

Abstract
Obesity and ageing are both characteristics of the human population that are on the increase across the globe. It has long been established that ageing is the major risk factor for neurodegenerative conditions such as Alzheimer's disease, and it is becoming increasingly evident that obesity is another such factor. Leptin resistance or insensitivity has been uncovered as a cause of obesity, and in addition the leptin signalling system is less potent in the elderly. Taken together, these findings reveal that this molecule may be a link between neurodegeneration and obesity or ageing. It is now known that leptin has beneficial effects on both the survival and neurophysiology of the neurons that are lost in Alzheimer's disease suggesting that it may be an important research target in the quest for strategies to prevent, halt, or cure this condition.
1. Introduction
It is a fact of life that as we age, changes occur to all body systems. The outward phenotype of the elderly is obvious with greying hair, wrinkling and thinning of the skin, and a change in posture and the fluidity of movement as joints and muscles stiffen. However, some of the more devastating changes associated with ageing are often hidden from the outside world, as neurological function declines caused by the death of critical neuronal populations. Neurodegenerative conditions arise when excessive neuronal loss occurs within a discrete region of the nervous system. Thus Parkinson's disease (PD) is linked to the large scale loss of neurons within the substantia nigra pars compacta, and Alzheimer's disease (AD) with degeneration in the hippocampus and cerebral cortex. Loss of neuronal function has a huge impact on the ability of an individual to interact with their environment, whether through the lack of motor control in PD, or though the decline in cognition, and as a consequence of this, a full range of social interaction, in AD. These changes affect not only the sufferers, but also their family and carers, and the whole of society, as the burden of care falls onto national healthcare and social care systems.
The major risk factor for the development of neurodegenerative conditions is ageing. Therefore, as the human population ages, it has been forecast that for dementia, between the years 2001 and 2040, western countries will see a 100% increase in the number of individuals afflicted, with a 300% increase predicted for India and China [1]. Currently, neurodegenerative conditions remain incurable. At the time of writing, nearly 41 000 articles are cited within the pubmed database for which neurodegeneration is a tagged search term [2]. And yet from all this knowledge, no cures have arisen. The reason for this is the complexity of the task of curing a neurodegenerative condition. Firstly, the neurons that have been lost need to be replaced, thereafter these replacement cells need to make the correct connections with other neurons and, if applicable, with target fields outside the nervous system. The functionality of the neuronal network must also be restored, and finally the replacement neurons must be protected from the factors that triggered neurodegeneration in the first instance, such that they too do not undergo physiological decline and cell death. Given the multistage process needed to cure a degenerative condition within the nervous system, an easier target may be to find strategies that prevent the occurrence of neurodegeneration in the first place.
A plethora of risk factors, in addition to ageing, have been identified that increase the incidence of neurodegenerative diseases. These include a sedentary lifestyle [3], blows to the head (in the case of PD) [4], exposure to heavy metals such as aluminium [5], elevated plasma levels of homocysteine [6], and obesity [7], to name but a few. This review will focus on obesity as a risk factor for the development of neurodegenerative conditions, in particular the loss of cognitive function associated with Alzheimer's disease and other dementias, with an emphasis on the antiobesity hormone leptin.
2. Obesity and Dementia
Obesity is the excessive accumulation of fat within an organism that may be caused by genetic factors, environmental factors, or a complex interplay between the two. The resulting energy imbalance between calories consumed in the food, against calories expended in day to day living, results in the excess nutrition being stored as fat. The World Health Organisation estimated that in 2010 there were 300 million obese adults in the global population and 42 million overweight children [8]. To clarify these figures further, it is known that in developed countries, obesity affects 25–30% of adults [9, 10]. Furthermore, the incidence of obesity in the elderly is rising. In a study of 65–74 year olds in the United States, the rate of obesity increased from 27% in 1988 to 39% in 2000 [10]. These figures give us an indication of the immense scale of the occurrence of obesity.
Investigations into a link between obesity and the onset of dementia have been widely reported in the literature. However, conflicting results have been presented. Thus, a systematic review of published papers was undertaken, and a definitive relationship between elevated body mass index (BMI) and the incidence of dementia emerged [11]. Furthermore, it has been demonstrated that carriers of the FTO allele have a slight increase in BMI as compared to noncarriers. Interestingly, possession of this allele is also linked to a decrease in brain volume in the healthy elderly [12], linking elevated BMI to central nervous system changes. It has also been established that there is a relationship between an elevated waist to hip ratio and the risk of developing a neurodegenerative condition [13], and that advancing age and elevated BMI are linked to a decrease in brain volume, but not cognition, in the middle aged [14]. Similarly a decrease in grey matter volume has been linked to adiposity in otherwise healthy postmenopausal women [15]. However, these studies did not reveal the underlying mechanisms that are activated by obesity, that then lead to degenerative changes in the brain. Furthermore, there is a lack of clarity in the published literature as to whether being overweight or just being obese is a risk factor for neurodegeneration and therefore further population-based studies, coupled with rigorous laboratory-based tests designed to tease out the underlying mechanisms linking obesity to dementia, need to be carried out.
Just as the causes of neurodegeneration per se are multiple, complex, and likely to be interlinked, the causes of the increased neurodegenerative risk induced by obesity are also multiple and complex. Roles for lipotoxicity [16], diabetes and insulin dysregulation or insensitivity [17, 18], and erroneous synthesis or response to a number of circulating factors have been postulated as potential mechanisms. It is likely that all these factors and more are important in the observed risk, but this review will solely focus on a potential role for the antiobesity hormone leptin.
3. The Antiobesity Hormone Leptin
Leptin is the product of the ob gene, first cloned in 1994 [19]. Expression has been detected in adipose tissue, placenta, the gastrointestinal system, and in a number of regions of the central nervous system [19–22]. It is well established that leptin plays a central role in the regulation of body weight, reducing appetite and thus an inverse relationship is established between plasma leptin levels and food intake [23, 24]. The leptin receptor, Ob-R is widely expressed in the body and is a member of the class I cytokine receptor family [25]. Alternate splicing of Ob-R gives rise to six isoforms (Ob-Ra, Ob-Rb, Ob-Rc, Ob-Rd, Ob-Re, Ob-Rf) all of which possess the extracellular leptin binding domain, but of which only Ob-Rb has signalling competency, due to the possession of the full intracellular domain [26–31].
Downstream of leptin binding to Ob-Rb, a number of intracellular signalling pathways, can be triggered (Figure 1). Ob-Rb does not possess intrinsic enzymatic activity and therefore in order to transduce the signals initiated by leptin binding, Janus kinase-2 (JAK-2) that is associated with Ob-Rb, is autophosphorylated and activated [25, 32]. Thereafter JAK-2 phosphorylates tyrosine residues within the Ob-Rb intracellular domain to mediate the downstream effects. Each tyrosine (Tyr) that is phosphorylated triggers a unique signalling event. As such, phosphorylation of Tyr985 leads to the recruitment of Src-homology-2 (SH2)-containing tyrosine phosphatase-2, triggering an extracellular signal-regulated kinase (ERK) signalling cascade [33]. It is of interest to note that mutagenesis of this tyrosine residue leads to adult-onset obesity linked to leptin resistance in mice [34]. Tyr1077 phosphorylation recruits and activates the transcription factor signal transducer and activator of transcription 5 (STAT5) [35], whilst STAT3 activation (and to a lesser extent STAT5 activation) has been linked to phosphorylation of Tyr1138 [33, 35]. Other signalling events known to be triggered by the binding of leptin to Ob-Rb include phosphatidylinositol 3-kinase (PI-3kinase), nuclear factor kappa B (NF-κB) [36], and adenosine monophosphate (AMP)-dependent protein kinase [37] activity although the enzymatic pathways linking them to receptor activation remain to be fully elucidated.
Figure 1.
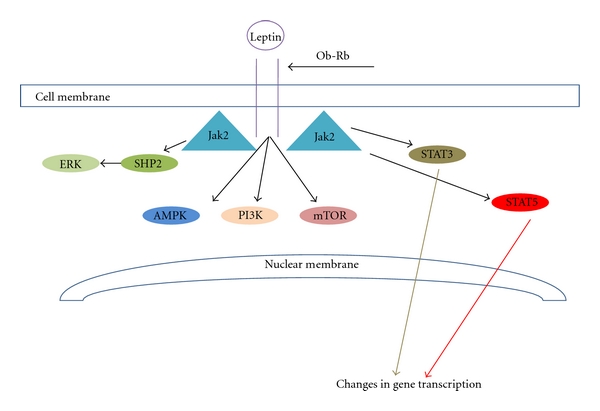
Signalling downstream of leptin binding to the Ob-R receptor. A number of pathways are activated. JAK-2-mediated phosphorylation of Tyr985 on Ob-Rb activates SHP2 and subsequently ERK. Phosphorylation of Tyr1077 and to a lesser extent Tyr1138 activates STAT5, whilst phosphorylation of Tyr1138 activates STAT3. Activation of STAT transcription factors leads to alterations in gene expression. The biochemical pathways leading to activation of PI3kinase, AMPK, and mTOR when leptin binds Ob-Rb are less well characterised.
Expression of the leptin receptor has been described in the brain, with transport of leptin into the brain believed to be achieved by leptin binding to the short forms of the receptor Ob-Ra and Ob-Rc that are present in the choroid plexus and the microvasculature of the brain [26]. The signalling of leptin within the central nervous system is believed to predominantly be via the full length Ob-Rb form of the Ob receptor thereafter activating the signalling pathways described in the previous paragraph. Expression of Ob-Rb has widely been identified within the central nervous system including the cortex and hippocampus that are the major site of neurodegeneration in AD and the substantia nigra that is the major site of neuronal loss in PD [27, 30, 38]. To date there is very little known about regulation of the leptin receptor under the specific circumstances of a given neurodegenerative disorder, and this will clearly be a very important area of research in the future. It is known that both a deficiency in circulating leptin, as observed in leptin-deficient animals, and an inability to respond to leptin, as observed in animals with deficits in the Ob receptor, lead to upregulation of Ob-Rb suggesting that there is close interplay between levels of leptin detected and expression of the full length receptor [39, 40].
A myriad of research has been conducted demonstrating that leptin, coupled with a correct response to the hormone, is central to the control of bodyweight and to other physiological processes. Thus adipocyte-generated leptin acts as a satiety signal inhibiting food intake [39] and in addition, evidence suggests that leptin plays a key role in energy expenditure. Thus, mice that fail to respond to leptin develop obesity even if fed a diet that restricts calorific intake to normal levels [41] implying that these animals are not expending the energy from their food efficiently. Therefore, a reduced ability to synthesise or respond to leptin is likely to lead to a failure to control appetite and reduced expenditure of energy, leading to an increase in weight and, in time, obesity. In ob/ob mice, a loss of function mutation in the ob gene that encodes leptin leads to obesity by around four weeks of age [42]. Likewise db/db mice or fa/fa rats that carry a loss of function mutation in the Ob-R gene are obese by a similar age [42]. It is therefore not surprising that failures in the leptin signalling system have been linked to obesity in humans [43–45]. Mechanisms by which this failure can occur include deficits in leptin transport across the blood-brain barrier, insufficient leptin release and mutations in the Ob-R receptor, or defects in its trafficking or downstream signalling, preventing the correct response to the circulating hormone [7].
4. Leptin and Neurodegeneration
Neurodegenerative diseases, as mentioned above, are characterised by the loss of neurons in a specific region of the nervous system and with abnormalities of neuronal function in the cells that remain. It has been widely reported that we face an increasing demographic shift across the globe to an ageing population [46], which will bring about an increase in the incidence of age-related neurodegeneration. Given that we are also facing increasing levels of obesity in the elderly [11], if there is a link between obesity and neurodegeneration, then we need to know what it is and how to deal with it. There is now a range of research data published that reveals that leptin is one molecule that is involved in linking obesity to neurodegeneration.
It has been demonstrated that aged rats have a lower response to leptin as compared to young rats with decreased signalling downstream of the Ob-R receptor identified as a reason for this, and in particular a decrease in STAT3 activation has been noted with age [47]. In addition to the age-linked reduced ability to respond to leptin, there is also an age-linked decrease in the uptake of leptin by hypothalamic nuclei, which correlates to a decrease in Ob-R expression [48]. Another mechanism that influences the age-related decline in leptin responsiveness is an increase in the levels of the suppressor of cytokine signalling-3 (SOCS-3) that inhibits the transduction of the signals triggered by leptin binding to Ob-R [49]. Thus, increased SOCS-3 expression has been identified in the aged hypothalamus [50]. Similarly, enhanced hypothalamic levels of protein tyrosine phosphatase 1B are believed to contribute to age-related leptin resistance [51]. Thus, the elderly are less able to respond to leptin for a variety of physiological reasons, which raises the question of what this decrease in leptin signalling might mean for neurons.
One of the physiological properties of leptin that is central to its potential role in neurodegeneration is the discovery that leptin can prevent neuronal death. Thus leptin can protect cultured dopaminergic neurons from the central nervous system, and trigeminal sensory neurons from the peripheral nervous system, from cell death in response to a number of different stimuli [36]. In accordance with this it has been noted that the brains of leptin-deficient mice are of lower weight than their wild-type counterparts, perhaps revealing that a lower number of neurons survive in these animals [52]. In cultured murine cortical preparations, leptin can prevent excitotoxic neuronal loss triggered by NMDA. Furthermore, systemically delivered leptin can prevent excitotoxic damage in vivo, reducing cortical lesions in mice given high doses of ibotenate [53]. Neuroprotective roles for leptin have also been found in animal models of stroke where leptin administration protects against both oxygen-glucose deprivation and middle cerebral artery occlusion [54]. Similarly leptin is neuroprotective in both in vivo and in vitro models of PD [36, 55], and it protects hippocampal neurons in models related to epilepsy [56]. In the case of AD, transgenic mice that exhibit neurodegeneration in brain areas linked to AD have a marked reduction in the number of dying neurons following treatment with a lentiviral vector encoding leptin [57]. Taken together there is a large body of evidence to suggest that leptin may be beneficial in the prevention of neuronal death in neurodegenerative situations.
In addition to neuronal death, conditions such as AD are characterised by alterations in neuronal function. Thus, leptin can modulate synaptic plasticity via enhancing N-Methyl-D-aspartate (NMDA) receptor function [58, 59]. Given that synaptic plasticity is studied as the cellular basis of memory formation, these findings raise the possibility that leptin can have beneficial effects on the memory loss that is perhaps the best known symptom of AD. Thus rodents that have mutations in Ob-R show memory impairment [60]. Furthermore, transgenic mice that exhibit deficits in memory and are used to model AD show a reduction in AD-linked pathological changes and an enhancement in memory if treated with exogenous leptin [61]. Whilst the discovery that leptin can influence both memory and neuronal viability in a laboratory setting strongly suggests that it may be important in either the pathogenesis of or the treatment of AD, the case for further study is only truly strengthened with human data.
Thus studies of the individuals that form the Framingham cohort have revealed that decreased circulating leptin levels are predictive of an increased risk of developing dementia or AD [62]. In contrast to this, cross-sectional studies have not revealed a link between leptin levels and vascular dementia [63], and carriers of an Ob-R mutation that reduces leptin binding do not have an enhanced risk of AD [64]. Another study looked at vascular dementia and AD in isolation. Once again this demonstrated a link between low leptin levels and the development of AD when compared to nondemented or vascular dementia patients. These latter two groups had similar leptin levels. This study exhibits that AD and vascular dementia [65], which are often grouped together as disorders, may have a very different pathogenesis with regards to the action of this antiobesity hormone.
It is becoming increasingly evident that neuroinflammation is an important part of the pathology of neurodegenerative conditions [66]. Thus, it is crucial to determine whether leptin influences the activity or activation of the astrocytes and microglia of the central nervous system. It has been determined that glial cells may play a central role in synaptic inputs into the hypothalamus and that leptin can influence this process. It is particularly interesting to note that chronic leptin administration leads to astrocyte activation suggesting that the potential role of these cells in modulating obesity and neurodegeneration needs to be more extensively investigated in the coming years [67].
5. Discussion
Articles in the press often highlight so-called “epidemics” that it is believed will greatly impact upon the way that healthcare and social care systems are organised as we move forward in time. One of these issues that is frequently mentioned is ageing, another is obesity. It is becoming increasingly apparent in the scientific literature that a common consequence of both of these so-called “epidemics” is neurodegeneration. Thus, it is worthwhile to explore whether there are shared mechanisms that link these two risk factors to neurodegeneration with a view to preventing or treating illnesses such as AD. Within this framework leptin appears as one of the candidate molecules that might link obesity and age to neurodegeneration.
It is clear that obesity increases with age and that one of the reasons for this is that the way in which the body responds to leptin changes with age. Thus there is a decrease in circulating leptin [68], Ob-R, and in leptin uptake [48], coupled with an increase in inhibitors of leptin signalling [49–51]. It is well established that leptin is a satiety signal and thus a decrease in the response to it will decrease satiety, increasing energy intake and thus risking obesity. It is also well documented that leptin has many beneficial effects on both neuronal survival and neurophysiology, thus this decrease in leptin signalling in the elderly will have a negative impact on the function of neuronal networks that are sensitive to its effects. Given that the hippocampus has been long established to be fundamental to memory processes that are lost in AD, the scientific evidence that leptin is important for both the viability [56] and functioning [58, 59] of hippocampal neurons strongly suggests that leptin may be important in the pathogenesis of AD. Coupled to this is the finding from the Framingham cohort that low levels of circulating leptin are a risk factor for the future development of AD [62].
Since it is clear that leptin is a strong candidate as one of the molecules that links obesity to neurodegeneration, and we know that obesity in midlife enhances the risk of AD [69], the possibility of manipulating leptin levels or leptin signalling in advance of the onset of AD symptoms arises. Thus as a preventative measure, adults with midlife obesity could be assessed for their levels of, and response to, leptin and strategies put in place to boost this signalling system. In addition, restoring leptin function could have beneficial effects outwith the central nervous system including energy homeostasis and neuroendocrine function [70]. However, leptin is just one factor relating to obesity that may be involved in AD pathogenesis. In addition to leptin, other obesity-related factors are linked to AD including insulin [18], changes to the cerebral vasculature [71], and direct lipotoxicity [17]. In support of the notion that leptin may not be a cure-all for all cases of dementia is the finding that there is no correlation between leptin levels and vascular dementia [63, 65]. Nonetheless, there is strong evidence that leptin signalling has beneficial consequences for neural networks, and therefore introducing corrective strategies in individuals in whom this is compromised can only be of benefit. Furthermore, given that leptin is only one of the links between obesity and AD, any targeting of leptin signalling in obese individuals who have leptin deficiency or reduced sensitivity is likely to be most successful when combined with lifestyle changes targeting a healthy weight and level of activity. Of course it should also be emphasised that individuals who exhibit a high level of leptin insensitivity may be completely insensitive to leptin or leptin-derived therapies if treatment is not carried out alongside therapies to reverse this state of insensitivity.
There are good arguments to be put to promote leptin and its downstream signalling pathways as possible drug targets for not only the treatment of AD but also of other neurodegenerative disorders [7, 72]. However, the ability of leptin to protect against cell death is not universal, and it has been demonstrated that leptin exerts neuroprotective effects on cerebellar Purkinje but not granule neurons in culture [73]. It is therefore crucial to investigate leptin's potential therapeutic effects extensively against each condition of interest. As such a word of caution must be raised against the current findings that imply that leptin is neuroprotective in models of AD. There are numerous mice models for AD, none of which provide a perfect recreation of the disease pathology observed in human AD patients. For example, although the Tg2576 transgenic mouse models do show the glial-mediated inflammatory response and elevated levels of Aβ and associated plaques that occur in the brains of AD sufferers [74], there are other pathologies associated with AD, such as neuronal loss and neurofibrillary tangles, that are not displayed in this model. Thus leptin's effects must be robustly tested in as many different AD rodent and in vitro models as possible before a definitive conclusion about how it might confer neuroprotection can be drawn. This would also help to elucidate where within the temporal progression of AD leptin might confer the maximal benefits.
In this regard, leptin's antiapoptotic actions would not offer any potential to cure AD. However, if given before symptoms occur, leptin-targeting medicines could prevent the loss of neurons associated with AD in the first instance. It could also be targeted to early stage sufferers in order to prevent further neuronal loss and therefore halt or slow down disease progression. Another possible benefit of leptin's antiapoptotic abilities is in protecting grafted tissue, whether from embryonic stem cells or fetal sources, once it is transplanted into the sufferer. Grafting of host tissue has been used with some success as a potential treatment for PD but one of the problems with this therapy is that a large number of the transplanted neurons degenerate within the host. Thus for just one PD patient, tissue from between 9 and 12 embryos is required for bilateral implantation [75]. For PD, interest has therefore turned to neurotrophins that can enhance dopaminergic neuron number in grafted tissue such as glial cell line-derived neurotrophic factor [76] or, for tissue from early embryos, tumour necrosis factor alpha (TNF-α) [77]. It is feasible therefore that leptin could offer neurotrophic support to grafted tissue in AD suffers if technology advances to a point where such transplants become a treatment option for this disease.
In addition to its antiapoptotic effects, leptin has also demonstrated beneficial effects on memory [78]. Thus leptin administration, again particularly early in disease progression, may help to alleviate the alterations in synaptic function associated with AD. There is a growing body of evidence that synaptic alterations may precede neuronal death in AD and therefore leptin's effects on these may be central to allowing individuals to halt the decline in memory function so strongly associated with AD. Thus leptin exerts its beneficial effects on the nervous system at multiple levels from neuronal survival to synaptic plasticity, suggesting that it could benefit AD sufferers in a number of ways. Specifically it has been determined that leptin inhibits hippocampal neurons, that are afflicted in AD, by activating BK channels leading to the initiation of a PI3-kinase signalling cascade [79]. Furthermore, leptin can evoke NMDA-receptor dependent-long-term depression under conditions of enhanced excitability [80]. Thus electrophysiological data suggests that leptin can enhance the cellular processes thought to underpin memory formation.
One of the major benefits that leptin offers as a potential therapeutic target is that it is currently licensed for human use as an antiobesity drug. The possession of this license would markedly speed up the drug discovery pipeline [7]. Furthermore, we all produce leptin in our bodies as a naturally occurring substance and as such it is well tolerated in a therapeutic setting and few side effects have been identified following its use as an antiobesity agent. However, leptin is not an easy drug to administer, and the current treatment for obesity involves giving subcutaneous injections of the drug. Pharmacological leptin has so far been given to patients for peripheral metabolic disorders and to modulate behaviours and endocrine pathways in the hypothalamus. Thus, it is unclear whether subcutaneous administration of leptin could act on the hippocampus and cortex that are affected in AD. In rodents intranasal administration of leptin successfully raises brain leptin levels, particularly in the hypothalamus [81, 82]. Also, a nonreplicative, nonimmunogenic and non-pathogenic recombinant adeno-associated virus encoding the ob gene has been used as a form of central gene therapy by direct injection into the affected brain area to provide a stable supply of leptin in that region. This has been used successfully to suppress weight gain in prepubertal rats [83], to inhibit fat deposition [84], and to block high fat diet-induced weight gain [85]. Thus, much important work still needs to be carried out to reveal the most effective method of leptin delivery into regions of the brain in humans.
In conclusion, with an increasing elderly population and an increasing rate of obesity within that population, it is imperative that we fully understand the noted links between obesity and neurodegeneration. Candidate molecules for explaining this connection are being discovered all the time, and it is within this framework that interest has turned to the antiobesity hormone leptin. The multilevel beneficial effects that are reported for leptin in neurons give hope that we may be on the road to uncovering pathways that can be targeted to halt, prevent, and perhaps eventually cure neurodegenerative disorders such as AD.
Acknowledgments
Dr Doherty holds an Alzheimer's Society personal research fellowship (Grant no. 93) with support from the Henry Smith Charity. Alzheimer's Society is a charity (Registration no. 296645) and a company registered in England and Wales (Registration no. 2115499). The University of St Andrews is a charity registered in Scotland (Registration no. SC013532).
Abbreviations
- AD:
Alzheimer's disease
- AMP:
Adenosine monophosphate
- BMI:
Body mass index
- ERK:
Extracellular signal-regulated kinase
- JAK-2:
Janus kinase-2
- NF-κB:
Nuclear factor kappa B
- NMDA:
N-Methyl-D-aspartate
- PD:
Parkinson's disease
- PI3-kinase:
Phosphatidylinositol 3-kinase
- SOCS-3:
Suppressor of cytokine signalling-3
- STAT
Signal transducer and activator of transcription
- TNF-α:
Tumour necrosis factor alpha
- Tyr:
Tyrosine.
References
- 1.Ferri CP, Prince M, Brayne C, et al. Global prevalence of dementia: a Delphi consensus study. The Lancet. 2005;366(9503):2112–2117. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. 2011, http://www.ncbi.nlm.nih.gov/pubmed?term=neurodegeneration.
- 3.Florencio Gama E, Maria Santarém J, Aparecido Liberti E, Jacob Filho W, de Souza RR. Exercise changes the size of cardiac neurons and protects them from age-related neurodegeneration. Annals of Anatomy. 2010;192(1):52–57. doi: 10.1016/j.aanat.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Dick FD, De Palma G, Ahmadi A, et al. Environmental risk factors for Parkinson’s disease and parkinsonism: the Geoparkinson study. Occupational and Environmental Medicine. 2007;64(10):666–672. doi: 10.1136/oem.2006.027003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawahara M, Kato-Negishi M. Link between aluminum and the pathogenesis of Alzheimer's disease: the integration of the aluminum and amyloid cascade hypotheses. International Journal of Alzheimer's Disease. 2011;2011 doi: 10.4061/2011/276393. Article ID 276393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doherty GH. Boom and bust for homocysteine? Central Nervous System Agents in Medicinal Chemistry. 2008;8(2):107–120. [Google Scholar]
- 7.Doherty GH. Leptin: a novel therapeutic target in the fight against neurodegeneration? In: Hemming RM, Belikin AT, editors. Leptin: Hormonal Functions, Dysfunctions and Clinical Uses. Nova; 2011. [Google Scholar]
- 8.WHO (World Health Organization) Obesity and overweight. 2010. 2011, http://www.who.int/dietphysicalactivity/media/en/gsfs_obesity.pdf.
- 9.Rennie KL, Jebb SA. National prevalence of obesity: prevalence of obesity in Great Britain. Obesity Reviews. 2005;6(1):11–12. doi: 10.1111/j.1467-789X.2005.00164.x. [DOI] [PubMed] [Google Scholar]
- 10.Centers for Disease Control and Prevention. 2011, http://www.cdc.gov/NCHS/data/hestat/obesity_adult_07_08/obesity_adult_07_08.pdf.
- 11.Gorospe EC, Dave JK. The risk of dementia with increased body mass index. Age and Ageing. 2007;36(1):23–29. doi: 10.1093/ageing/afl123. [DOI] [PubMed] [Google Scholar]
- 12.Ho AJ, Stein JL, Hua X, et al. A commonly carried allele of the obesity-related FTO gene is associated with reduced brain volume in the healthy elderly. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(18):8404–8409. doi: 10.1073/pnas.0910878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jagust W, Harvey D, Mungas D, Haan M. Central obesity and the aging brain. Archives of Neurology. 2005;62(10):1545–1548. doi: 10.1001/archneur.62.10.1545. [DOI] [PubMed] [Google Scholar]
- 14.Ward MA, Carlsson CM, Trivedi MA, Sager MA, Johnson SC. The effect of body mass index on global brain volume in middle-aged adults: a cross sectional study. BMC Neurology. 2005;5, article no. 23 doi: 10.1186/1471-2377-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soreca I, Rosano C, Richard Jennings J, et al. Gain in adiposity across 15 years is associated with reduced gray matter volume in healthy women. Psychosomatic Medicine. 2009;71(5):485–490. doi: 10.1097/PSY.0b013e3181a5429d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martínez de Morentin PB, Varela L, Fernø J, Nogueiras R, Diéguez C, López M. Hypothalamic lipotoxicity and the metabolic syndrome. Biochimica et Biophysica Acta. 2010;1801(3):350–361. doi: 10.1016/j.bbalip.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 17.Manolopoulos KN, Klotz LO, Korsten P, Bornstein SR, Barthel A. Linking Alzheimer’s disease to insulin resistance: the FoxO response to oxidative stress. Molecular Psychiatry. 2010;15(11):1046–1052. doi: 10.1038/mp.2010.17. [DOI] [PubMed] [Google Scholar]
- 18.Luchsinger JA. Diabetes, related conditions, and dementia. Journal of the Neurological Sciences. 2010;299(1-2):35–38. doi: 10.1016/j.jns.2010.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 20.Hoggard N, Mercer JG, Rayner DV, Moar K, Trayhurn P, Williams LM. Localization of leptin receptor mRNA splice variants in murine peripheral tissues by RT-PCR and in situ hybridization. Biochemical and Biophysical Research Communications. 1997;232(2):383–387. doi: 10.1006/bbrc.1997.6245. [DOI] [PubMed] [Google Scholar]
- 21.Mix H, Widjaja A, Jandl O, et al. Expression of leptin and leptin receptor isoforms in the human stomach. Gut. 2000;47(4):481–486. doi: 10.1136/gut.47.4.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morash B, Li A, Murphy PR, Wilkinson M, Ur E. Leptin gene expression in the brain and pituitary gland. Endocrinology. 1999;140(12):5995–5998. doi: 10.1210/endo.140.12.7288. [DOI] [PubMed] [Google Scholar]
- 23.Halaas JL, Gajiwala KS, Maffei M, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269(5223):543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 24.Pelleymounter MA, Cullen MJ, Baker MB, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269(5223):540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 25.Tartaglia LA, Dembski M, Weng X, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83(7):1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 26.Bjørbæk C, Elmquist JK, Michl P, et al. Expression of leptin receptor isoforms in rat brain microvessels. Endocrinology. 1998;139(8):3485–3491. doi: 10.1210/endo.139.8.6154. [DOI] [PubMed] [Google Scholar]
- 27.Elmquist JK, Bjørbæk C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. Journal of Comparative Neurology. 1998;395(4):535–547. [PubMed] [Google Scholar]
- 28.Fei H, Okano HJ, Li C, et al. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(13):7001–7005. doi: 10.1073/pnas.94.13.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395(6704):763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 30.Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Trayhurn P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Letters. 1996;387(2-3):113–116. doi: 10.1016/0014-5793(96)00473-5. [DOI] [PubMed] [Google Scholar]
- 31.Wang MY, Zhou YT, Newgard CB, Unger RH. A novel leptin receptor isoform in rat. FEBS Letters. 1996;392(2):87–90. doi: 10.1016/0014-5793(96)00790-9. [DOI] [PubMed] [Google Scholar]
- 32.Kloek C, Haq AK, Dunn SL, Lavery HJ, Banks AS, Myers MG. Regulation of Jak kinases by intracellular leptin receptor sequences. Journal of Biological Chemistry. 2002;277(44):41547–41555. doi: 10.1074/jbc.M205148200. [DOI] [PubMed] [Google Scholar]
- 33.Gong Y, Ishida-Takahashi R, Villanueva EC, Fingar DC, Münzberg H, Myers MG. The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. Journal of Biological Chemistry. 2007;282(42):31019–31027. doi: 10.1074/jbc.M702838200. [DOI] [PubMed] [Google Scholar]
- 34.You J, Yu Y, Jiang L, et al. Signaling through Tyr985 of leptin receptor as an age/diet-dependent switch in the regulation of energy balance. Molecular and Cellular Biology. 2010;30(7):1650–1659. doi: 10.1128/MCB.01307-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hekerman P, Zeidler J, Bamberg-Lemper S, et al. Pleiotropy of leptin receptor signalling is defined by distinct roles of the intracellular tyrosines. FEBS Journal. 2005;272(1):109–119. doi: 10.1111/j.1742-4658.2004.04391.x. [DOI] [PubMed] [Google Scholar]
- 36.Doherty GH, Oldreive C, Harvey J. Neuroprotective actions of leptin on central and peripheral neurons in vitro. Neuroscience. 2008;154(4):1297–1307. doi: 10.1016/j.neuroscience.2008.04.052. [DOI] [PubMed] [Google Scholar]
- 37.Cota D, Proulx K, Blake Smith KA, et al. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312(5775):927–930. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- 38.Burguera B, Couce ME, Long J, et al. The long form of the leptin receptor (OB-Rh) is widely expressed in the human brain. Neuroendocrinology. 2000;71(3):187–195. doi: 10.1159/000054536. [DOI] [PubMed] [Google Scholar]
- 39.Mercer JG, Moar KM, Rayner DV, Trayhurn P, Hoggard N. Regulation of leptin receptor and NPY gene expression in hypothalamus of leptin-treated obese (ob/ob) and cold-exposed lean mice. FEBS Letters. 1997;402(2-3):185–188. doi: 10.1016/s0014-5793(96)01525-6. [DOI] [PubMed] [Google Scholar]
- 40.Bennett PA, Lindell K, Karlsson C, Robinson ICAF, Carlsson LMS, Carlsson B. Differential expression and regulation of leptin receptor isoforms in the rat brain: effects of fasting and oestrogen. Neuroendocrinology. 1998;67(1):29–36. doi: 10.1159/000054295. [DOI] [PubMed] [Google Scholar]
- 41.Thurlby PL, Trayhurn P. The role of thermoregulatory thermogenesis in the development of obesity in genetically-obese (ob/ob) mice pair-fed with lean siblings. British Journal of Nutrition. 1979;42(3):377–385. doi: 10.1079/bjn19790127. [DOI] [PubMed] [Google Scholar]
- 42.Garris DR, Garris BL. Cytochemical analysis of pancreatic islet hypercytolipidemia following diabetes (db/db) and obese (ob/ob) mutation expression: influence of genomic background. Pathobiology. 2004;71(5):231–240. doi: 10.1159/000080056. [DOI] [PubMed] [Google Scholar]
- 43.Farooqi IS, Keogh JM, Kamath S, et al. Metabolism: partial leptin deficiency and human adiposity. Nature. 2001;414(6859):34–35. doi: 10.1038/35102112. [DOI] [PubMed] [Google Scholar]
- 44.Montague CT, Farooqi IS, Whitehead JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387(6636):903–908. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 45.Strobel A, Issad T, Camoin L, Ozata M, Strosberg AD. A leptin missense mutation associated with hypogonadism and morbid obesity. Nature Genetics. 1998;18(3):213–215. doi: 10.1038/ng0398-213. [DOI] [PubMed] [Google Scholar]
- 46.Doherty GH. Homocysteine: more than just a matter of life and death. Experimental Neurology. 2007;205(1):5–8. doi: 10.1016/j.expneurol.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 47.Scarpace PJ, Matheny M, Moore RL, Tümer N. Impaired leptin responsiveness in aged rats. Diabetes. 2000;49(3):431–435. doi: 10.2337/diabetes.49.3.431. [DOI] [PubMed] [Google Scholar]
- 48.Fernández-Galaz C, Fernández-Agulló T, Campoy F, et al. Decreased leptin uptake in hypothalamic nuclei with ageing in Wistar rats. Journal of Endocrinology. 2001;171(1):23–32. doi: 10.1677/joe.0.1710023. [DOI] [PubMed] [Google Scholar]
- 49.Wang Z, Zhou YT, Kakuma T, et al. Leptin resistance of adipocytes in obesity: role of suppressors of cytokine signaling. Biochemical and Biophysical Research Communications. 2000;277(1):20–26. doi: 10.1006/bbrc.2000.3615. [DOI] [PubMed] [Google Scholar]
- 50.Peralta S, Carrascosa JM, Gallardo N, Ros M, Arribas C. Ageing increases SOCS-3 expression in rat hypothalamus: effects of food restriction. Biochemical and Biophysical Research Communications. 2002;296(2):425–428. doi: 10.1016/s0006-291x(02)00906-3. [DOI] [PubMed] [Google Scholar]
- 51.Morrison CD, White CL, Wang Z, et al. Increased hypothalamic protein tyrosine phosphatase 1B contributes to leptin resistance with age. Endocrinology. 2007;148(1):433–440. doi: 10.1210/en.2006-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahima RS, Bjorbæk C, Osei S, Flier JS. Regulation of neuronal and glial proteins by leptin: implications for brain development. Endocrinology. 1999;140(6):2755–2762. doi: 10.1210/endo.140.6.6774. [DOI] [PubMed] [Google Scholar]
- 53.Dicou E, Attoub S, Gressens P. Neuroprotective effects of leptin in vivo and in vitro. NeuroReport. 2001;12(18):3947–3951. doi: 10.1097/00001756-200112210-00019. [DOI] [PubMed] [Google Scholar]
- 54.Zhang F, Wang S, Signore AP, Chen J. Neuroprotective effects of leptin against ischemic injury induced by oxygen-glucose deprivation and transient cerebral ischemia. Stroke. 2007;38(8):2329–2336. doi: 10.1161/STROKEAHA.107.482786. [DOI] [PubMed] [Google Scholar]
- 55.Weng Z, Signore AP, Gao Y, et al. Leptin protects against 6-hydroxydopamine-induced dopaminergic cell death via mitogen-activated protein kinase signaling. Journal of Biological Chemistry. 2007;282(47):34479–34491. doi: 10.1074/jbc.M705426200. [DOI] [PubMed] [Google Scholar]
- 56.Guo Z, Jiang H, Xu X, Duan W, Mattson MP. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. Journal of Biological Chemistry. 2008;283(3):1754–1763. doi: 10.1074/jbc.M703753200. [DOI] [PubMed] [Google Scholar]
- 57.Pérez-González R, Antequera D, Vargas T, Spuch C, Bolós M, Carro E. Leptin induces proliferation of neuronal progenitors and neuroprotection in a mouse model of alzheimer's disease. Journal of Alzheimer's Disease. 2011;24(supplement 2):17–25. doi: 10.3233/JAD-2011-102070. [DOI] [PubMed] [Google Scholar]
- 58.Shanley LJ, Irving AJ, Harvey J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. The Journal of Neuroscience. 2001;21(24):p. RC186. doi: 10.1523/JNEUROSCI.21-24-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oomura Y, Hori N, Shiraishi T, et al. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides. 2006;27(11):2738–2749. doi: 10.1016/j.peptides.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 60.Li XL, Aou S, Oomura Y, Hori N, Fukunaga K, Hori T. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience. 2002;113(3):607–615. doi: 10.1016/s0306-4522(02)00162-8. [DOI] [PubMed] [Google Scholar]
- 61.Greco SJ, Bryan KJ, Sarkar S, et al. Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer’s disease. Journal of Alzheimer’s Disease. 2010;19(4):1155–1167. doi: 10.3233/JAD-2010-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lieb W, Beiser AS, Vasan RS, et al. Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. Journal of the American Medical Association. 2009;302(23):2565–2572. doi: 10.1001/jama.2009.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ban Y, Watanabe T, Suguro T, et al. Increased plasma urotensin-II and carotid atherosclerosis are associated with vascular dementia. Journal of Atherosclerosis and Thrombosis. 2009;16(3):179–187. doi: 10.5551/jat.e608. [DOI] [PubMed] [Google Scholar]
- 64.Utsunomiya K, Shinkai T, Sakata S, et al. Lack of association between the leptin receptor gene (LEPR) Gln223Arg polymorphism and late-onset alzheimer disease. Alzheimer Disease and Associated Disorders. 2010;24(1):101–103. doi: 10.1097/WAD.0b013e3181b982dc. [DOI] [PubMed] [Google Scholar]
- 65.Power DA, Noel J, Collins R, O’Neill D. Circulating leptin levels and weight loss in Alzheimer’s disease patients. Dementia and Geriatric Cognitive Disorders. 2001;12(2):167–170. doi: 10.1159/000051252. [DOI] [PubMed] [Google Scholar]
- 66.Li C, Zhao R, Gao K, et al. Astrocytes: implications for neuroinflammatory pathogenesis of Alzheimer's disease. Current Alzheimer Research. 2011;8(1):67–80. doi: 10.2174/156720511794604543. [DOI] [PubMed] [Google Scholar]
- 67.García-Cáceres C, Fuente-Martán E, Burgos-Ramos E, et al. Differential acute and chronic effects of leptin on hypothalamic astrocyte morphology and synaptic protein levels. Endocrinology. 2011;152(5):1809–1818. doi: 10.1210/en.2010-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arai Y, Takayama M, Abe Y, Hirose N. Adipokines and aging. Journal of Atherosclerosis and Thrombosis. 2011;18(7):545–550. doi: 10.5551/jat.7039. [DOI] [PubMed] [Google Scholar]
- 69.Whitmer RA, Gunderson EP, Barrett-Connor E, Quesenberry CP, Yaffe K. Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. British Medical Journal. 2005;330(7504):1360–1362. doi: 10.1136/bmj.38446.466238.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mantzoros CS, Magkos F, Brinkoetter M, et al. Leptin in human physiology and pathophysiology. American Journal of Physiology-Endocrinology and Metabolism. 2011;301(4):E567–E584. doi: 10.1152/ajpendo.00315.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kalaria RN. Vascular basis for brain degeneration: faltering controls and risk factors for dementia. Nutrition Reviews. 2010;68(2):s74–s87. doi: 10.1111/j.1753-4887.2010.00352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harvey J. Leptin: the missing link in Alzheimer disease? Clinical Chemistry. 2010;56(5):696–697. doi: 10.1373/clinchem.2010.144006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oldreive CE, Harvey J, Doherty GH. Neurotrophic effects of leptin on cerebellar Purkinje but not granule neurons in vitro. Neuroscience Letters. 2008;438(1):17–21. doi: 10.1016/j.neulet.2008.04.045. [DOI] [PubMed] [Google Scholar]
- 74.Benzing WC, Wujek JR, Ward EK, et al. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiology of Aging. 1999;20(6):581–589. doi: 10.1016/s0197-4580(99)00065-2. [DOI] [PubMed] [Google Scholar]
- 75.Olanow CW, Kordower JH, Freeman TB. Fetal nigral transplantation as a therapy for Parkinson’s disease. Trends in Neurosciences. 1996;19(3):102–109. doi: 10.1016/s0166-2236(96)80038-5. [DOI] [PubMed] [Google Scholar]
- 76.Meyer M, Matarredona ER, Seiler RW, Zimmer J, Widmer HR. Additive effect of glial cell line-derived neurotrophic factor and neurotrophin-4/5 on rat fetal nigral explant cultures. Neuroscience. 2001;108(2):273–284. doi: 10.1016/s0306-4522(01)00418-3. [DOI] [PubMed] [Google Scholar]
- 77.Doherty GH. Developmental switch in the effects of TNFα on ventral midbrain dopaminergic neurons. Neuroscience Research. 2007;57(2):296–305. doi: 10.1016/j.neures.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 78.Harvey J. Leptin regulation of neuronal excitability and cognitive function. Current Opinion in Pharmacology. 2007;7(6):643–647. doi: 10.1016/j.coph.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shanley LJ, Irving AJ, Rae MG, Ashford MLJ, Harvey J. Leptin inhibits rat hippocampal neurons via activation of large conductance calcium-activated K+ channels. Nature Neuroscience. 2002;5(4):299–300. doi: 10.1038/nn824. [DOI] [PubMed] [Google Scholar]
- 80.Durakoglugil M, Irving AJ, Harvey J. Leptin induces a novel form of NMDA receptor-dependent long-term depression. Journal of Neurochemistry. 2005;95(2):396–405. doi: 10.1111/j.1471-4159.2005.03375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schulz C, Paulus K, Lehnert H. Central nervous and metabolic effects of intranasally applied leptin. Endocrinology. 2004;145(6):2696–2701. doi: 10.1210/en.2003-1431. [DOI] [PubMed] [Google Scholar]
- 82.Fliedner S, Schulz C, Lehnert H. Brain uptake of intranasally applied radioiodinated leptin in Wistar rats. Endocrinology. 2006;147(5):2088–2094. doi: 10.1210/en.2005-1016. [DOI] [PubMed] [Google Scholar]
- 83.Beretta E, Dube MG, Kalra PS, Kalra SP. Long-term suppression of weight gain, adiposity, and serum insulin by central leptin gene therapy in prepubertal rats: effects on serum ghrelin and appetite-regulating genes. Pediatric Research. 2002;52(2):189–198. doi: 10.1203/00006450-200208000-00010. [DOI] [PubMed] [Google Scholar]
- 84.Boghossian S, Ueno N, Dube MG, Kalra P, Kalra S. Leptin gene transfer in the hypothalamus enhances longevity in adult monogenic mutant mice in the absence of circulating leptin. Neurobiology of Aging. 2007;28(10):1594–1604. doi: 10.1016/j.neurobiolaging.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 85.Dube MG, Beretta E, Dhillon H, Ueno N, Kalra PS, Kalra SP. Central leptin gene therapy blocks high-fat diet-induced weight gain, hyperleptinemia, and hyperinsulinemia: increase in serum ghrelin levels. Diabetes. 2002;51(6):1729–1736. doi: 10.2337/diabetes.51.6.1729. [DOI] [PubMed] [Google Scholar]