., et al. 2010. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J. Virol. 84:11905–11915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Helle F., et al. 2006. Cyanovirin-N inhibits hepatitis C virus entry by binding to envelope protein glycans. J. Biol. Chem. 281:25177–25183 [DOI] [PubMed] [Google Scholar]
- 21. Hunt L. A., Etchison J. R., Summers D. F. 1978. Oligosaccharide chains are trimmed during synthesis of the envelope glycoprotein of vesicular stomatitis virus. Proc. Natl. Acad. Sci. U. S. A. 75:754–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jones C. T., et al. 2010. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat. Biotechnol. 28:167–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kouokam J. C., et al. 2011. Investigation of griffithsin's interactions with human cells confirms its outstanding safety and efficacy profile as a microbicide candidate. PLoS One 6:e22635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Law M., et al. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14:25–27 [DOI] [PubMed] [Google Scholar]
- 25. Leroux-Roels G. 2005. Development of prophylactic and therapeutic vaccines against hepatitis C virus. Expert Rev. Vaccines 4:351–371 [DOI] [PubMed] [Google Scholar]
- 26. Lindenbach B. D., et al. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626 [DOI] [PubMed] [Google Scholar]
- 27. Lindenbach B. D., et al. 2006. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. U. S. A. 103:3805–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mercer D. F., et al. 2001. Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 7:927–933 [DOI] [PubMed] [Google Scholar]
- 29. Meuleman P., et al. 2011. In vivo evaluation of the cross-genotype neutralizing activity of polyclonal antibodies against hepatitis C virus. Hepatology 53:755–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meuleman P., et al. 2008. Anti-CD81 antibodies can prevent a hepatitis C virus infection in vivo. Hepatology 48:1761–1768 [DOI] [PubMed] [Google Scholar]
- 31. Meuleman P., et al. 2005. Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology 41:847–856 [DOI] [PubMed] [Google Scholar]
- 32. Meuleman P., Vanlandschoot P., Leroux-Roels G. 2003. A simple and rapid method to determine the zygosity of uPA-transgenic SCID mice. Biochem. Biophys. Res. Commun. 308:375–378 [DOI] [PubMed] [Google Scholar]
- 32a. Meuleman P., et al. 27 September 2011, posting date. A human monoclonal antibody targeting SR-BI precludes hepatitis C virus infection and viral spread in vitro and in vivo. Hepatology doi:10.1002/hep.24692. [DOI] [PMC free article] [PubMed]
- 33. Meunier J. C., et al. 2005. Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc. Natl. Acad. Sci. U. S. A. 102:4560–4565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mori T., et al. 2005. Isolation and characterization of griffithsin, a novel HIV-inactivating protein, from the red alga Griffithsia sp. J. Biol. Chem. 280:9345–9353 [DOI] [PubMed] [Google Scholar]
- 35. Moulaei T., et al. 2010. Monomerization of viral entry inhibitor griffithsin elucidates the relationship between multivalent binding to carbohydrates and anti-HIV activity. Structure 18:1104–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakabayashi H., Taketa K., Miyano K., Yamane T., Sato J. 1982. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 42:3858–3863 [PubMed] [Google Scholar]
- 37. O'Keefe B. R., et al. 2010. Broad-spectrum in vitro activity and in vivo efficacy of the antiviral protein griffithsin against emerging viruses of the family Coronaviridae. J. Virol. 84:2511–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O'Keefe B. R., et al. 2009. Scaleable manufacture of HIV-1 entry inhibitor griffithsin and validation of its safety and efficacy as a topical microbicide component. Proc. Natl. Acad. Sci. U. S. A. 106:6099–6104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Op De Beeck A., et al. 2004. Characterization of functional hepatitis C virus envelope glycoproteins. J. Virol. 78:2994–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pawlotsky J. M. 2011. The results of Phase III clinical trials with telaprevir and boceprevir presented at the Liver Meeting 2010: a new standard of care for hepatitis C virus genotype 1 infection, but with issues still pending. Gastroenterology 140:746–754 [DOI] [PubMed] [Google Scholar]
- 41. Pietschmann T. 2009. Virology: final entry key for hepatitis C. Nature 457:797–798 [DOI] [PubMed] [Google Scholar]
- 42. Popescu C. I., Dubuisson J. 2009. Role of lipid metabolism in hepatitis C virus assembly and entry. Biol. Cell 102:63–74 [DOI] [PubMed] [Google Scholar]
- 43. Scheel T. K., et al. 2008. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc. Natl. Acad. Sci. U. S. A. 105:997–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schiano T. D., et al. 2006. Monoclonal antibody HCV-AbXTL68 in patients undergoing liver transplantation for HCV: results of a phase 2 randomized study. Liver Transpl. 12:1381–1389 [DOI] [PubMed] [Google Scholar]
- 45. Syder A. J., et al. 2011. Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. J. Hepatol. 54:48–55 [DOI] [PubMed] [Google Scholar]
- 46. Tateno C., et al. 2004. Near completely humanized liver in mice shows human-type metabolic responses to drugs. Am. J. Pathol. 165:901–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Timpe J. M., et al. 2008. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 47:17–24 [DOI] [PubMed] [Google Scholar]
- 48. Vanwolleghem T., et al. 2008. Polyclonal immunoglobulins from a chronic hepatitis C virus patient protect human liver-chimeric mice from infection with a homologous hepatitis C virus strain. Hepatology 47:1846–1855 [DOI] [PubMed] [Google Scholar]
- 49. Vanwolleghem T., et al. 2010. Factors determining successful engraftment of hepatocytes and susceptibility to hepatitis B and C virus infection in uPA-SCID mice. J. Hepatol. 53:468–476 [DOI] [PubMed] [Google Scholar]
- 50. Verna E. C., Brown R. S., Jr 2008. Hepatitis C and liver transplantation: enhancing outcomes and should patients be retransplanted. Clin. Liver Dis. 12:637–659 [DOI] [PubMed] [Google Scholar]
- 51. Watt K., Veldt B., Charlton M. 2009. A practical guide to the management of HCV infection following liver transplantation. Am. J. Transplant. 9:1707–1713 [DOI] [PubMed] [Google Scholar]
- 52. Witteveldt J., et al. 2009. CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J. Gen. Virol. 90:48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ziolkowska N. E., et al. 2006. Domain-swapped structure of the potent antiviral protein griffithsin and its mode of carbohydrate binding. Structure 14:1127–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ziolkowska N. E., et al. 2007. Crystallographic, thermodynamic, and molecular modeling studies of the mode of binding of oligosaccharides to the potent antiviral protein griffithsin. Proteins 67:661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]