

Abstract
A large number of G-protein-coupled receptors (GPCRs) show varying degrees of basal or constitutive activity. This constitutive activity is usually minimal in natural receptors but is markedly observed in wild type and mutated (naturally or induced) receptors. According to conventional two-state drug receptor interaction model, binding of a ligand may initiate activity (agonist with varying degrees of positive intrinsic activity) or prevent the effect of an agonist (antagonist with zero intrinsic activity). Inverse agonists bind with the constitutively active receptors, stabilize them, and thus reduce the activity (negative intrinsic activity). Receptors of many classes (α-and β-adrenergic, histaminergic, GABAergic, serotoninergic, opiate, and angiotensin receptors) have shown basal activity in suitable in vitro models. Several drugs that have been conventionally classified as antagonists (β-blockers, antihistaminics) have shown inverse agonist effects on corresponding constitutively active receptors. Nearly all H1 and H2 antihistaminics (antagonists) have been shown to be inverse agonists. Among the β-blockers, carvedilol and bucindolol demonstrate low level of inverse agonism as compared to propranolol and nadolol. Several antipsychotic drugs (D2 receptors antagonist), antihypertensive (AT1 receptor antagonists), antiserotoninergic drugs and opioid antagonists have significant inverse agonistic activity that contributes partly or wholly to their therapeutic value. Inverse agonism may also help explain the underlying mechanism of beneficial effects of carvedilol in congestive failure, naloxone-induced withdrawal syndrome in opioid dependence, clozapine in psychosis, and candesartan in cardiac hypertrophy. Understanding inverse agonisms has paved a way for newer drug development. It is now possible to develop agents, which have only desired therapeutic value and are devoid of unwanted adverse effect. Pimavanserin (ACP-103), a highly selective 5-HT2A inverse agonist, attenuates psychosis in patients with Parkinson's disease with psychosis and is devoid of extrapyramidal side effects. This dissociation is also evident from the development of anxioselective benzodiazepines devoid of habit-forming potential. Hemopressin is a peptide ligand that acts as an antagonist as well as inverse agonist. This agent acts as an antinociceptive agent in different in vivo models of pain. Treatment of obesity by drugs having inverse agonist activity at CB1/2 receptors is also underway. An exciting development is evaluation of β-blockers in chronic bronchial asthma—a condition akin to congestive heart failure where β-blockade has become the standard mode of therapy. Synthesis and evaluation of selective agents is underway. Therefore, inverse agonism is an important aspect of drug–receptor interaction and has immense untapped therapeutic potential.
Keywords: Constitutive activity, G-protein-coupled receptors, inverse agonism, inverse agonists
Introduction
Drug–receptor interaction has fascinated researchers since antiquity. It has been evolved from a concept of “Lock and Key” to the use of Furchgott's method (1966) to quantify dissociation constants and relative efficacies of agonist–receptor complexes. In the last 40 years, great strides in developments in molecular pharmacology have resulted in the accumulation of a vast amount of knowledge. This article briefly summarizes the recent developments in drug–receptor interactions with focus on inverse agonism and its potential therapeutic utility.
Inverse Agonism
The two-state receptor model (binary model) shows that a receptor remains in an inactive (Ri) or active (R*) state and there is an equilibrium, which can be shifted by ligands that bind to receptors [Figure 1]. The profile of a ligand, traditionally, endogenous or synthetic, is characterized by its “affinity” toward the receptor and intrinsic activity or “efficacy.” It expresses the degree to which different ligands produce varying biological responses while occupying same number of receptors. The drug or a ligand avidly binds with the receptor (affinity) and produces conformational changes, which shift the balance toward active state [Figure 1]. Thus, a drug-activated receptor complex may initiate an activity (intrinsic activity or efficacy), resulting in a response. The ultimate response can be stimulatory or inhibitory depending on the activation of subcellular effector mechanisms.
Figure 1.
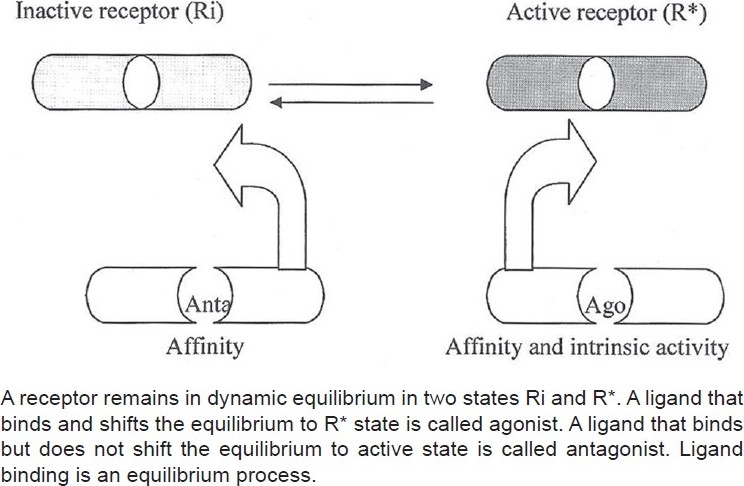
Conventional two-state model of drug–receptor interaction
According to this hypothesis, receptors are quiescent at rest (Ri) and are stimulated by drugs (R*, receptors are switched on) known as agonists. Antagonists do not have intrinsic activity and therefore oppose the effects of agonists by preventing agonist binding and activation (receptors are switched off). On the other hand, partial agonists have varying degrees of affinity and intrinsic activities and produce submaximal tissue responses at any degree of receptor occupancy (usually less than 100%). This simple mechanism of “on–off switch” remained elusive for a longer period of time to pharmacologists who could not correlate certain discrepant experimental findings in cell systems, using isolated and mutated receptors with this model of drug–receptor interaction. For example, experimental evidence indicates that all agonists do not produce the same active state of G-protein-coupled receptors (GPCRs).[1] The receptors for benzodiazepines, opiates, serotonin, cannabinoids, histamine, and a variety of other (heptahelical) GPCRs show activity even when not occupied by agonists, that is, they have constitutive activity independent of ligand activation [Figure 2]. This means that a proportion of the receptor population spontaneously undergoes a conformational change that can bind and activate G-proteins or the alternative pathways. In addition, wild-type or experimentally mutated receptors also exhibit varying degrees of spontaneous activation. Under physiological conditions, this activity is usually low because the fraction of unoccupied receptors in active conformational state is minimal but when the population of constitutive receptors is overexpressed in the in vitro systems, such as when expressed in high amounts in cultured cells, they exhibit significant and measurable spontaneous activity.[2] Interestingly, when certain ligands bind these constitutively activated receptors in suitable experimental settings, the overall effects are opposite to pure or full agonists. There is a shift of equilibrium from activation (R*) to quiescence (Ri), as shown in Figures 2–4. Such ligands are called as “inverse agonists.” Inverse agonists preferentially bind and stabilize receptors in the inactive (Ri) state, and thus have negative intrinsic activity [Figures 2 and 5]. This results in a reduction in spontaneous receptor activity. If receptors do not exhibit constitutive activity, the same inverse agonist may behave as competitive antagonist. Neutral antagonists have equal preferences for both Ri and R* states, lack any intrinsic activity, and are able to block actions produced by either agonists or inverse agonists. Many conventional antagonists, such as antihistaminics are now considered to be inverse agonists.[1,3] As described above, a ligand must recognize at least two receptor conformational species as being identical: RFNx01 and Ri. For constitutively active receptors (which couple with G-proteins at rest), a three-state model [Figure 4] is best suited to explain this interaction, that is, R#G (constitutively activated GPCR), R*G (agonist-activated GPCR), and RiG (resting or inactive state), any of these 3 states are available for ligand (L) binding and forming ternary complexes as R#G–L, R*G–L, or RiG–L. The degree of observed inverse agonism depends on the relative affinity of the inverse agonist for the various receptor species and the degree of constitutive activity in the system. Therefore, there are partial inverse agonists and full inverse agonists [Figure 5].[1–3]
Figure 2.
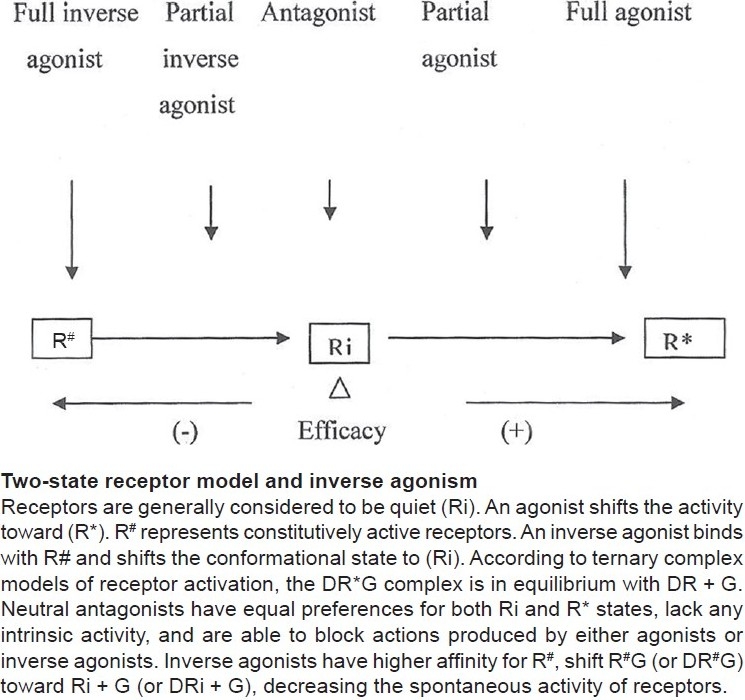
Intrinsic activities of full agonist, antagonist, and inverse agonists
Figure 4.
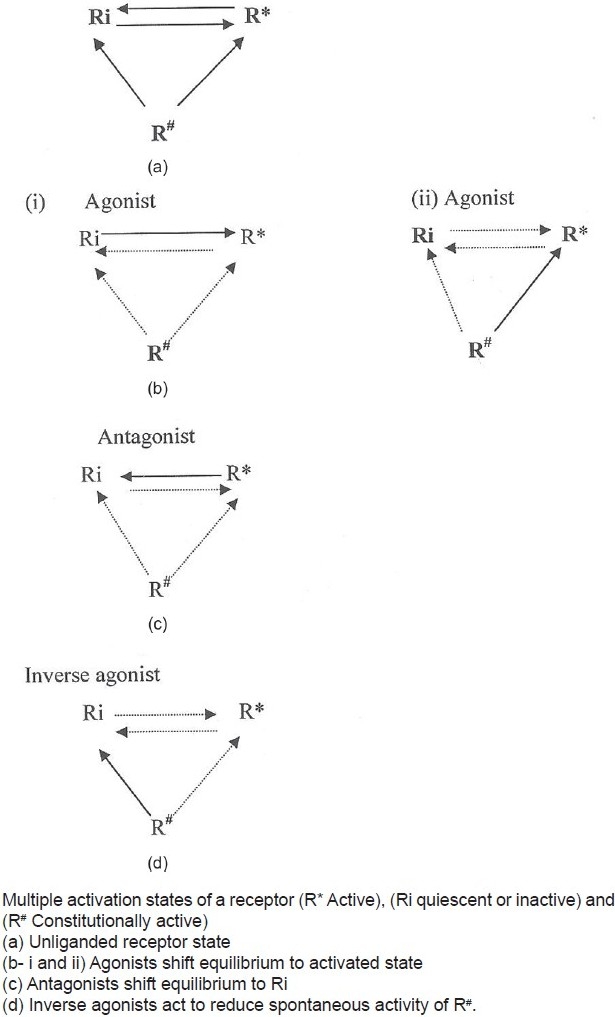
Proposed drug–receptor model to explain inverse agonism
Figure 5.
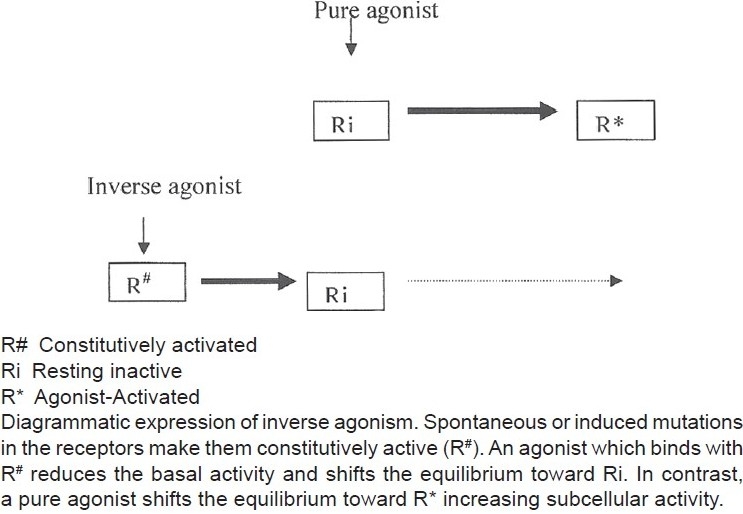
Inverse agonist-receptor interaction
Figure 3.
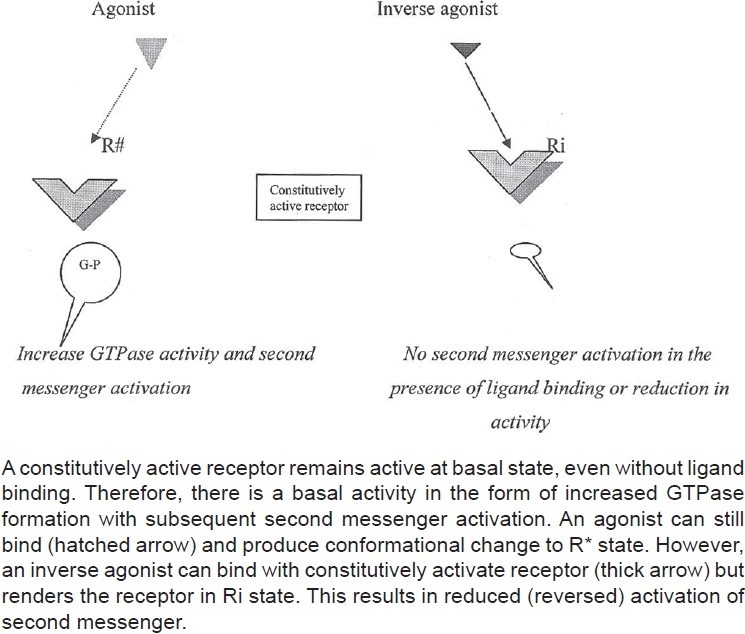
Constitutively active receptor and inverse agonism
G-Protein-Coupled Receptors and Inverse Agonism
The GPCRs have the ability to undergo conformational changes upon ligand binding. The properties include existence in various conformations, ability to alter receptor activity, and production of multiple active receptor states. Response emanates from the hydrolysis of GTP by the G-protein resulting from activation by R*. As stated above, some receptors can spontaneously interact with G-proteins to initiate GTPase activity, in the absence of agonist ligands [Figure 2]. This constitutive activity can be suppressed in suitable in vitro experimental settings by inverse agonists either partially (partial inverse agonists) or fully (full inverse agonists), resulting in action exactly opposite to agonists. Methods to detect inverse agonism are based on the determination of ligand affinity at Ri and R* with binding experiments, on the modulation of G-protein activity (GTP binding and hydrolysis) and change in effectors activity. The receptor agonists (ligands that bind and activate surface receptors) and direct G-protein agonists increase basal effectors activity. Receptor inverse agonists, such as G-protein antagonists and GTPase inhibitors, decrease spontaneous G-protein activity. It is interesting to note that neutral antagonists block the effects of inverse agonists. A term “protean agonist” is used to describe a theoretical class of agonists that produce receptor activation of lower magnitude than that emanating from spontaneous receptor constitutive activity. These kinds of ligands were given the name protean agonists after Proteus, the Greek God who could change shape and appearance at will. This class of ligands shows positive agonism in some GPCR systems and inverse agonism in others.[4]
Evidence of Constitutive Activity of GPCRs
Constitutive activity of GPCRs is demonstrated by measuring cyclic AMP (cAMP) levels, GTPase activity or by inositol phosphate (IP3) production in in vitro overexpressed receptor models [Box 1]. The constitutional activity was first demonstrated at δ-opioid receptors, which paved the ground for many further studies, all pointing to the existence of constitutive receptor activity in benzodiazepine (BDZ) receptors and GPCRs.[5] The constitutive activation of G-protein-coupled receptors (GPCRs) is now well recognized and more than 80% of the classical GPCR antagonists have been found to be inverse agonists.[2,5] The constitutive activity may be present in native or mutated strains (constitutively activated mutants). Such receptors may be underactive and lead to inherited diseases, such as diabetes insipidus, male pseudohermaphroditism, congenital hypothyroidism, retinitis pigmentosa, and congenital night blindness. The mutation may also render the receptors overactive and this may also lead to diseases, such as Leydig cell tumor and familial hypocalciuria.[6] The constitutive activation may also be seen in genetic variations in receptors (polymorphisms) that alter coupling efficiency. This is typified by allelic variants of human beta-adrenergic receptors (β-ARs). The Ile164Thr allele of the human β2-AR or the Arg389Gly allele of the human β1–ARs show spontaneous activity, which is not seen in naturally stable β-ARs.[7] The spontaneous activity so obtained may result in ligand-induced opening of L-type Ca2+ channels. Binding of “inverse agonists” on these constitutively active receptors reduces their activity. Inverse agonists may initiate signal transduction cascade activity different from GTPase activity and cAMP production. Antagonists to [cysteine–leukotriene receptor (CysLT) receptors] montelukast and zafirlukast are actually inverse agonists because these antagonists decrease the basal IP3 production in cells expressing CysLT receptors.[8]
Box 1.

Transfection: An in-vitro method to study receptor function
Inverse Agonistic Activity on Various Receptors and its Significance
β-adrenoceptors
Spontaneous activity is demonstrated in β1-ARs and β2-ARs by several scientists in a variety of models.[9–11] Engelhardt et al.[9] studied constitutive activity of the human β1-ARs and β2-ARs in transgenic mice. An increased basal cAMP (about 5-fold higher for β2 than for β1 receptors) output was recorded and the rise was proportional to the receptor density, thus demonstrating constitutive activity for both the subtypes. Chidiac et al.[10] studied agonist-independent properties of the human β2-ARs using the baculovirus expression system in Chinese hamster fibroblasts expressing β2-ARs. The amount of cAMP production correlated with the activity of β2-ARs. The increase in the production of cAMP in the absence of catecholamines and its reversal by beta-blockers reflected spontaneous β2-ARs activity. In the model for enhanced beta-adrenergic signaling (transgenic mice with heart-specific overexpression of the human β1-ARs), the β1-ARs displayed constitutive activity as evidenced by a higher spontaneous beating rate of isolated right atria of β1-transgenic mice (which overexpress β1-ARs) as compared to that of wild-type mice (which do not express β1-ARs).[12] Zhou et al.[13] showed β1-AR spontaneous activity using the highly novel system of adenovirus-mediated gene transfer of -AR subtypes into isolated ventricular myocytes obtained from hearts of β1/β2-AR double knockout mice.
A β-blocker does not simply “block” the receptor, but further inactivates receptor activity beyond its baseline value, depending on its degree of inverse agonist activity. Antagonists (at both β1-ARs and β2-ARs), such as metoprolol, carvedilol, propranolol, nebivolol, and bisoprolol, have shown inverse agonist activity.[14,15] The following rank order of “inverse efficacy” is reported; timolol ≥ propranolol > alprenolol ≥ pindolol > labetalol > dichloroisoproterenol (DCI). The same rank order was observed using membranes prepared from Chinese hamster fibroblasts expressing β2-ARs. The effect of timolol was partly blocked by labetalol and DCI, in an apparently competitive manner. The intracellular cAMP content of serum-free medium in which cells were cultured for assessment of receptor activity was also increased by the expression of β2-ARs, and that increase was reversed by timolol and propranolol. The notable observation was that DCI, labetalol, and pindolol produced increase in cAMP concentration in the normal cells. However, when the cell membranes were prepared from Sf-9 cells (these cells are devoid of GTP), the system became constitutively active due to removal of GTP and, under these circumstances, and these ligands produced the depression of the basal cAMP levels. These two observations show that these beta-blockers are actually inverse agonists.[10–12]
Other β-blockers, such as bisoprolol, bucindolol and metoprolol, and propranolol, were also shown to have inverse agonist activity.[16,17] Studies show that propranolol reduces basal cAMP accumulation (via β2-mediated inverse agonism) but stimulates β2-mediated mitogen-activated protein kinase (MAP) activity.[18] MAP activation is known to occur with cytokines, growth factors, and adrenergic overactivity. Therefore, propranolol can simultaneously act as an inverse agonist through a Gs-coupled mechanism (reduced cAMP) while stimulating an alternative G-protein-independent mechanism (MAP kinase activation).[18,19] In overexpressed human β2receptors, bucindolol has a low inverse agonist profile that correlates with its minimal reduction in Holter-monitored lowest 24-h heart rate—a clinical measure of β-AR inverse agonism.[20] Carvedilol and metoprolol exert different inverse agonist activity at β-ARs. Metoprolol is a strong inverse agonist that reduces basal β-AR activity to a higher degree than the weak inverse agonist carvedilol. Thus, carvedilol and metoprolol have different negative inotropic effects at equivalent levels of β-AR occupation. This is indicated by experiments on isolated muscle strip preparations from human ventricular myocardium. While at 100% β-AR occupation, force of contraction is reduced by 27% with carvedilol and by 89% by metoprolol.[19,20]
This information can be useful in selecting a particular β-blocking agent in cardiovascular disease. β-blockers have 5 ancillary properties, such as partial agonism, cardioselectivity, and membrane stabilizing effect, lipophilicity/hydrophility, and vasodilating actions.[18] To these, the property of inverse agonist effects can be added that may determine the utility of β-blockers’ in cardiovascular disorders.
In spontaneously hypertensive rats with cardiac hypertrophy, binding characteristics of β-blockers on β1-ARs change in a way that the inverse agonist activity of metoprolol is reduced.[21] In congestive heart failure(CHF), β1-ARs are reduced in number and β2activity is overexpressed[22,23] and inverse agonists may be excellent drugs to treat this condition. Inverse agonists enhance baseline atrial tension more than neutral antagonists. Inverse agonist activity is of importance for β-AR regulation also. In chronic heart failure, β-ARs are downregulated due to chronic sympathetic activation. Inactivation of β-ARs by inverse agonists inhibits phosphorylation of receptors, and thus desensitization and downregulation. Strong inverse agonist, such as metoprolol, but not the weak inverse agonist carvedilol, leads to upregulation of ventricular β-AR density.[21,24]
Therefore, different pharmacologic profiles of carvedilol and metoprolol lead to different effects on hemodynamics, especially in situations of β-adrenergic stimulation. In this respect, at a submaximal level of stimulation (low-grade exercise), carvedilol may have advantages compared with metoprolol because cardiac output is increased to a similar degree with a lesser increase of heart rate. This signifies a more economical mechanism to respond to exercise. In contrast, at a maximal exercise level, metoprolol is advantageous because maximum heart rate and oxygen consumption are improved by metoprolol to a higher degree than by carvedilol. This may explain metoprolol-induced improvement in submaximal and maximal exercise tolerance, whereas carvedilol improves only submaximal exercise tolerance in clinical studies.[18,19,24] Thus the role of inverse agonism in cardiovascular hemodynamics facilitates future selection of a β-blocker in CHF.
α2A-Adrenergic receptors (α2A-ARs)
Constitutive activity in α1- and α2-ARs have been shown recently.[25] Isolation of receptors in stable Chinese hamster ovary cell lines expressing the porcine α2A-ARs in its wild-type and constitutively activated mutant (CAM)T373K forms have enabled researchers to study inverse agonism at α2A-adrenergic receptors.[25] Activation of both G i and G swas enhanced for CAM receptors. cAMP production was suppressed in cells with CAM α2A-ARs and this suppression was reversed by some α2-adrenergic antagonists (indicating that these have inverse agonist action). The order of inverse efficacy is shown to be rauwolscine > yohimbine > RX821002 > MK912.[25–27] Whereas, phentolamine and idazoxan are reported to be without inverse agonism, that is, are neutral antagonists.[28] This study shows that some classical α2A-antagonists are actually inverse agonists. Medetomidine has 2 enantiomers. Dexmedetomidine behaves as an agonist (it causes Ca++mobilization and depression of forskolin-stimulated cAMP production), whereas levomedetomidine, a weak agonist, reduces intracellular Ca++ levels and further increases forskolin-stimulated cAMP production and therefore, it can be classified as an inverse agonist.[25]
It has been shown that chronic elevation of second messengers produced by constitutive G-protein activity can lead to cell transformation. The α1-ARs have been shown to be agonist-independent proto-oncogenes.[26,29] GTPase-inhibiting mutations constitutively activate alpha subunits of signal-transducing guanine nucleotide-binding proteins (G-proteins). Constitutive GPCR activity leading to chronic elevation of cell metabolism may also have a role in promoting the growth of tumors.[28,29] Overexpression of specific GPCRs in tumor cells have been reported to have proliferative properties. Also there is evidence to show that inhibition of these cellular effects can inhibit tumor growth, and therefore α-AR inverse agonists may play role as potential antitumor agents.[30] Rauwolscine (an inverse agonist) treatment reduced mitotic index and enhanced apoptosis in mouse mammary tumor cell lines.[31]
Benzodiazepine receptors
BDZ–GABA receptor complex is a pentameric structure having 3 different subunits α, β, and γ in different proportions; the usual subunits being α1β2γ2. The BDZ–GABA receptor complex is the site of action for several central nervous system depressants, such as general anesthetics, benzodiazepines, barbiturates, non-BDZ hypnotics, and antiepileptics. Depending on the affinity and efficacy, these agents act as partial/full inverse agonists or antagonists. BDZ inverse agonists are the compounds that produce paradoxical anxiety and have proconvulsive effects and can be partial or full inverse agonists. RO15-4513 is partial inverse agonist at recombinant α1, α2, α3, α5GABAA receptors and produces severe anxiety rather than the sedative effect of the BDZs.[32] Synthetic BDZ ligand, FG-7142, induces sweating, nausea, palpitations, tightness in the chest, restlessness, tremors, and feelings of worry; all markers of increased anxiety. Interestingly, it also antagonizes several behavioral and neurochemical effects of ethanol. Similarly, RO19-4603 an inverse agonist, antagonizes effects of ethanol on locomotor behavior and suppresses ethanol intake in selectively bred alcohol-preferring (P) rats.[32] These exciting discoveries are paving a way for the development of newer analogs for the treatment of chronic alcoholism. Other examples of BDZ-inverse agonists are DMCM hydrochloride (4-Ethyl-6,7-dimethoxy-9H-pyrido[3 , 4-b]indole-3-carboxylic acid methyl ester hydrochloride) and butyl β-carboline-3-carboxylate β-CCB, both having anxiogenic and proconvulsant activity.[32]
The α1 subunits are involved in sedation and amnesia, α2 subunits subserve anxiolytic and muscle relaxant effects and α5 subunits are involved in memory impairment.[33] Therefore, compounds that have different efficacies at the various subtypes would show dissociation of anxiolytic action from sedation. Attempts have been made to dissociate anxiolytic effects from sedative, dependence, and proconvulsive actions. Neuroactivity of a number of flavonoids (which act as partial agonists) is mediated by modulation of GABAA receptor function via BDZ sites. In whole-cell patch clamp studies on neuroblastoma IMR-32 cells expressing native GABAA receptors, 6,2’-dihydroxyflavone (DHF), and FG-7142 decreased GABA-induced currents, which could be blocked by flumazenil, a BDZ site antagonist.[34] In mouse behavioral models, DHF elicited significant anxiety-like effects in the elevated plus-maze test and enhanced cognitive performance in the passive avoidance test, suggesting that they have inverse agonist activity. However, no proconvulsant effect was shown by DHF. In electrophysiological studies on subtypes of recombinant GABAA receptors expressed in HEK 293T cells (human embryonic kidney cells easily express receptor activity), DHF decreased GABA-induced currents in some pentmeric isoforms of GABA-receptors, such as α1β3γ2, α2β3γ2, or α5β3γ2, but not in α3β3γ2 and normal β1β2γ2 receptors. The results demonstrated DHF as a partial inverse agonist-like modulator of GABAA receptors with selectivity in receptor subtypes. The DHF subtype-selectivity suggested that α3-containing subtypes could be a mediator of the convulsive activities of GABAA receptors.[34] Another implication is development of anxioselective compounds, which have low potential of withdrawal syndrome after prolonged use. Abrupt termination of the treatment of humans with BDZs leads to a rapid onset of discontinuation syndrome characterized by anxiety, muscle spasms, and occasionally convulsions. In a study, mice withdrawn from chronic treatment with alprazolam showed anxiety, muscle rigidity, and seizures between days 1 and 28 after termination of the treatment. Replacement of alprazolam with a β-carboline selective agonist, abecarnil for 7 days, prevented the occurrence of the signs of dependence.[35]
Opioid receptors
Constitutive activity at μ receptors is shown by various workers in different in vitro models and chronic exposure to morphine enhances this constitutional activation at μ receptors. It is hypothesized that this constitutional activation is important in the development of physical dependence by morphine.[28] Naloxone has been shown to act as an inverse agonist at the μ receptors, stimulating cAMP levels and inhibiting GTPγS binding in morphine-pretreated, but not untreated, tissues.[36] Opioid dependence can be viewed as an overexpression of the active receptors and withdrawal as an abrupt change from the active to the inactive state.[36,37]
Gi/o-coupled δ-opioid receptors were the first GPCRs to be described as having constitutive activity.[1] Ligand-independent activity of δ receptors is measured by [35S]GTPγS binding assay (An assay in which G-protein activation is assessed with [35S]guanosine-5’-O-(3-thio)triphosphate (GTPγS) binding). Such ligand-independent activity of this receptor was shown in mouse neuroblastoma-rat glioma hybrid cells (NG108-15 cells), which endogenously express the murine δ-opioid receptor.[37] The constitutional activity of these δ receptors can be selectively manipulated by inverse agonists. Treatment with inverse agonists upregulates the surface δ receptors. There are two types of inverse agonists acting on d receptors. Alkaloid inverse agonists (RTI-5989-1, RTI-5989-23, RTI-5989-25) are more potent than the peptide inverse agonist ICI-174864 (N, N-diallyl-Tyr-Aib-Aib-Phe-Leu). These developments are used to understand the complex effects of opiates on behavior. The basal (constitutional) activity at μ receptors may play a role in opioid dependence. Agonists cause opioid receptor internalization and downregulation and treatment with inverse agonists would result in upregulation of opioid receptors.
In a study, the intrinsic effects of ligands were tested by measuring GTPγS binding to cell membranes and cAMP levels in intact cells. Some ligands, such as nalmefene, were identified as inverse agonists (suppressing basal signal activity). Naloxone and naltrexone were neutral antagonists (not affecting basal signaling) in untreated cells, but had inverse agonistic effects in morphine-pretreated mice. In contrast, 6α- and 6β-naltrexol and 6β-naloxol, and 6β-naltrexamine were neutral antagonists regardless of morphine pretreatment. In acute and chronic mouse models of morphine-dependence, 6β-naltrexol caused significantly reduced withdrawal jumping compared to naloxone and naltrexone, at doses effective in blocking morphine antinociception.[38] This supports the hypothesis that naloxone-induced withdrawal symptoms result at least in part from suppression of basal signaling activity of μ receptors in morphine-dependent animals.
Histaminergic receptors (H1;H2;H3)
All the 3 histaminergic receptors exhibit constitutional activity. Transient expression of the wild-type human histamine H1 receptor in SV40-(simian virus infected) immortalized African green monkey kidney cells reveals an agonist-independent elevation of the basal levels of the second messenger inositol trisphospate, suggestive of constitutional activity. Several histamine H1receptor antagonists (cetirizine and loratidine) reduced this constitutive histamine H1 receptor activity. Inverse agonism, that is, stabilization of an inactive conformation of the human histamine H1 receptor, may therefore be a key component of the antiallergic mechanism of action of clinically used antihistamines. The older antihistaminic, mepyramine, was also found to reduce basal constitutional activity of H1 receptors. The effect of mepyramine on ATP-induced signaling was specifically neutralized by Gall overexpression, indicating that mepyramine is able to reduce G-protein availability.[39] Furthermore, the cardiotoxic and anti-inflammatory effects of certain H1 antihistaminics, such as inhibition of ICAM-1 (intercellular adhesion molecule-1) expression and the reducing effects of bradykinin are attributed to the inverse agonist action.[40] Since all H1 -antihistamines examined to date have shown inverse agonists, it is suggested that the term “H1 -receptor antagonists” be replaced by “H1 -antihistamines.”[41,42]
Constitutional activity is also demonstrated at H2 receptors. Many H2receptor antagonists, such as cimetidine, ranitidine, tiotidine, and famotidine, described previously as pure H2 antagonists, actually behave as inverse agonists and diminish basal cAMP levels. By using transfected Chinese hamster ovarian (CHO) cells expressing different densities of wild-type H2receptors or uncoupled H2 (Leu124Ala) receptors, considerable agonist-independent H2 receptor activity was found.[43] Ranitidine and cimetidine acted as inverse agonists (both induced H2 receptor upregulation), whereas burimamide was shown to be a neutral antagonist. The inverse agonism is specific at H2 receptors because no change in receptor density was observed after H1 or H3 antagonist treatment or after incubation with the structural analog of cimetidine, VUF 8299, which has no H2 antagonistic effects.[44] The displayed inverse agonism of H2 antagonists offer a plausible explanation for the observed development of tolerance after prolonged clinical use.[45]
Similarly, constitutive activity is also demonstrated in 2 isoforms of H3 receptors in rodent brain. H3 receptors control histaminergic neuron activity in vivo.[44,46] Ciproxifan, a H3 inverse agonist/antagonist is shown to modulate dopaminergic striatal activity by this mechanism.[46] Other studies have shown that intraperitoneal injections of thioperamide an imidazole-based H3 receptor inverse agonist, enhances histamine release in the brain and potentiates cocaine-induced locomotion hyperactivity in mice by this action.[47]
5-Hydroxytryptamine receptors (5HT1, 5HT2, 5HT6, 5HT7)
The heterogeneity of 5-HT receptors is well known. These receptors play diverse roles. Receptor Selection and Amplification Technology (RSAT) is used to study of the 5-HT2 subclass of serotonin receptors. RSAT is a phenotypic assay of receptor function that involves the heterologous expression of receptors in mammalian fibroblasts. Using this technology, it is demonstrated that native 5-HT2A receptors possess significant constitutive, or agonist-independent, receptor activity.[48–50] The activating mutations in 5-HT2A receptors may be causative or predisposing to neuropsychiatric disease, or modulate response to treatment with antipsychotics.[48] The creation of a transgenic mouse model that exhibits increased 5-HT2A receptor activity may provide an excellent model of preclinical evaluation of antipsychotic drugs having 5-HT2A receptor activity. Many compounds used in psychiatry are found to be potent 5-HT2A inverse agonists.[48] Thus 5-HT2A receptor inverse agonism may be the molecular mechanism of antipsychotic efficacy in humans. 5-HT system also plays a role in learning and memory. Meneses[49] reported that post-training injection of SB-224289 (5-HT1B receptor inverse agonist) facilitated learning and this effect was partially reversed by GR127935 (a 5-HT1B/1D receptor antagonist), but not by MDL100907 (a 5-HT2A receptor antagonist) or ketanserin (a 5-HT1D/2A/7 receptor antagonist) at low doses. Hence, 5-HT1B receptor inverse agonists or antagonists could represent drugs for the treatment of learning and memory dysfunctions.[49] In order to determine the possible relationship between antipsychotic drug properties and inverse agonist activity at 5HT6 and 5HT7 receptors, constitutively activated forms of these receptors were created by site-specific mutagenesis. Typical and atypical antipsychotic drugs were assayed for their potencies as inverse agonists at these mutated receptors.[49] The inverse agonist mechanism-of-action of the atypical antipsychotic drugs showed greater inverse agonism at the 5HT7 receptors as compared to typical agents.[50]
An important implication of inverse agonism at serotonin receptors is development of compounds which have antipsychotic and antiparkinsonian activities. Selective 5HT2A/2C receptor inverse agonists have therapeutic potential for neurodegenerative diseases. These demonstrate in vivo efficacy in models of psychosis and dyskinesias. This includes activity in reversing MK-801 (dizocilpine, also known as MK-801, is a noncompetitive antagonist of the N-methyl-D-aspartate receptors)-induced locomotor dysfunction and also has antidyskinetic activity against MPTP (1-methyl-4 phenyl-1,2,3,6,tetrahydropyridine) primate model of extrapyramidal disorder, suggesting that this compound may be an efficacious antipsychotic and antidyskinesia agent.[50] Pimavanserin (ACP-103), a highly selective 5-HT2A inverse agonist, which attenuates psychosis in patients with Parkinson's disease with psychosis and is devoid of worsening of motor and nonmotor side effects.[51] In addition, APD125, a selective serotonin 5-HT2A receptor inverse agonist, significantly improves sleep maintenance in primary insomnia.[52]
Cannabinoid receptors (CB1;CB2)
Many cannabinoid effects are now known to be mediated by CB1 receptors that are present in the central nervous system as well as in certain neuronal and nonneuronal peripheral tissues or by CB2 receptors that are found mainly in cells of the immune system. Both these receptors are GPCRs and thus exhibit significant constitutional activity. SR141716A is a selective CB1 receptor inverse agonist and SR144528 has selective CB2 receptor inverse agonist activity.[53] AM630 is a CB2-selective inverse agonist and has a weak partial agonist at CB1 receptors. L759633 and L759656 are both potent CB2-selective agonists.[53]
There are endogenous ligands derived from membrane lipids, which bind with CB1 and CB2 receptors.[54] Hemopressin is such a peptide ligand that acts as an antagonist as well as inverse agonist. This agent acts as an antinociceptive in different in vivo models of pain. This has opened new avenues for newer analgesic agents.[54] The central cannabinoid (CB1) receptors play a role in controlling food consumption and are therefore targets for evaluation of antiobesity medications.[55] Rimonaband, an antagonist at CB1 receptors and taranaband, a highly selective inverse agonist at CB1 receptors were developed for their use in obesity. Unfortunately both these agents have been withdrawn due to serious psychiatric manifestations. AM251, an inverse agonist at CB1 receptors, has antidepressant and anorectic action in mice. The tail-suspension test (TST) and forced-swim test (FST) are used as tests sensitive to antidepressant compounds. AM251 significantly reduced immobility at 10 mg/kg in the TST and at 1 and 10 mg/kg in the FST. In addition, it reduced fasting-induced hyperphagia in mice.[55] These observations suggest that it has significant antidepressant and anorectic action. CB2 receptors have immunoregulatory role. An inverse agonist at CB2 receptors, Sch414319 is a potent modulator of immune cell mobility in vivo.[56]
Angiotensin receptors (AT1)
Constitutional activation is demonstrated at AT1 receptors. Losartan, candesartan, and olmesartan have been shown to possess inverse agonist effects in assays evaluating c-fos gene expression and phosphorylated extracellular signal-regulated protein kinases.[57] Mechanical stretching of cardiac cells activates the AT1 receptors without the involvement of angiotensin II and that is inhibited by the inverse agonistic activity of candesartan and olmesartan. It is conceptually novel that AT1 receptor directly mediates mechanical stress-induced cellular responses, and inverse-agonist activity of AT1 -receptor blockers (ARBs), such as candesartan, emerges as an important pharmacological parameter that determines its efficacy in preventing organ damage (stretch-induced hypertrophy) in cardiovascular diseases.[58]
β-Adrenergic receptor (β-AR)
β2-Adrenergic agonists are sheet anchor in therapy of bronchial asthma. Short-acting agents, such as salbutamol and terbuline, are used by inhalation to terminate an acute attack. Long-acting β2-adrenergic agonists (LABAs) are also given by inhalation to prevent recurrent bronchospasm. There have been concerns about increased risk of asthma-related deaths with the use of LABAs.[59] Specific label warnings have been recommended by the Food and Drug Administration recently indicating judicious use of these agents. In addition, chronic administration of β2-adrenergic agonists has been demonstrated to lead to reduced responsiveness resulting from desensitization, sequestration, and downregulation of receptors.[60] This consequence may be an observed loss of responsiveness seen in asthmatic patients on long-acting β-agonists leading to tolerance or tachyphylaxis. Furthermore, there is also an increased hyper-responsiveness of the pulmonary airways in response to provocative allergens. One of the reasons for this is the proinflammatory activity of LABAs. The proinflammatory role of LABAs is attributed to activation of inflammatory cells, such as type-2 T cells which exhibit β2AR-agonist-promoted proinflammatory properties and/or to activation of alternative signaling pathways independent of cAMP activation.[60] This proinflammatory activity is also attributed to the constitutive activity of β2ARs. Nguyen et al.[61] proposed that β2ARs activation, resulting from constitutive β2AR activity, endogenous catecholamines, or inhaled β-agonists, has a positive effect on airway inflammation that generates intermediates, which cause airway smooth muscle contraction and promotes airway mucus secretion.
Inverse agonistic activity is demonstrated in some beta-receptor antagonists, such as nadolol.[10] It is hypothesized that inverse agonism may reduce airway inflammation. β2AR inverse agonists by inhibiting β2AR activity may reduce allergic inflammation and nullify or modestly inhibit the bronchoprotective/relaxing effect of β2AR activity in bronchial smooth musculature.[61–63]
Chronic administration of cardioselective β inverse agonists do not change pulmonary function in patients with congective heart failure (CHF) and chronic obstructive pulmonary disease (COPD) or asthma.[62] Forced expiratory volume (FEV1), a standard measure of pulmonary function, was unchanged in patients treated with cardioselective β inverse agonists. Therefore, chronic administration of cardioselective β inverse agonists are safe in patients with CHF and pulmonary airway disease. Chronic administration of an inverse agonist has the effect of upregulating the population of active β-adrenergic receptors.[62–64]
Nguyen et al. have recently studied the effects of beta-blockers—ICI 118,551 and nadolol (full inverse agonists at β2-ARs)—on airway inflammation in rats and showed that chronic administration of beta-blockers decreased airway hyper-responsiveness in a murine model of asthma. Administration of beta-blockers in a murine model of allergic asthma resulted in a reduction in total cell counts, eosinophils, and the cytokines IL-13, IL-10, IL-5, and TGF-β1 in bronchoalveolar lavage, and attenuated epithelial mucin content and morphologic changes. These results indicate that in a murine model of asthma, chronic administration of beta-blockers reduce inflammation and mucous production.[61]
β-blockers are presently contraindicated for asthmatics, but might actually be able to prevent serious attacks when used judiciously on long-term basis.[63] Bond and colleagues proposed that duration of therapy might be a key determinant of whether β-adrenoceptor ligands have a positive or negative effect. They suggested that beta-blockers, which have been shown to be harmful to asthmatics in the short-term by heightening airway constriction, might actually have beneficial effects when administered on long-term basis. To investigate this hypothesis, airway sensitivity to methacholine, which elicits airway narrowing, is commonly used as provocative test. It was monitored in mouse models of asthma after chronic (daily dosing over 28 days) and acute (single dose 15 min before methacholine) administration of a range of β2-ligands; salbutamol (a partial β2-agonist), alprenolol (a β-antagonist with partial β2-agonist activity), carvedilol and nadolol (both β1/β2-antagonists with inverse agonist activity at the β2-adrenoceptor). Acute administration of salbutamol decreased airway responsiveness to methacholine compared with nontreated asthmatic mice, whereas chronic dosing had no effect. Similar results were observed for alprenolol, indicating that it was behaving as a β2-agonist. Carvedilol and nadolol, on the other hand, although acutely detrimental, significantly reduced airway sensitivity after chronic administration. The authors then used radioligand-binding assays to examine β-adrenoceptor density in mouse lungs. They found the density of β-adrenoceptors to be significantly lower in asthmatic mice compared with controls. They also reported that the capacity to reduce airway responsiveness in asthmatic mice correlated with a ligand's ability to increase β-adrenoceptor density. Salbutamol and alprenolol increased β-adrenoceptor density in the lungs after an acute dose but had no effect after chronic administration, whereas carvedilol and nadolol had no acute effect on receptor density, but significantly increased β-adrenoceptor levels with chronic dosing. In view of these findings the β-adrenoceptor blockers with inverse agonist activity are attractive long-term therapeutic options in chronic airway obstruction. There are theoretical advantages in the use. There will not be tachyphylaxis on long-term use and improvement in signal mechanism will counteract the inflammation underlying the airway narrowing. At present it remains to be known as to how and when a β-blocker be started in asthma.[63,65]
Dopaminergic receptors (D1, D2, D3, D4)
Constitutional activation is demonstrated in dopaminergic receptors. The antipsychotic drugs are conventionally thought to act as antagonists at D2 dopamine receptors but recently these drugs have been shown to possess inverse agonist properties at this receptor.[66] Interestingly, several compounds previously classified as D2 receptor antagonists behaved as inverse agonists at the D3 receptor also, that is, they inhibited the basal [35S] GTPγS binding in a dose-dependent fashion.[66] Haloperidol was full inverse agonist but clozapine was a partial inverse agonist.[66] In NG 108-15 cells but not in wild-type cells, haloperidol, fluphenazine, and various other antipsychotics inhibited basal [3H] thymidine incorporation in a concentration-dependent manner. In contrast, other dopamine antagonists, such as nafadotride, a D3-preferring antagonist, was without effect.[67] The concentration–response curve of haloperidol was shifted to the right in the presence of nafadotride. These data indicate that some dopamine antagonists behave as inverse agonists, and thus appear to inhibit an agonist-independent activity of the D3receptor on [3H]thymidine incorporation pathway, but not on the cAMP pathway.[67] Robert and Strang showed that some inverse agonists {(+)-butaclamol, spiperone} act by stabilizing the uncoupled form of the receptor at the expense of the coupled form.[68]
Using overexpression of Gaoin cell-based functional assay, which induces constitutive activity in the human D2-like receptors (D2, D3, and D4), many antidopaminergic antipsychotics showed D2and D3inverse agonism. Aripiprazole and the principle active metabolite of clozapine-NDMC [8-chloro-11-(1-piperazinyl)-5H-dibenzo [b, e] [1,4] diazepine] were identified as partial agonists at D2 and D3 receptors, although clozapine itself was a full inverse agonist at these receptors.[66,67] It is proposed that the low incidence of extrapyramida syndrome associated with clozapine and aripiprazole used may be due, in part, to these partial inverse agonist properties of NDMC and aripiprazole. It is possible to use NDMC in place of clozapine so as to avoid unpleasant extrapyramidal effects.[66,68]
Conclusion
Hence it is clear that constitutional activity at GPCRs and non-GPCRs is a clinically relevant phenomenon. Many therapeutically useful antagonists are in fact either full or partial inverse agonists. If a disease is induced by constitution activation of receptors, it would be possible to use inverse agonists to subdue this constitutive activity. An exciting area is hypertension, where constitutive activation of beta-adrenergic receptors may lead to a rise in blood pressure. Potential new antihypertensive with inverse agonism can normalize the receptor activity, providing a rational therapeutic approach to essential hypertension. In future it will also be possible for a clinician to have highly selective agents with varying degrees of therapeutically useful inverse agonistic activity for the treatment of chronic disorders, such as asthma and chronic congestive heart failure.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Kenakin TP. Drug efficacy at G Protein-Coupled receptors. Annu Rev Pharmacol Toxicol. 2002;42:349–79. doi: 10.1146/annurev.pharmtox.42.091401.113012. [DOI] [PubMed] [Google Scholar]
- 2.Bond RA, Ijzerman AP. Recent developments in constitutive receptor activity and inverse agonism, and their potential for GPCR drug discovery. Trends Pharmacol Sci. 2006;27:92–6. doi: 10.1016/j.tips.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 3.Labarre M, Butterworth J, St-Onge S, Payza K, Schmidhammer H. Inverse agonism by Dmt-Tic analogues and HS 378, a naltrindole analogue. Eur J Pharmacol. 2000;406:R1–3. doi: 10.1016/s0014-2999(00)00636-1. [DOI] [PubMed] [Google Scholar]
- 4.Kenakin T. Inverse, protean, and ligand-selective agonism: Matters of receptor conformation. FASEB J. 2001;15:598–611. doi: 10.1096/fj.00-0438rev. [DOI] [PubMed] [Google Scholar]
- 5.Braestrup C, Schmiechen R, Neef G, Nielsen RA, Petersen EN. Interaction of convulsive ligands with benzodiazepine receptors. Science. 1982;216:1241–3. doi: 10.1126/science.6281892. [DOI] [PubMed] [Google Scholar]
- 6.Spiegel AM, Weinstein LS. Inherited diseases involving G Proteins and G Protein-coupled receptors. Annu Rev Med. 2004;55:27–39. doi: 10.1146/annurev.med.55.091902.103843. [DOI] [PubMed] [Google Scholar]
- 7.Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD, Mitarai S, et al. Early and delayed consequences of beta 2-adrenergic receptor over expression in mouse hearts: Critical role for expression level. Circulation. 2000;101:1707–14. doi: 10.1161/01.cir.101.14.1707. [DOI] [PubMed] [Google Scholar]
- 8.Dupré DJ, Le Gouill C, Gingras D, Rola-Pleszczynski M, Stanková J. Inverse agonist activity of selected ligands of the cysteinyl-leukotriene receptor. J Pharmacol Exp Ther. 2004;309:102–8. doi: 10.1124/jpet.103.059824. [DOI] [PubMed] [Google Scholar]
- 9.Engelhardt S, Grimmer Y, Fan GH, Lohse MJ. Constitutive activity of the human β1-adrenergic receptor in β1-receptor transgenic mice. Mol Pharmacol. 2001;60:712–7. [PubMed] [Google Scholar]
- 10.Chidiac P, Hebert TE, Valiquette M, Dennis M, Bouvier M. Inverse agonist activity of beta-adrenergic antagonists. Mol Pharmacol. 1994;45:490–9. [PubMed] [Google Scholar]
- 11.Baker JG, Hall IP, Hill SJ. Agonist and inverse agonist actions of β-Blockers at the human β2-ARs provide evidence for agonist-directed signaling. Mol Pharmacol. 2003;64:1357–69. doi: 10.1124/mol.64.6.1357. [DOI] [PubMed] [Google Scholar]
- 12.Gurdal H. Inverse agonism at b-adrenergic receptors: Therapeutic implications. Expert Rev Neurother. 2002;2:261–9. doi: 10.1586/14737175.2.2.261. [DOI] [PubMed] [Google Scholar]
- 13.Zhou YY, Yang D, Zhu WZ, Zhang SJ, Wang DJ, Rohrer DK, et al. Spontaneous activation of β2- but not β1-adrenoceptors expressed in cardiac myocytes from β1β2 double knockout mice. Mol Pharmacol. 2000;58:887–94. doi: 10.1124/mol.58.5.887. [DOI] [PubMed] [Google Scholar]
- 14.Nagaraja S, Iyer S, Liu X, Eichberg J, Bond RA. Treatment with inverse agonists enhances baseline atrial contractility in transgenic mice with chronic beta2-adrenoceptor activation. Br J Pharmacol. 1999;127:1099–104. doi: 10.1038/sj.bjp.0702645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keating GM, Jarvis B. Carvedilol, A review of its use in chronic congestive heart failure. Drugs. 2003;63:1697–741. doi: 10.2165/00003495-200363160-00006. [DOI] [PubMed] [Google Scholar]
- 16.Maack C, Cremers B, Flesch M, Hoper A, Sudkamp M, Bohm M. Different intrinsic activities of bucindolol, carvedilol and metoprolol in human failing myocardium. Br J Pharmacol. 2000;130:1131–9. doi: 10.1038/sj.bjp.0703400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beta-Blocker Evaluation of Survival Trial Investigators. A trial of the beta-blocker bucindolol in patients with advanced chronic heart failure. N Engl J Med. 2001;344:1659–67. doi: 10.1056/NEJM200105313442202. [DOI] [PubMed] [Google Scholar]
- 18.Bristow MR. What type of beta-blocker should be used to treat chronic heart failure? Circulation. 2000;102:484–6. doi: 10.1161/01.cir.102.5.484. [DOI] [PubMed] [Google Scholar]
- 19.Gilbert EM, Abraham WT, Olsen S, Hattler B, White M, Mealy P, et al. Comparative hemodynamic, left ventricular functional, and antiadrenergic effects of chronic treatment with metoprolol versus carvedilol in the failing heart. Circulation. 1996;94:2817–25. doi: 10.1161/01.cir.94.11.2817. [DOI] [PubMed] [Google Scholar]
- 20.Lowes BD, Chidiac P, Olsen S, Port JD, Bouvier M, Gilbert EM, et al. Clinical relevance of inverse agonism and guanine nucleotide modulatable binding properties of β-adrenergic receptor blocking agents. Circulation. 1994;90:I–543. [Google Scholar]
- 21.Verniero C, Höcht C, Opezzo JA, Taira C. Changes in the in vitro Pharmacodynamic properties of metoprolol in atrial myocardium isolated from spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 2007;34:161–5. doi: 10.1111/j.1440-1681.2007.04566.x. [DOI] [PubMed] [Google Scholar]
- 22.Liggett SB. ß-Adrenergic receptors in the failing heart: The good, the bad, and the unknown. J Clin Invest. 2001;107:947–8. doi: 10.1172/JCI12774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lefkowitz RJ, Rockman HA, Koch WJ. Catecholamines, cardiac beta-adrenergic receptors, and heart failure. Circulation. 2000;101:1634–7. doi: 10.1161/01.cir.101.14.1634. [DOI] [PubMed] [Google Scholar]
- 24.Yoshikawa T, Port JD, Asano K, Chidiak P, Bouvier M, Dutcher D, et al. Cardiac adrenergic receptor effects of carvedilol. Eur Heart J. 1996;17(Suppl B):8–16. doi: 10.1093/eurheartj/17.suppl_b.8. [DOI] [PubMed] [Google Scholar]
- 25.Wade SM, Lan KL, Moore DJ, Neubig RR. Inverse agonist activity at the α2A-adrenergic receptor. Mol Pharmacol. 2001;59:532–42. doi: 10.1124/mol.59.3.532. [DOI] [PubMed] [Google Scholar]
- 26.Allen LF, Lefkowitz RJ, Caron MG, Cotecchia S. G-protein-coupled receptor genes as proto-oncogenes: Constitutively activating mutation of the α1B-adrenergic receptor enhances mitogenesis and tumorigenicity. Proc Natl Acad Sci U S A. 1992;88:11354–8. doi: 10.1073/pnas.88.24.11354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vazquez SM, Mladovan AG, Perez A, Baldi A, Luthy IA. Human breast cell lines exhibit functional alpha2-adrenoceptors. Cancer Chemother Pharmacol. 2006;58:50–61. doi: 10.1007/s00280-005-0130-4. [DOI] [PubMed] [Google Scholar]
- 28.Liu JG, Prather PL. Chronic exposure to mu-opioid agonists produces constitutive activation of mu-opioid receptors in direct proportion to the efficacy of the agonist used for pretreatment. Mol Pharmacol. 2001;60:53–62. doi: 10.1124/mol.60.1.53. [DOI] [PubMed] [Google Scholar]
- 29.Gonzalez-Cabrera PJ, Shi T, Yun J, McCune DF, Rorabaugh BR, Perez DM. Differential regulation of the cell cycle by alpha1-adrenergic receptor subtypes. Endocrinology. 2004;145:5157–67. doi: 10.1210/en.2004-0728. [DOI] [PubMed] [Google Scholar]
- 30.Lyons J, Landis CA, Harsh G, Vallar L, Grünewald K, Feichtinger H, et al. Two G-protein oncogenes in human endocrine tumors. Science. 1990;249:655–9. doi: 10.1126/science.2116665. [DOI] [PubMed] [Google Scholar]
- 31.Bruzzone A, Pinero PC, Castillo LF, Sarappa MG, Rojas P, Lanari C, et al. α2-Adrenoceptor action on cell proliferation and mammary tumour growth in mice. Br J Pharmacol. 2008;155:494–504. doi: 10.1038/bjp.2008.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy JM, Jackie J, Mellor-Burke J, Lumeng L, Li T. BDZ inverse agonist RO15-4513 exerts prolonged and selective suppression of ethanol intake in alcohol preferring Rats. Psychopharmacology. 1994;115:325–31. doi: 10.1007/BF02245073. [DOI] [PubMed] [Google Scholar]
- 33.Sieghart W. Pharmacology of benzodiazepine receptors: An update. J Psychiatry Neurosci. 1994;19:24–9. [PMC free article] [PubMed] [Google Scholar]
- 34.Wang F, Xu Z, Yen CT, Chow CY, Lui YL, Tsang Y, et al. 6,2’ Dihydroxyfl avone, a subtype-selective partial inverse agonist of GABAA receptor benzodiazepine site. Neuropharmacology. 2007;53:574–82. doi: 10.1016/j.neuropharm.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 35.Pinna G, Galici R, Herbert H, Schneider H, Stephens DN, Turski L. Alprazolam dependence prevented by substituting with the β-carbolineabecarnil. Proc Natl Acad Sci U S A. 1997;94:2719–23. doi: 10.1073/pnas.94.6.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cruz SL, Villarreal JE, Volkow ND. Further evidence that naloxone acts as an inverse opiate agonist: Implications for drug dependence and withdrawal. Life Sci. 1996;26:PL381–9. doi: 10.1016/0024-3205(96)00250-0. [DOI] [PubMed] [Google Scholar]
- 37.Costa T, Herz A. Antagonists with negative intrinsic activity at δ opioid receptors coupled to GTP-binding proteins. Proc Natl Acad Sci U S A. 1989;86:7321–5. doi: 10.1073/pnas.86.19.7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z, Bilsky EJ, Porreca F, Sadee W. Constitutive mu opioid receptor activation as a regulatory mechanism underlying narcotic tolerance and dependence. Life Sci. 1994;54:L339–50. doi: 10.1016/0024-3205(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 39.Leurs R, Church MK, Taglialatela M. H1-antihistamines: Inverse agonism, anti-inflammatory actions and cardiac effects. Clin Exp Allergy. 2002;32:489–98. doi: 10.1046/j.0954-7894.2002.01314.x. [DOI] [PubMed] [Google Scholar]
- 40.Fitzsimons CP, Monczor F, Fernandez N, Shayo C, Davio C. Mepyramine, a histamine H1 receptor inverse agonist, binds preferentially to a G protein-coupled form of the receptor and sequesters G protein. J Biol Chem. 2004;279:34431–9. doi: 10.1074/jbc.M400738200. [DOI] [PubMed] [Google Scholar]
- 41.Simons FE. Advances in H1 antihistamines. N Engl J Med. 2004;351:2203–17. doi: 10.1056/NEJMra033121. [DOI] [PubMed] [Google Scholar]
- 42.Leurs R, Church MK, Taglialatela M. Antihistamines: Inverse H1-agonism, anti-inflammatory actions and cardiac effects. Clin Exp Allergy. 2002;32:489–98. doi: 10.1046/j.0954-7894.2002.01314.x. [DOI] [PubMed] [Google Scholar]
- 43.Monczor F, Fernandez N, Legnazzi BL, Riviero ME, Baldi A, Shayo C, et al. Tiotidine, a histamine H2 receptor inverse agonist that binds with high affinity to an inactive G-protein–coupled form of the receptor. Experimental support for the cubic ternary complex model. Mol Pharmacol. 2003;64:512–20. doi: 10.1124/mol.64.2.512. [DOI] [PubMed] [Google Scholar]
- 44.Morisset S, Rouleau A, Ligneau X, Gbahou F, Tardivel-Lacombe J, Stark H, et al. High constitutive activity of native H3 receptors regulates histamine neurons in brain. Nature. 2000;408:860–4. doi: 10.1038/35048583. [DOI] [PubMed] [Google Scholar]
- 45.Netzer P, Gaia C, Sandoz M, Huluk T, Gut A, Halter F, et al. Effect of repeated injection and continuous infusion of omeprazole and ranitidine on intragastric pH over 72 hours. Am J Gastroenterol. 1999;94:351–7. doi: 10.1111/j.1572-0241.1999.857_y.x. [DOI] [PubMed] [Google Scholar]
- 46.Pillot C, Heron A, Schwartz JC, Arrang JM. Ciproxifan, a histamine H3-receptor antagonist/inverse agonist, modulates the effects of methamphetamine on neuropeptide mRNA expression in rat striatum. Eur J Neurosci. 2003;2:307–14. doi: 10.1046/j.1460-9568.2003.02422.x. [DOI] [PubMed] [Google Scholar]
- 47.Brabant C, Alleva L, Grisar T, Quertemont E, Lakaye B, Ohtsu H, et al. Effects of the H3 receptor inverse agonist thioperamide on cocaine-induced locomotion in mice: Role of the histaminergic system and potential pharmacokinetic interactions. Psychopharmacology (Berl) 2009;202:673–87. doi: 10.1007/s00213-008-1345-y. [DOI] [PubMed] [Google Scholar]
- 48.Herrick-Davis K, Grinde E, Teitler M. Inverse agonist activity of atypical antipsychotic drugs at human 5-hydroxytryptamine2C receptors. J Pharmacol Exp Ther. 2000;295:226–32. [PubMed] [Google Scholar]
- 49.Meneses A. Could the 5-HT1B receptor inverse agonism affect learning consolidation? Neurosci Biobehav Rev. 2001;2:193–201. doi: 10.1016/s0149-7634(01)00007-0. [DOI] [PubMed] [Google Scholar]
- 50.Purohit A, Smith C, Herrick-Davis K, Teitler M. Stable expression of constitutively activated mutant h5HT6 and h5HT7 serotonin receptors: Inverse agonist activity of antipsychotic drugs. Psychopharmacology (Berl) 2005;179:461–9. doi: 10.1007/s00213-004-2057-6. [DOI] [PubMed] [Google Scholar]
- 51.Meltzer HY, Roger-Mills R, Revell S, Williams H, Johnson A, Bahr D. Pimavanserin, a Serotonin2A Receptor Inverse Agonist, for the Treatment of Parkinson's Disease Psychosis. Neuropsychopharmacology. 2010;35:881–92. doi: 10.1038/npp.2009.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosenberg R, Seiden DJ, Hull SG, Erman M, Schwartz H, Anderson C, et al. APD125, a selective serotonin 5-HT(2A) receptor inverse agonist, significantly improves sleep maintenance in primary insomnia. Sleep. 2008;31:1663–71. doi: 10.1093/sleep/31.12.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, et al. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656 and AM630. Br J Pharmacol. 1999;126:665–72. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heimann AS, Gomes I, Dale CS, Pagano RL, Gupta A, de Souza LL, et al. Hemopressin is an inverse agonist of CB 1 cannabinoid receptors. Proc Natl Acad Sci U S A. 2007;104:20588–93. doi: 10.1073/pnas.0706980105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shearman LP, Rosko KM, Fleischer R, Wang J, Xu S, Tong XS, et al. Antidepressant-like and anorectic effects of the cannabinoid CB1 receptor inverse agonist AM251 in mice. Behav Pharmacol. 2003;14:573–82. doi: 10.1097/00008877-200312000-00001. [DOI] [PubMed] [Google Scholar]
- 56.Lunn CA, Reich EP, Fine JS, Lavey B, Kozlowski JA, Hipkin RW, et al. Biology and therapeutic potential of cannabinoid CB2 receptor inverse agonists. Br J Pharmacol. 2008;153:226–39. doi: 10.1038/sj.bjp.0707480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qin Y, Yasuda N, Akazawa H, Ito K, Kudo Y, Liao CH, et al. Multivalent ligand-receptor interactions elicit inverse agonist activity of AT(1) receptor blockers against stretch-induced AT(1) receptor activation. Hypertens Res. 2009;32:875–83. doi: 10.1038/hr.2009.117. [DOI] [PubMed] [Google Scholar]
- 58.Yasuda N, Akazawa H, Qin Y, Zou Y, Komuro I. A novel mechanism of mechanical stress-induced angiotensin II type 1-receptor activation without the involvement of angiotensin II. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:393–9. doi: 10.1007/s00210-007-0215-1. [DOI] [PubMed] [Google Scholar]
- 59.Chowdhury BA, Pan GD. The FDA and safe use of long-acting beta-agonists in the treatment of asthma. N Engl J Med. 2010;362:1169–71. doi: 10.1056/NEJMp1002074. [DOI] [PubMed] [Google Scholar]
- 60.Callaerts-Vegh Z, Evans KL, Dudekula N, Cuba D, Knoll BJ, Callaerts PF, et al. Effects of acute and chronic administration of beta-adrenoceptor ligands on airway function in a murine model of asthma. Proc Natl Acad Sci U S A. 2004;101:4948–53. doi: 10.1073/pnas.0400452101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nguyen LP, Omoluabi O, Parra S, Joanna M, Frieske JM, Clement C, et al. Chronic exposure to beta-blockers attenuates inflammation and mucin content in a murine asthma model. Am J Respir Cell Mol Biol. 2008;38:256–62. doi: 10.1165/rcmb.2007-0279RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salpeter S, Ormiston T, Salpeter E. Cardioselective beta-blockers for reversible airway disease. Cochrane Database Syst Rev. 2002:CD002992. doi: 10.1002/14651858.CD002992. [DOI] [PubMed] [Google Scholar]
- 63.Bond RA. Is paradoxical pharmacology a strategy worth pursuing? Trends Pharmacol Sci. 2001;22:273–6. doi: 10.1016/s0165-6147(00)01711-9. [DOI] [PubMed] [Google Scholar]
- 64.Penn RB. Embracing emerging paradigms of G protein-coupled receptor agonism and signaling to address airway smooth muscle pathobiology in asthma. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:149–69. doi: 10.1007/s00210-008-0263-1. [DOI] [PubMed] [Google Scholar]
- 65.Singh BN, Whitlock RM, Comber RH, Williams FH, Harris EA. Effects of cardioselective beta adrenoceptor blockade on specific airways resistance in normal subjects and in patients with bronchial asthma. Clin Pharmacol Ther. 1976;19:493–501. doi: 10.1002/cpt1976195part1493. [DOI] [PubMed] [Google Scholar]
- 66.Griffon N, Pilon C, Sautel F, Schwartz C, Sokoloff P. Antipsychotics with inverse agonist actions on D3 receptors. J Neural Transm. 1996;103:1163–75. doi: 10.1007/BF01271201. [DOI] [PubMed] [Google Scholar]
- 67.Malmerg A, Mikaels A, Mohell N. Agonist and inverse agonist activity at the dopamine D3 receptor measured by guanosine 5’-[γ-Thio]triphosphate-[35S] binding. J Pharmacol Exp Ther. 1998;285:119–26. [PubMed] [Google Scholar]
- 68.Roberts DJ, Strange PG. Mechanisms of inverse agonist action at D2 dopamine receptors. Br J Pharmacol. 2005;145:34–42. doi: 10.1038/sj.bjp.0706073. [DOI] [PMC free article] [PubMed] [Google Scholar]