

Abstract
Background:
Researchers have determined that Indians face a higher risk of heart disease, despite the fact that nearly half of them are vegetarians and lack many of the other traditional risk factors. In the below-30 age group, coronary artery disease mortality among Indians is three-fold higher than in the whites in United Kingdom and ten-fold higher than the Chinese in Singapore. High levels of homocysteine have been widely linked to the early onset of heart diseases in other populations, although a definite proof among Indians is lacking, which needs to be investigated by way of screening for factors responsible for high homocysteine levels.
Objective:
To screen for genetic factors responsible for hyperhomocysteinemia and the risk for premature coronary artery disease.
Materials and Methods:
A total of 100 individuals with proven premature coronary artery disease and 200 age-and-sex matched controls were screened for polymorphisms in Methylenetetrahydrofolate reductase (MTHFR) (C677T) Methionine synthase (MS) genes (A2756G, C2758G), and the B12 and Folate levels were estimated.
Results:
Results from the mutational analysis revealed that in the study group, seven individuals had a polymorphism for the C677T allele in the MTHFR gene (one homozygous and six heterozygous) (Fischer's Exact test P > 0.046) (OR: 0.2711 95% CI 0.0774 to 0.9491). Six were heterozygous for the A2756G polymorphism in the MS gene (Fischer's Exact test P > 0.0012). None showed a polymorphism at the C2758G allele in the MS gene. Four controls showed heterozygosity for the C677T polymorphism and none for the MS gene. The B12 and Folate levels were significantly lower in the study group as compared to the controls.
Conclusions:
It is important to know which factors determine the total homocysteine concentrations. In the general population, the most important modifiable determinants of tHcy are folate intake and coffee consumption. Smoking and alcohol consumption are also associated with the total homocysteine concentrations, but more research is necessary to elucidate whether these relations are not originating from residual confounding due to other lifestyle factors.
Keywords: Hyperhomocysteinemia, methionine synthase, methylenetetrahydrofolate reductase, polymorphisms, premature coronary artery disease
INTRODUCTION
Indians have the highest rate of heart disease, usually two-to-three times higher than the Americans, Europeans and Japanese, according to a study.[1] About 25% of heart attacks among men and women of Indian descent occur when they are younger than 40. This is unheard of in other populations. In a significant study, in view of the propensity of Indians for heart diseases, scientists have recorded high levels of homocysteine, which is emerging as an ‘independent risk factor’. The elevated levels of this compound, homocysteine, may add to the already existing high risk factors.[2] High levels of homocysteine have been widely linked to early onset of heart diseases in other populations, although a definite proof among Indians is lacking, which needs to be investigated by way of screening for factors responsible for high homocysteine levels.[3]
Cardiovascular disease remains the major cause of morbidity and death in developed countries. The major classic risk factors like diabetes, hypertension, and the like, and non-modifiable risk factors such as age, sex, and family history cannot fully explain why some individuals are prone to coronary artery disease and others are not. Pathological and epidemiological studies suggest that only about one-half to two-thirds of the variation in anatomic extent of atherosclerosis and risk for atherosclerotic vascular disease can be explained by the classic risk factors. Therefore, many emerging risk factors have been investigated, and among these, elevated plasma homocysteine levels (hyperhomocysteinemia) is of particular interest. Recent epidemiological studies have shown that moderately elevated plasma homocysteine levels are highly prevalent in the general population and are associated with an increased risk for fatal and non-fatal cardiovascular disease, independent of classic cardiovascular risk factors.
Within the last decade, homocysteine has arguably attracted more attention from biochemists than any other amino acid. Homocysteine is a sulfur-containing amino acid produced during the metabolism of the essential amino acid methionine. On account of this, homocysteine may be renewed into methionine. It may also be converted into either cysteine or taurine, via transsulfuration. If specific cofactors are lacking, homocysteine may not be converted into any beneficial compounds within the body.
Homocysteine is toxic in nature and the accumulation (or build up) of this amino acid will often result in plasma toxicity. Various epidemiological studies indicate that this homocysteine toxicity, in concurrence with genetic and dietary factors, accounts for the elevated risks of coronary heart disease, stroke, and peripheral vascular disease.[4]
Hyperhomocysteinemia is an independent risk factor for cardiovascular disease. Although there is a growing body of evidence that homocysteine plays a causal role in atherogenesis, specific mechanisms to explain the underlying pathology have remained elusive.
Homocysteine (Hcy) is a sulfhydryl-containing amino acid, formed during the metabolism of methionine, found in abundance in proteins of animal origin. Homocysteine is metabolized via remethylation or transsulfuration, and there are two alternate pathways of remethylation in humans. The predominant vitamin B12-dependent re-synthesis of methionine from Hcy occurs in all human tissues and comprises of the transfer of the methyl group during the conversion of 5-methyltetrahydrofolate to tetrahydrofolate. Tetrahydrofolate binds the carbon unit from serine to nitrogen N5 and N10 forming 5,10-methylenetetrahydrofolate that is later converted to 5-methly tetrahydrofolate in the reaction catalyzed by the 5,10-MTHFR. An additional remethylation reaction of minor physiological importance requires the presence of betaine as the methyl group donor and betaine-homocysteine methyl transferase as a catalyzing enzyme. In the transsulfuration pathway Hcy combines with serine forming cystathione in a vitamin-dependent reaction, catalyzed by cystathione beta-synthase (CBS). Cystathione is later hydrolyzed to cysteine and alpha-ketobutyrate in the reaction catalyzed by gamma-cystathionase, with pyridoxal phosphate as a cofactor. Cysteine is eventually metabolized to taurine and sulfates.[5]
It is well-accepted that the elevated blood levels of homocysteine (hyperhomocysteinemia) increase the risk of cardiovascular disease. High homocysteine is a strong predictor for cardiovascular events and is a risk factor for mortality in patients with coronary artery disease.[6,7]
The understanding of homocysteine-induced vascular dysfunction is still incomplete from a mechanistic point of view. Homocysteine may alter the normally anti-coagulant phenotype of blood vessels to a pro-coagulant phenotype, along with the activation of platelets. In homocystinurics with severe hyperhomocysteinemia, this leads to premature thromboembolism. The movement of monocytes into the neo-intima may be stimulated by the homocysteine-induced expression of the monocyte chemoattractant protein 1 (MCP-1) in the endothelium. Homocysteine may also be mitogenic to smooth muscle cells that invade the neo-intima of the developing lesion. Recent studies suggest that hyperhomocysteinemia in humans and animals cause endothelial dysfunction, which results in impaired relaxation of the blood vessels. Although the mechanism is still not clear, many of these studies suggest that homocysteine limits the bioavailability of endothelial-derived nitric oxide, a potent vasodilator. The limited bioavailability of nitric oxide may be due to the ability of homocysteine to generate reactive oxygen species, and hence, oxidative stress.[8]
Factors that determine plasma Hcy levels can be genetic or environmental. The former include a rare cystathionine beta-synthase (CBS) deficiency and homozygous forms of the MTHFR C677T mutation. A vascular event occurs in half of the untreated patients, of less than 30 years.
The MTHFR gene is located on chromosome 1 at 1p 36.3. The cDNA sequence is 2.2 kilo bases long and appears to consist of 11 exons.[9] The C677T allele is a single base pair mutation in which a cytosine is converted to thymine at basepair 677, resulting in an amino acid substitution of alanine to valine, in the enzyme.[10,11] The molecular genetics of the mutation have been recently reviewed. Functionally, the encoded protein has a reduced protein activity at 37 degrees[12] and higher, so the C677T mutation is often termed ‘thermolabile’. For instance, compared to the controls treated similarly, samples from the C677T homozygotes have 50 - 60% lower MTHFR activity at 37°C and about 65% lower activity at 46°C. Heterozygotes are in the intermediate range.
Methionine synthase, one of the B12-dependent mammalian enzymes, catalyzes the remethylation of homocysteine to methionine and the concurrent demethylation of 5-methylenetetrahydrofolate to tetrahydrofolate. Under conditions of B12-depletion, such as pernicious anemia, loss of the methione synthase activity leads to a ‘methyl folate trap’. The depletion of other folate coenzymes result in defective DNA synthesis and the development of megablastic anemia. Of late homocysteine has received considerable attention as elevations in plasma homocysteine have been implicated as a risk factor for vascular disease.[13]
The methionine synthase gene is located on chromosome 1q43, producing methionine and tetrahydrofolate. The gene coding for MS has common polymorphic forms (A2756G, D919G, and C2758G H920D). The base transition, converting an aspartic acid into glycine, occurs near the crucial vitamin B12 binding site, and therefore, might influence the enzyme secondary structure with possible functional consequences.[14] The present study was undertaken to screen SNP's of MTHFR, MS genes and levels of vitamin B12 and Folate in individuals with premature coronary artery disease from India.
MATERIALS AND METHODS
A total of 100 individuals with proven premature cardiovascular disease, that is, individuals with myocardial infarction or angina, below 45 years of age, who were non-hypertensive, non-diabetic, and with normal lipid profiles, and 200 age- and sex-matched controls were studied for the stated polymorphisms. Blood was collected from the individuals with prior informed written consent and was used for polymorphism and biochemical analyses. Peripheral blood was collected in 5-mL EDTA container for isolation of genomic DNA. The white blood cells were isolated from the collected blood samples using erythrocyte lysis buffer. Cells were incubated at 55°C for 2 hours with 20 mg/mL proteinase K and 20% sodium lauryl sulphate. The cells were added to 5 mL phenol (Tris saturated, pH 8) and centrifuged at 10,000rpm for 10 minutes. The supernatant was collected and 5 mL phenol-chloroform-isoamyl alcohol (25:24:1) was added and centrifuged at 10,000g for 10 minutes. To the collected supernatant, 5 mL chloroform was added and centrifuged at 10,000rpm. Absolute alcohol was added to the supernatant to precipitate the DNA. The DNA was dissolved in 150 μL Millipore water and stored at –20°C.
C677T mutational analysis of the Methylene tetrahydrofolate reductase (MTHFR) gene was carried out by the method of Frosst et al., 1995. The primers used were Forward Primer : 5’ TGA AGG AGA AGG TGT CTG CGG GA 3’ and Reverse Primer: 5’ AGG ACG GTG CGG TGA GAG TG 3’. A2756G mutational analysis of the Methionine synthase (MS) gene was carried out by the method of D. Leclerc et al, using Forward Primer : 5’CAT GGA AGA ATA TGA AGA TAT TAG AC 3’ and Reverse Primer: 5’ GAA CTA GAA GAC AGA AAT TCT CTA 3’. C2758G mutational analysis of the Methionine synthase (MS) gene was carried out by the method of Leclerc et al., (1996), using Forward Primer: 5’ TGA AGG AGA AGG TGT CTG CGG GA 3’ and Reverse Primer: 5’AGG ACG GTG CGG TGA GAG TG 3’. The concentration of the primers used was 20 pmoles, 10 mM of dNTPs, 1 unit of Taq enzyme, 10X concentration of Taq buffer, and 200 ng of template DNA.
Thermal cycling conditions for C677T MTHFR were 95°C for five minutes and 95°C for one minute, 59°C for one minute and 72°C for two minutes, followed by 72°C for seven minutes, for a total of 35 cycles, which yielded a product size of 198 bp. Thermal cycling conditions for A2756G and C2758G MS were 95°C for five minutes and 95°C for one minute, 61°C for one minute, 72°C for two minutes, followed by 72°C for seven minutes for a total of 35 cycles, which yielded a product size of 189 bp.
The MTHFR mutation creates a Hinf I restriction site at 677 position in the gene. The polymerase chain reaction (PCR) products were subjected to restriction digestion using 5 units of the Hinf I restriction enzyme at 37°C for 12 hours. Homozygote C677T yielded 175 bp; 23 bp, the heterozygote 198 bp, 175 bp, and 23 bp, and the wild type yielded 198 bp. The methionine synthase A2756G mutation created a Hae III restriction site at the 2756 position in the gene. The PCR products were subjected to restriction digestion using five units of BsuRI, an isoschizomer of the Hae III restriction enzyme, at 37°C for 12 hours. Homozygote A2756G yielded 150 bp; 39 bp, the heterozygote yielded 189 bp, 150 bp, and 39 bp, and the wild type yielded 189 bp. The methionine synthase C2758G mutation abolished the Sau96I restriction site at 2758 position in the gene. The PCR products were subjected to restriction digestion using five units of Cfr13I, an isoschizomer of the Sau96 I restriction enzyme, at 37°C for three hours. The homozygote C2758G yielded 189 bp, the heterozygote yielded 189 bp, 150 bp, and 39 bp, and the wild type yielded 150 bp, 39 bp. All the products were resolved on 2% agarose gel.
Estimation of serum B12 levels
Estimation of serum vitamin B12 levels was done using a competitive protein binding chemiluminescent assay.(IMMULITE; Diagnostic Products Corporation).
The serum was diluted with Vitamin B12 sample diluent. Two hundred microliters of the diluted serum sample or standard were added to the prepared tubes, as also 1000 μl working solution (Borate KCN buffer solution and Dithiothreitol Solution), and it was vortexed. All the test tubes were loosely capped and placed in a covered, boiling water bath for 15 - 20 minutes. The tubes were removed from the boiling water bath, and cooled in an ambient water bath for five minutes. Three hundred and fifty microliters of the treated sample was added to the each IMMULITE sample cup, and incubated for 30 minutes at 37°C, with intermittent agitation. During this incubation, an alkaline phosphatase-labeled, anti-hog intrinsic factor was introduced and the test unit incubated at 28°C for a 30 minute cycle. The unbound enzyme conjugate was removed with the help of a centrifugal wash. The intensity of color was monitored, to estimate B12.
Estimation of serum folic acid levels
The serum samples were diluted with the folic acid diluent and 200 μl of sample was added to each prepared tube containing 1000μl of the working solution (Borate KCN buffer solution, ligand labeled folate, and Dithiothreitol Solution). All test tubes were loosely capped and placed in a covered, boiling water bath for 15 - 20 minutes. The tubes were removed from the boiling water bath, and cooled in an ambient water bath for five minutes. Three hundred and fifty microliters of the treated sample, ligand labeled folic acid, and folic acid binding protein were simultaneously introduced into the each IMMULITE sample cup, and incubated for 30 minutes at 37 °C, with intermittent agitation. The intensity of color was monitored to estimate folate.
Statistical analysis
Statistical analyses were performed using Fischer's Exact test and Odd's ratio, P < 0.05 was considered statistically significant.
RESULTS
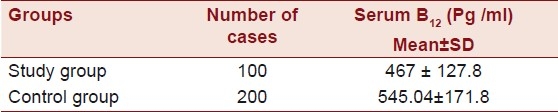
The mean age of the individuals was found to be 37.85 ± 6.01 years (age range 23 - 45) and that of the controls was 38.7 ± 5.25 years (age range 22 - 45 years). Of the 100 individuals that comprised the study group, 10 (10 %) were of highly mobile group, and the majority comprised of the sedentary category, which was found to be 90 (90 %). Of the 100 individuals, the majority had anterior wall MI incidence, 63 (63%), followed by the other categories like angina, nine (9%), Inferior wall MI, nine (9%), posterior wall MI, nine (9%), lateral wall MI, nine (9%), and LV dysfunction one (1%). The majority of the study group comprised of individuals from the urban populace, 75 (75 %), and 25 (25 %) individuals were from the rural background. Of the 100 individuals 48 (48%) were smokers, none were obese, 16 (16%) consumed oral tobacco of different kinds (Gutka, Zarda, etc.), 45 (45 %) were alcoholics, and a majority of 75 (75 %) were non-vegetarians. The majority in the study group were found not to consume fruits and salads. Only five (5 %) were found to consume fruits and salads out of the 100 that comprised the study group. Of the 100 individuals five individuals (5 %) had a history of consanguinity [Table 1]. The Mean ± SD of homocysteine was 36.80 ± 14.69 in the patients and 16.25 ± 12.30 in the controls, serum triglycerides were found to be 143.18 ± 66.02 in the patients and 140.25 ± 68.05 in the controls. Cholesterol was 158.13 ± 28.62 in the patients and 150.10 ± 30.2 in the controls, HDL was 38.5 ± 5.86 in the patients and 38.9 ± 6.16 in the controls, and the LDL was found to be 101.66 ± 26.39 in the patients and 109.5 ± 7.15 in the controls [Table 2]. The Mean ± SD of B12 levels in the affected individuals was 467 ± 127.8 pg/ml and it was 545.04 ± 171.8 pg/ml in the controls, and the Folate levels were 7.50 ± 2.0 ng/ml in the affected individuals and 11.85 ± 2.7 ng/ml in the controls [Table 3].
Table 1.
Clinical and relevant data of individuals with pre-CAD and controls
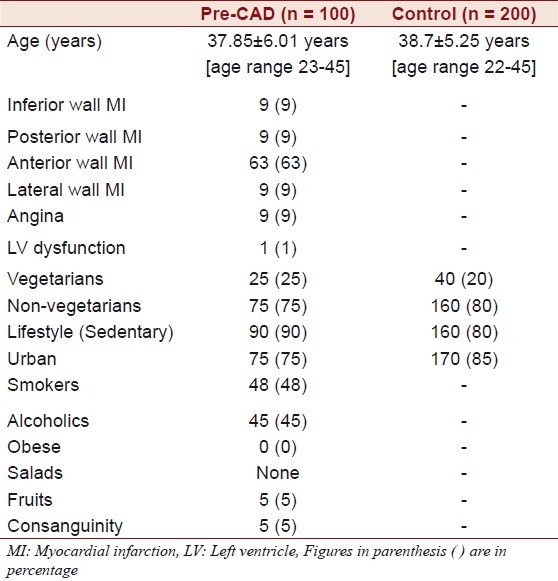
Table 2.
Biochemical data of individuals with pre-CAD and controls
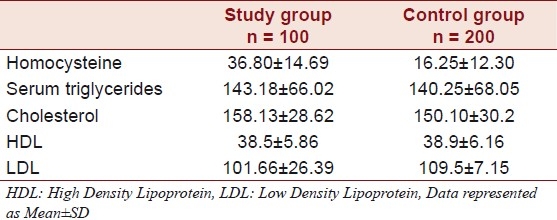
Table 3.
B12 levels in individuals with pre-CAD and controls
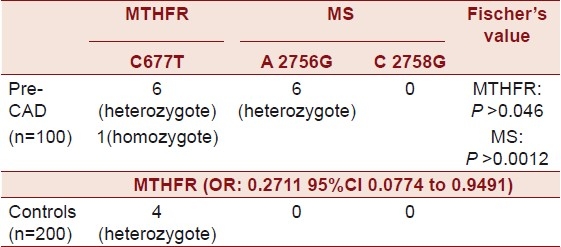
Seven patients showed 677C→T mutation; restriction fragment length polymorphism (RFLP) analysis revealed one was Homozygous for TT [Figure 1] and six were heterozygous [Figure 2] for CT. Four of the 200 controls revealed CT (heterozygous) polymorphism (Fischer's Exact test P < 0.046) (OR: 0.2711 95% CI 0.0774 to 0.9491). Six patients showed 2756A→G mutation [Figure 3], RFLP analysis revealed Heterozygosity in all the six (Fischer's Exact test P > 0.0012). None of the controls revealed the stated polymorphism. None of the 100 individuals or 200 controls had shown this variant 2758C→G in the MS gene [Figure 3].
Figure 1.
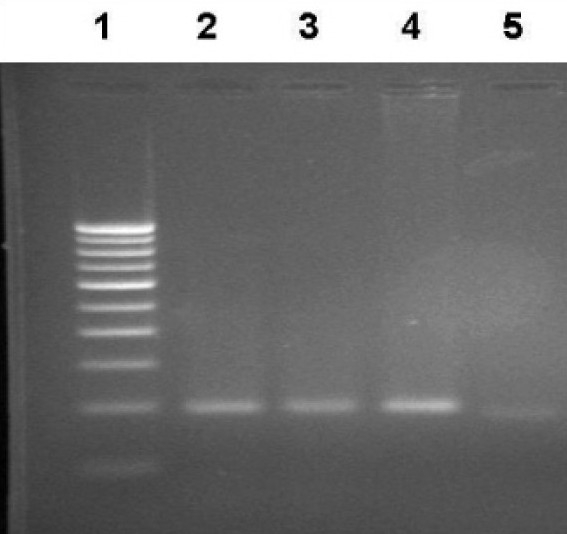
Representative photograph showing RFLP analysis of the MTHFR C677T gene using Hinf I. Wild type gives a single band at 198 bp. Homozygosity for the variant yields two fragments of 175 bp and 23 bp. The 23 bp is undetectable in 2% agarose gel. Lane 1: Marker lane; Lane 2,3 - Wild type; Lane 4 - PCR product (198 bp), Lane 5: Homozygous variant
Figure 2.
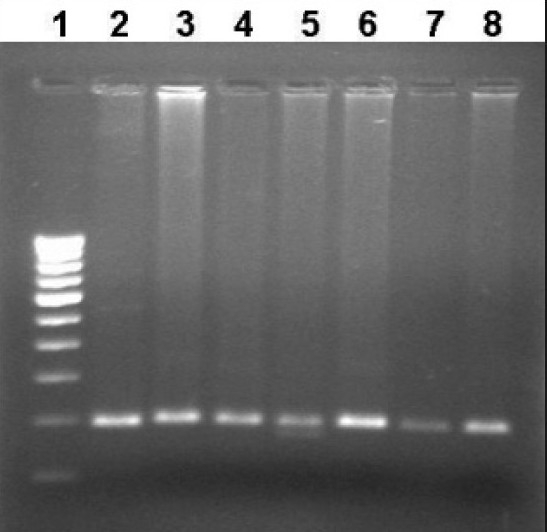
Representative photograph showing RFLP analysis of the MTHFR C677T gene using Hinf I. Wild type gives a single band at 198 bp. Heterozygosity for the variant yields three fragments of 198 bp, 175 bp, and 23 bp. The 23 bp is undetectable in 2% agarose gel. Lane 1. Marker lane; Lane 2- PCR Product (198bp); Lane 3,4 6-8- Wild type (198bp), Lane 5: Heterozygous variant
Figure 3.
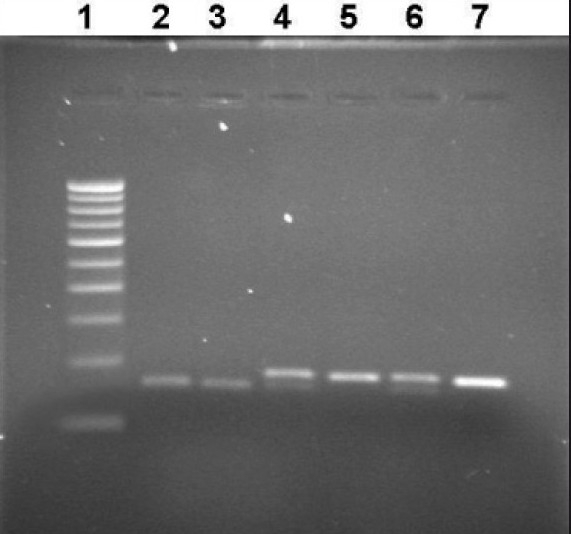
Representative photograph showing RFLP analysis of MS A2756G gene using BsuRI and MS C2758G gene using Cfr13I. Wild type for 2756 variant yields a single band of 189 bp, heterozygote yields three bands of 189 bp, 150 bp, and 39 bp. The homozygote yields two bands of 150 bp and 39 bp. Wild type for 2758 yields two fragments of 150 bp and 39 bp, heterozygote yields three fragments of 189 bp, 150 bp, and 39 bp, and the homozygote yields an intact band of 189 bp. Lane 1: Marker lane, Lanes 2 and 3: Normal 2758 allele (150 bp), Lane 4 and 6: Heterozygote for 2756 allele, Lane 5: Normal for 2756 allele, and Lane 7: PCR product (189 bp)
DISCUSSION
Cardiovascular disease is the leading cause of death in developed and developing countries. A person's genetic makeup, reflected by his or her family history, may influence the risk of various forms of cardiovascular disease. Although classic Mendelian patterns of inheritance have been observed for certain less common cardiovascular disorders, more common diseases, such as atherosclerotic disease, have atypical patterns of inheritance and a multifactorial pathogenesis. It is a difficult challenge to relate the study of the genetic basis of complex causes of common cardiovascular disease, such as atherosclerosis.[14]
Hyperhomocysteinemia is an independent risk factor for cardiovascular disease. There is growing evidence that homocysteine plays a causal role in atherogenesis. Homocysteine is a sulfhydryl-containing amino acid, formed during the metabolism of methionine, found in abundance in proteins of animal origin. Homocysteine is metabolized via the remethylation or transulfuration. The predominant Vitamin-B12-dependent resynthesis of methionine from homocysteine occurs in human tissues and comprises the transfer of the methyl group during the conversion of 5-methyltetrahydrofolate to tetrahydrofolate. Tetrahydrofolate binds to the carbon unit from serine to the nitrogen N5 and N10, forming 5,10-methylenetetrahydrofolate in the reaction catalyzed by 5,10-methylenetetrahydro folate reductase (MTHFR).[5]
The non-significant relationship between the C677T MTHFR polymorphism and CVD observed in this study does not contradict the homocysteine theory, because of the genetic and nutrient interactions. The C677T polymorphism may predict CVD risk only in certain ethnic groups[15] and the significant association of polymorphisms in the MTHFR gene has not been observed in this group.
The difference in the frequency of the C677T polymorphism in the MTHFR in different populations, suggests that in the past, the mutation had the advantage of selection. It has been suggested that in times of famine, reduced MTHFR activity led to decreased remethylation of homocyeteine and thus preserved the available one-carbon moieties of the tetrahydrofolate metabolism for the vital synthesis of purines and thymidine.[16–18]
A review on the role of nutritional supplementation in reducing the levels of homocysteine, had suggested that in the Indian population, apart from low dietary folate intake, the intake of riboflavin, vitamin B12, and vitamin B6 are also low when compared with the Western population.[19] Other studies[20,21] that mentioned the ‘Indian dietary paradox’, which has assumed that many Indians, by virtue of their vegetarian diet, do receive fairly adequate amounts of folate and explained the paradox for this as prolonged cooking, heating, frying of vegetables, which is a common practice among Indian households, and can destroy up to 90% of the folate content in the vegetables. This assumption could probably explain the low frequency of polymorphisms seen in the present study.
There is no significant association between C677T polymorphism and CAD[22] (OR: 0.2711 95% CI 0.0774 to 0.9491) [Table 4]. There is a higher frequency of T alleles in females than in males,[23] and the present study comprised of all males, below the age of 45 years. The MTHFR C677T mutation, identified as a major determinant of homocysteine concentrations in Europeans, is less prevalent and does not influence homocysteine concentrations in South Asians.[24] A clear association exists between tHcy-levels, but not between C677T in the MTHFR and the two polymorphisms in MS and CAD, as stated by Rothenbacher et al.[25]
Table 4.
Frequency of different genotypes in individuals with pre-CAD and controls
Methionine synthase (5-methyltetrahydrofolate homocysteine methyl transferase) catalyzes the re-methylation of homocysteine to methionine in a reaction wherein methylcobalamin serves as the intermediate methyl carrier. This occurs by transfer of the methyl group of 5-methyl tetrahydrofolate to the enzyme bound cob(I)alamin, which forms methylcobalamin, with subsequent transfer of the methyl group to the homocysteine, to form methionine. Over time cob(I)alamin may become oxidized to cob(II)alamin, reducing the enzyme and making it inactive. Regeneration of the functional enzyme occurs through methionine synthase mediated methylation of the cob(II) alamin in which S-adenosyl methionine is used as a methyl donor. Deficiency of methionine synthase activity results in hyperhomocysteinemia.
Two polymorphisms namely 2756 A→G (D919G) and 2758 C→G (H920D) have been identified in the MS gene. The two identified polymorphisms cause cblG disease and are located in the vicinity of the cobalimin-binding domain. H920D substitution is found in a region that is in the α helix at the C-terminal, and a region in the α/β domain, and the genetic variants of methionine synthase may lead to mild hyperhomocysteinemia, with a consequent impact on cardiovascular disease. Although, in the present study, a low frequency (six heterozygotes), A2756G polymorphism was observed; it was significantly associated with hyperhomocysteinemia, as the polymorphisms were seen in hyperhomocysteinemic individuals and not in individuals with normal homocysteine levels. The other polymorphism C2758G has not been observed in the study group and it can be concluded by the present study that it is not a risk factor in the Indian population.[26]
Vitamin B(12) and folate deficiency, reflected by increased Methylmalonic Acid (MMA) concentration, is an important risk factor both for the development of hyperhomocysteinemia and CAD.[27] The B12 levels were significantly lower (467 ± 127.8 pg / ml) in the study group as compared to the control group (545.04 ± 171.8) [Table 3] and the folate levels were also significantly lower in the study group (7.50 ± 2.0 ng / ml) as compared to the control group (11.85 ± 2.7) [Table 5].
Table 5.
Serum folate in individuals with pre-CAD and controls

Studies have shown an inverse association between fruit, vegetable, and fiber consumption and the risk for coronary artery disease. Majority of the individuals in the study group rarely consumed fruits or vegetables [Table 1], which not only reduced the risk for coronary artery disease, but was also known to quench the free radicals.[28] The free radical nitric oxide levels in the present study group were significantly raised compared to those in the controls, the study was published elsewhere.[29]
According to a recent study, both cigarette smoking and coffee consumption were associated with increased homocysteine levels.[30] In the present study both smoking and caffeine consumption was significantly high and seems to be associated with an increased risk of cardiovascular disease.
Majority of the individuals in the present study had sedentary lifestyles (90 % Table 1). Prospective studies strongly support the view that a sedentary lifestyle is associated with an increased risk of CAD.[31] Adopting a lifestyle that includes moderate physical activity in middle age appears to have a beneficial effect on that risk, with a reduction in total and LDL cholesterol and a concomitant increase in HDL cholesterol.[32] All the Biochemical Parameters like LDL, HDL, and Cholesterol were normal in the study group [Table 2].
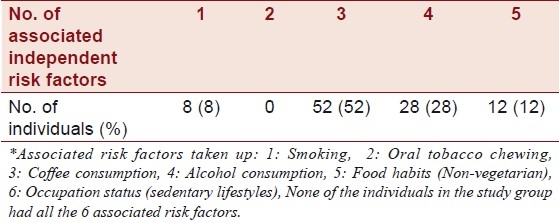
A significant increase in homocysteine levels among patients with CAD was seen with every additional risk factor. For the risk factors that were considered refer to Table 6. A majority (92%) of the individuals who comprised the study group had more that three associated additional risk factors, as stated by Abraham et al.[33]
Table 6.
Number of associated independent risk factors* for pre-CAD in individuals with hyperhomocystenemia
It is important to know which factors determine the total homocysteine concentrations, as it seems to be a significant risk factor for CAD. In the general population, the most important modifiable determinants of tHcy are folate intake and coffee consumption. Smoking and alcohol consumption are also associated with the total homocyeteine concentrations, but more research is necessary to elucidate whether these relations are not originating from residual confounding factors due to other lifestyle factors.
It is difficult to predict the usefulness of the increasing number of these polymorphisms in the manifestation of the disease, because the interaction of these genes with others would be difficult to establish, and it requires large population studies to sort out both the genetic and environmental interactions. It seems reasonable to have data on these polymorphisms in the clinical record as part of the investigation of coronary artery disease or of hyperhomocysteinemia. At this point, the presence or absence of a particular polymorphism in a gene of the homocysteine pathway is not a complete explanation of an individual phenotype for one of the most common multifactorial disease, such as the cardiovascular disease. As the genetics of atherosclerosis continues to evolve, these factors along with newer emerging factors may become a part of the routine assessment, aiding prediction of future coronary events.
To conclude, hyperhomocysteinemia is a risk factor for CAD, but not the polymorphisms in the studied genes in this study group, although many contributory factors namely low B12 and Folate levels are responsible for hyperhomocysteinemia. Low consumption of fruits and salads, sedentary lifestyles, smoking, and high levels of free radical nitric oxide are the other prominent risk factors for the causation of premature coronary artery disease. In the presence of mutations, either in the MTHFR or MS genes, there is a significant increase in homocysteine levels, so it would be useful to routinely genotype the polymorphisms in high-risk individuals. Adequate consumption of fruits and salads, foods fortified with B12 and folate, physical exercise, abstaining from smoking and consumption of tobacco products, is highly advised for a disease-free life.
ACKNOWLEDGMENTS
Ravi Kanth VV, Jaya Prakash Golla acknowledge Senior Research Fellowship from Indian Council of Medical Research and also acknowledge all the individuals who participated in the study.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Wild S, McKeigue P. Cross sectional analysis of mortality by country of birth in England and Wales, 1970-92. BMJ. 1997;314:705–10. doi: 10.1136/bmj.314.7082.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chambers JC, Kooner JS. Homocysteine: a novel risk factor for coronary heart disease in UK Indian Asians. Heart. 2001;86:121–2. doi: 10.1136/heart.86.2.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gauthier GM, Keevil JG, McBride PE. The association of homocysteine and coronary artery disease. Clin Cardiol. 2003;26:563–8. doi: 10.1002/clc.4960261204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malinow MR, Bostom AG, Krauss RM. Homocyst(e)ine, diet, and cardiovascular diseases: a statement for healthcare professionals from the Nutrition Committee, American Heart Association. Circulation. 1999;99:178–82. doi: 10.1161/01.cir.99.1.178. [DOI] [PubMed] [Google Scholar]
- 5.Undas A, Brozek J, Szczeklik A. Homocysteine and thrombosis: from basic science to clinical evidence. Thromb Haemost. 2005;94:907–15. doi: 10.1160/TH05-05-0313. [DOI] [PubMed] [Google Scholar]
- 6.Nygård O, Vollset SE, Refsum H, Brattström L, Ueland PM. Total homocysteine and cardiovascular disease. J Intern Med. 1999;246:425–54. doi: 10.1046/j.1365-2796.1999.00512.x. [DOI] [PubMed] [Google Scholar]
- 7.Nygård O, Nordrehaug JE, Refsum H, Ueland PM, Farstad M, Vollset SE. Plasma homocysteine levels and mortality in patients with coronary artery disease. N Eng J Med. 1997;337:230–6. doi: 10.1056/NEJM199707243370403. [DOI] [PubMed] [Google Scholar]
- 8.Jacobsen DW, Catanescu O, Dibello PM, Barbato JC. Molecular targeting by homocysteine: a mechanism for vascular pathogenesis. Clin Chem Lab Med. 2005;43(10):1076–83. doi: 10.1515/CCLM.2005.188. [DOI] [PubMed] [Google Scholar]
- 9.Goyette P, Pai A, Milos R, Frosst P, Tran P, Chen Z, et al. Gene structure of human and mouse methylenetetrahydrofolate reductase (MTHFR) Mamm Genome. 1998;9:652–6. doi: 10.1007/s003359900838. [DOI] [PubMed] [Google Scholar]
- 10.Rozen R. Genetic predisposition to hyperhomocysteinemia: deficiency of methylenetetrahydrofolate reductase (MTHFR) Thromb Haemost. 1997;78:523–6. [PubMed] [Google Scholar]
- 11.Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Matthews RG, et al. A candidate genetic risk factor for vascular disease: a common methylenetetrahydrofolate reductase. Nat Genet. 1995;10:111–3. doi: 10.1038/ng0595-111. [DOI] [PubMed] [Google Scholar]
- 12.Kang SS, Wong PW, Susmano A, Sora J, Norusis M, Ruggie N. Thermolabile methylenetetrahydrofolate reductase: an inherited risk factor for coronary artery disease. Am J Hum Genet. 1991;48:536–45. [PMC free article] [PubMed] [Google Scholar]
- 13.Chen LH, Liu ML, Hwang HY, Chen LS, Korenberg J, Shane B. Human methionine synthase cDNA cloning, gene localization and expression. J Biol Chem. 1997;272:3628–34. [PubMed] [Google Scholar]
- 14.Dietz HC, Pyeritz RE. Molecular Biology- To the Heart of the Matter. N Engl J Med. 1994;330:930–2. doi: 10.1056/NEJM199403313301311. [DOI] [PubMed] [Google Scholar]
- 15.Ueland PM, Refsum H, Beresford SA, Vollset SE. The controversy over homocysteine and cardiovascular risk. Am J Clin Nutr. 2000;72:324–32. doi: 10.1093/ajcn/72.2.324. [DOI] [PubMed] [Google Scholar]
- 16.Födinger M, Hörl WH, Sunder-Plassmann G. Molecualar biology of 5,10-methylenetetrahydrofolate reductase. J Nephrol. 2000;13:20–33. [PubMed] [Google Scholar]
- 17.Isotalo PA, Wells GA, Donnelly JG. Neonatal and Fetal methylen-etetrahydrofolate reductase genetic polymorphisms: an examination of C677T and A1298C mutations. Am J Hum Genet. 2000;67:986–90. doi: 10.1086/303082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muñoz-Moran E, Dieguez-Lucena JL, Fernandez-Arcas N, Peran-Mesa S, Reyes-Engel A. Genetic selection and folate intake during pregnancy. Lancet. 1998;352:1120–1. doi: 10.1016/s0140-6736(05)79761-0. [DOI] [PubMed] [Google Scholar]
- 19.Krishnaswamy K, Lakshmi AV. Role of nutritional supplementation in reducing the levels of homocysteine. J Assoc Physicians India. 2002;50:36–42. [PubMed] [Google Scholar]
- 20.Mathews JH, Wood JK. Megaloblastic anaemia in vegetarian Asians. Clin Lab Haematol. 1984;6:1–7. doi: 10.1111/j.1365-2257.1984.tb00519.x. [DOI] [PubMed] [Google Scholar]
- 21.Dawson DW, Waters HM. Malnutrition, folate and cobalamine deficiency. Br J Biomed Sci. 1994;51:221–7. [PubMed] [Google Scholar]
- 22.Vasisht S, Gulati R, Narang R, Srivastava N, Srivastava LM, Manchanda SC, et al. Polymorphism (C677T) in the 5, 10-methylenetetrahydrofolate reductase (MTHFR) gene: a preliminary study on North Indian men. Indian J Clin Biochem. 2002;1:99–107. doi: 10.1007/BF02867949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radha Rama Devi V, Govindaiah GR, Naushad SM. Prevalence of methylene tetrahydrofolate reductase polymorphism in South Indian population. Curr Sci. 2004;86:440–3. [Google Scholar]
- 24.Chambers JC, Ireland H, Thompson E, Reilly P, Obeid OA, Refsum H, et al. Methylenetetrahydrofolate reductase 677 C-->T mutation and coronary heart disease risk in UK South Asians. Arterioscler Thromb Vasc Biol. 2000;20:2448–52. doi: 10.1161/01.atv.20.11.2448. [DOI] [PubMed] [Google Scholar]
- 25.Rothenbacher D, Fischer HG, Hoffmeister A, Hoffmann MM, März W, Bode G, et al. Homocysteine and methylenetetrahydrofolate reductase genotype: association with risk of coronary heart disease and relation to inflammatory, hemostatic, and lipid parameters. Atherosclerosis. 2002;162:193–200. doi: 10.1016/s0021-9150(01)00699-2. [DOI] [PubMed] [Google Scholar]
- 26.Leclerc D, Campeau E, Goyette P, Adjalla CE, Christensen B, Ross M, et al. Human methionine synthase: cDNA cloning and identification of mutations in patients of the cblG complementation group of folate / cobalmin disorders. Human Mol Genet. 1996;5:1867–74. doi: 10.1093/hmg/5.12.1867. [DOI] [PubMed] [Google Scholar]
- 27.Biselli PM, Guerzoni AR, de Godoy MF, Eberlin MN, Haddad R, Carvalho VM, et al. Genetic polymorphisms involved in folate metabolism and concentrations of methylmalonic acid and folate on plasma homocysteine and risk of coronary artery disease. J Thromb Thrombolysis. 2010;29:32–40. doi: 10.1007/s11239-009-0321-7. [DOI] [PubMed] [Google Scholar]
- 28.La Vecchia C, Decarli A, Pagano R. Vegetable consumption and risk of chronic disease. Epidemiology. 1998;9:208–10. [PubMed] [Google Scholar]
- 29.Ravi Kanth VV, Prakash GJ, Naik S, Kabra N, Sujatha M. Premature coronary artery disease: role of free radical nitric oxide. Indian Heart J. 2008;60:45–9. [PubMed] [Google Scholar]
- 30.Nygård O, Refsum H, Ueland PM, Vollset SE. Major lifestyle determinants of plasma total homocysteine distribution: the Hordaland Homocysteine Study. Am J Clin Nutr. 1998;67:263–70. doi: 10.1093/ajcn/67.2.263. [DOI] [PubMed] [Google Scholar]
- 31.Sandvik L, Erikssen J, Thaulow E, Erikssen G, Mundal R, Rodahl K. Physical fitness as a predictor of mortality among healthy, middle-aged Norwegian men. N Engl J Med. 1993;328:533–7. doi: 10.1056/NEJM199302253280803. [DOI] [PubMed] [Google Scholar]
- 32.Berlin JA, Colditz GA. A meta-analysis of physical activity in the prevention of coronary heart disease. Am J Epidemiol. 1990;132:612–28. doi: 10.1093/oxfordjournals.aje.a115704. [DOI] [PubMed] [Google Scholar]
- 33.Abraham R, John MJ, Calton R, Dhanoa J. Raised serum homocysteine levels in patients of coronary artery disease and the effect of vitamin B12 and folate on its concentration. Indian J Biochem. 2006;21:95–100. doi: 10.1007/BF02913073. [DOI] [PMC free article] [PubMed] [Google Scholar]