

Abstract
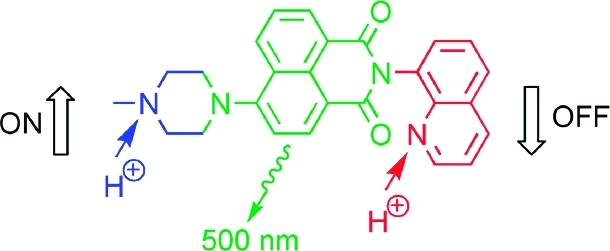
A novel molecular system with characteristics of an OFF-ON-OFF fluorescence switch was designed to integrate the function of a T-latch. In detail, a receptor1-fluorophore-receptor2 architecture was adopted to achieve fluorescence switching upon addition of protons.
The idea to use molecular systems for information processing has attracted a great deal of interest during the recent years.1−5 This has been manifested by the availability of molecular mimics for all essential logic gates (AND, OR, NOR, NAND, INH, XOR, etc.)1,3 and for rather complex logic devices such as adders/subtractors, encoders/decoders, and multiplexers/demultiplexers.2,6−10 Such logic functions are of elevated interest for applications such as object coding,(11) intelligent materials,12−14 pro-drug activation,15−17 and diagnostics/actuation.18−20 While these systems work independently of the order of input application (combinational logic), the molecular memorization of information is a precondition for applications which profit from a sequential application of input signals.21,22 This behavior is reflected in the function of molecular keypad locks10,23−28 and memory devices.29−35
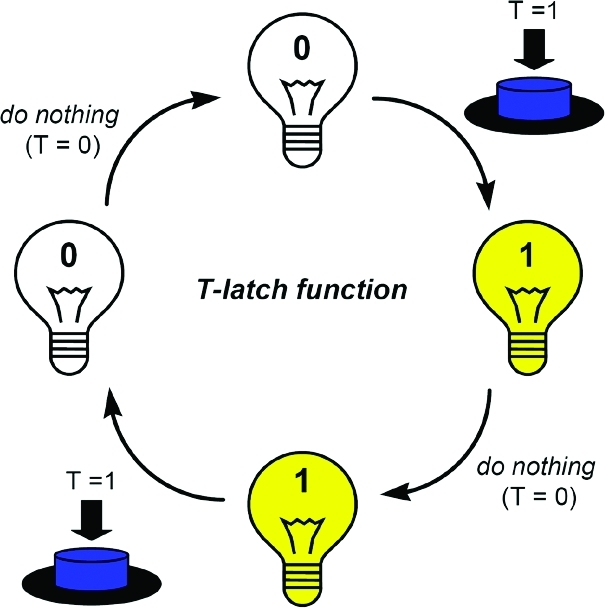
The set-reset (S-R) latch was one of the first memory devices that was implemented at the molecular level by using electrochemical, chemical, and photonic signaling.29,31−35 The device is characterized by a high state (binary 1) whenever the set input is applied (S = 1) and which upon reset (R = 1) has a binary 0 (low) state. The herein described toggle-latch (T-latch) is a different logic switch with memory capacity. Its working principle is well illustrated with the function of a conventional light switch or of the push button of a ballpoint pen: every time the toggle input is activated, the state Q of the system changes (see Scheme 1). The device “remembers” if a 0 or a 1 state was memorized (Qcurrent) and upon each T input application, the new state (Qnext) has the opposite value (0 → 1 and 1 → 0). The “do nothing” situation leaves the system state unchanged.
Scheme 1. Presentation of the T-Latch Function.
We anticipated that a molecular OFF-ON-OFF fluorescent switch could integrate this function. In detail, we needed a switch which upon single application of an input changes to the ON state and is set back to the OFF state by a second equal input. Fluorescent systems, which change their emission properties upon application of chemical input information, have been often explored in the design of logic switches and chemical sensors.3,4,36−38 The integrated receptor1-fluorophore-receptor2 architecture 2 (Scheme 2a) was identified as an excellent candidate to put the molecular T-latch function into practice.
Scheme 2. (a) Structures of Triad 2 and the Naphthalimide Models 3 and 5 with One Receptor Unit and (b) Synthesis of Triad 2 (for Compound 4, an Intermediary Product, see Supporting Information).

The synthesis of the new triad 2 is briefly sketched in Scheme 2b. The sequence started with the commercial 4-bromo-1,8-naphthalic anhydride, which was condensed with 8-aminoquinoline (74% yield). Further aromatic nucleophilic substitution of the intermediary 4-bromo-1,8-naphthalimide derivative with N-methylpiperazine resulted in the final product 2 with a yield of 52%. The synthesis of the naphthalimide derivatives 3 and 5 (Scheme 2a), which served herein as model structures, is described in the Supporting Information.
Triad 2 contains two proton receptors: a piperazinyl and a quinolinyl moiety. The receptors have sufficiently different pKa values (7.78 for N-benzoylpiperazine versus 4.60 for 8-methylquinoline as models)(39) so that they can be stepwise protonated. In accordance with this assumption and as shown in Figures 1 and 2, the addition of 1 equiv of protons (triflic acid; CF3SO3H) yielded a pronounced fluorescence enhancement (fluorescence quantum yield Φf = 0.67 versus 0.017 for 2H+ and 2, respectively) of the 4-amino-1,8-naphthalimide chromophore (λfluo,max = 504 nm for 2 and 499 nm for 2H+). However, the subsequent addition of a second equivalent of CF3SO3H caused practically quantitative fluorescence quenching (98% quenching). The photophysical properties of all investigated compounds and their protonated forms are summarized in Table 1. The independent actuation of both receptors in 2 was supported by the observation of the same differential photophysical effects upon protonation of the model compounds 3 and 5, which contain each only one of the two receptors (Scheme 2a). In accordance with the fluorescence response of 2 upon stepwise protonation, 3 showed quenching and 5 enhancement of the emission for the addition of 1 equiv of protons (Supporting Information). The superposition of the photophysical trends of the model compounds in the triad was also noted for the absorption spectra.(40)
Figure 1.

Relative absorption spectra (dashed lines) and normalized fluorescence spectra (solid lines) for 2 (red), 2H+ (blue), and 2H22+ (black). Note that the low fluorescence emissions of 2 and 2H22+ are hardly distinguishable.
Figure 2.

Fluorescence titration curve (λexc = 388 nm, λobs = 499 nm) of 2 (12.5 μM in acetonitrile) upon CF3SO3H addition.
Table 1. Photophysical Properties of Compounds 2, 3, and 5 and Their Protonated Forms in Aerated Acetonitrile Solution.
| λabs,max/nm | ε/M–1 cm–1 | λfluo,max/nm | Φf | τf/ns | |
|---|---|---|---|---|---|
| 2 | 401 | 10100 | 504 | 0.017 | a |
| 2H+ | 377 | 10500 | 499 | 0.67 | 8.90 |
| 2H22+ | 383 | 11100 | 499 | 0.017 | a |
| 3 | 433 | 13000 | 520 | 0.56 | 10.00 |
| 3H+ | 441 | 13400 | 519 | 0.006 | a |
| 5 | 398 | 9700 | 502 | 0.018 | a |
| 5H+ | 374 | 10100 | 498 | 0.62 | 9.06 |
Not determined due to low signal intensity.
The fluorescence switching of triad 2 can be mechanistically rationalized as follows. The electron-donating methyl-substituted piperazinyl nitrogen atom is protonated upon the addition of the first equivalent of protons, which leads to blocking of photoinduced electron transfer (PET) and consequently fluorescence ON switching.41−43 The second equivalent of protons serves to transform the quinolinyl residue into a quinolinium cation. The hydrogen-bonding interaction of NH+ with the imide carbonyl C=O is assumed to be at the origin of the fluorescence quenching of the 4-amino-1,8-naphthalimide derivative.(41) However, PET from the singlet-excited fluorophore to the electron-accepting quinolinium cation may also be involved in the observed fluorescence OFF switching.44,45 Noteworthy, the control of 4-aminonaphthalimide fluorescence by a receptor linked to the “imide side” of the fluorophore has been rarely observed.41,42,46−48
The first three columns of the truth table (Table 2) describe the implementation of the T-latch function, which is mimicked by the above-discussed fluorescence switching. Starting with the triad in its unprotonated state (2), only low fluorescence is observed for T = 0 (no addition of acid). This situation corresponds to Qcurrent = Qnext = 0. However, protonation of 2 with 1 equiv of acid (T = 1) leads to 2H+ and consequently a high fluorescence output (toggling from Qcurrent = 0 to Qnext = 1). Again, the “do nothing situation” (T = 0) preserves the Q state (i.e., Qcurrent = Qnext = 1 in this case). The second addition of 1 equiv of acid (T = 1) to 2H+ yields 2H22+ and concomitant fluorescence quenching, corresponding to a switching from Qcurrent = 1 to Qnext = 0.
Table 2. Truth Table for the Implemented Molecular T-Latch.
| T input (1 equiv H+) | Qcurrent (fluo) | Qnext (fluo) | control channel (abs, 313 nm) |
|---|---|---|---|
| 0 | 0 | 0 | 0 |
| 1 | 0 | 1 | 0 |
| 0 | 1 | 1 | 0 |
| 1 | 1 | 0 | 1 |
The protonation state of the triad can be easily reset by application of a strong base (P2-Et phosphazene), leading to the inverse titration curve (see Supporting Information). The consecutive protonation/deprotonation of 2 with acid/base can be repeated for at least five cycles without significant loss of the dynamic fluorescence switching range (Figure 3). The correct functioning of the T-latch requires that the initial device state is represented by the unprotonated triad 2. However, by solely reading the fluorescence output Q, it cannot be decided whether at a random point of operation Qcurrent = 0 corresponds to 2 or 2H22+. The unambiguous assignment of an output to a concrete input situation can be resolved by reading a control channel, as has been shown previously for the implementation of reversible logic functionality.10,28,49,50 This control signal is provided herein by the absorption of the quinolinium cation at ca. 313 nm (fourth column in Table 2).(51) This spectral signature only evolves when the quinoline unit becomes protonated (Supporting Information). Hence, when the fluorescence is low and the absorbance at 313 nm is high, 2 equiv of base is needed to reset the system to its initial state (unprotonated 2). If the fluorescence output and the absorbance at 313 nm are both low, then the system is already in its initial state. Hence, the two Q = 0 situations are now clearly distinguishable.
Figure 3.

Recycling of fluorescence switching (λobs = 499 nm) of 2 (8.7 μM in acetonitrile) upon consecutive addition of 1 equiv of CF3SO3H followed by 1 equiv of P2-Et phosphazene base. The dashed line marks the threshold. A conservative estimation yields that up to 10 cycles are possible, maintaining a dynamic switching range of I(ON)/I(OFF) ≥ 2.
In summary, we have shown that an OFF-ON-OFF fluorescence switch with two degenerate proton inputs can integrate the function of a molecular T-latch. The photophysical design of the switch is based on the control of electron transfer and hydrogen-bond interaction. Work on an all-optical version, exploring photoinduced proton transfer as relay mechanism, is underway.
Acknowledgments
Financial support by the Spanish MICINN (CTQ2008-06777-C02-02 for U.P., CTQ2010-20303 for E.P.-I.), the Consejería de Economía, Innovación y Ciencia–Junta de Andalucía (FQM-3685 for U.P.), FEDER, the Swedish Research Council (622-2010-280 for J.A.), and the European Research Council (FP7/2007-2013 No. 203952 for J.A.) is acknowledged.
Supporting Information Available
Details on the synthesis of 1–5, 1H and 13C NMR spectra, additional spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- de Silva A. P.; Uchiyama S. Nat. Nanotechnol. 2007, 2, 399–410. [DOI] [PubMed] [Google Scholar]
- Pischel U. Angew. Chem., Int. Ed. 2007, 46, 4026–4040. [DOI] [PubMed] [Google Scholar]
- Szaciłowski K. Chem. Rev. 2008, 108, 3481–3548. [DOI] [PubMed] [Google Scholar]
- Andréasson J.; Pischel U. Chem. Soc. Rev. 2010, 39, 174–188. [DOI] [PubMed] [Google Scholar]
- Pischel U. Aust. J. Chem. 2010, 63, 148–164. [Google Scholar]
- Margulies D.; Melman G.; Shanzer A. J. Am. Chem. Soc. 2006, 128, 4865–4871. [DOI] [PubMed] [Google Scholar]
- Amelia M.; Baroncini M.; Credi A. Angew. Chem., Int. Ed. 2008, 47, 6240–6243. [DOI] [PubMed] [Google Scholar]
- Pérez-Inestrosa E.; Montenegro J.-M.; Collado D.; Suau R. Chem. Commun. 2008, 1085–1087. [DOI] [PubMed] [Google Scholar]
- Ceroni P.; Bergamini G.; Balzani V. Angew. Chem., Int. Ed. 2009, 48, 8516–8518. [DOI] [PubMed] [Google Scholar]
- Andréasson J.; Pischel U.; Straight S. D.; Moore T. A.; Moore A. L.; Gust D. J. Am. Chem. Soc. 2011, 133, 11641–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Silva A. P.; James M. R.; McKinney B. O. F.; Pears D. A.; Weir S. M. Nat. Mater. 2006, 5, 787–790. [DOI] [PubMed] [Google Scholar]
- Motornov M.; Zhou J.; Pita M.; Gopishetty V.; Tokarev I.; Katz E.; Minko S. Nano Lett. 2008, 8, 2993–2997. [DOI] [PubMed] [Google Scholar]
- Angelos S.; Yang Y.-W.; Khashab N. M.; Stoddart J. F.; Zink J. I. J. Am. Chem. Soc. 2009, 131, 11344–11346. [DOI] [PubMed] [Google Scholar]
- Tokarev I.; Gopishetty V.; Zhou J.; Pita M.; Motornov M.; Katz E.; Minko S. ACS Appl. Mater. Interfaces 2009, 1, 532–536. [DOI] [PubMed] [Google Scholar]
- Amir R. J.; Popkov M.; Lerner R. A.; Barbas C. F. III; Shabat D. Angew. Chem., Int. Ed. 2005, 44, 4378–4381. [DOI] [PubMed] [Google Scholar]
- Ozlem S.; Akkaya E. U. J. Am. Chem. Soc. 2009, 131, 48–49. [DOI] [PubMed] [Google Scholar]
- Hammarson M.; Andersson J.; Li S.; Lincoln P.; Andréasson J. Chem. Commun. 2010, 46, 7130–7132. [DOI] [PubMed] [Google Scholar]
- Konry T.; Walt D. R. J. Am. Chem. Soc. 2009, 131, 13232–13233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies D.; Hamilton A. D. J. Am. Chem. Soc. 2009, 131, 9142–9143. [DOI] [PubMed] [Google Scholar]
- Privman M.; Tam T. K.; Bocharova V.; Halámek J.; Wang J.; Katz E. ACS Appl. Mater. Interfaces 2011, 3, 1620–1623. [DOI] [PubMed] [Google Scholar]
- Raymo F. M.; Alvarado R. J.; Giordani S.; Cejas M. A. J. Am. Chem. Soc. 2003, 125, 2361–2364. [DOI] [PubMed] [Google Scholar]
- Pischel U. Angew. Chem., Int. Ed. 2010, 49, 1356–1358. [DOI] [PubMed] [Google Scholar]
- Guo Z.; Zhu W.; Shen L.; Tian H. Angew. Chem., Int. Ed. 2007, 46, 5549–5553. [DOI] [PubMed] [Google Scholar]
- Margulies D.; Felder C. E.; Melman G.; Shanzer A. J. Am. Chem. Soc. 2007, 129, 347–354. [DOI] [PubMed] [Google Scholar]
- Suresh M.; Ghosh A.; Das A. Chem. Commun. 2008, 3906–3908. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Luxami V.; Saini R.; Kaur D. Chem. Commun. 2009, 3044–3046. [DOI] [PubMed] [Google Scholar]
- Andréasson J.; Straight S. D.; Moore T. A.; Moore A. L.; Gust D. Chem.—Eur. J. 2009, 15, 3936–3939. [DOI] [PubMed] [Google Scholar]
- Remón P.; Hammarson M.; Li S.; Kahnt A.; Pischel U.; Andréasson J. Chem.—Eur. J. 2011, 17, 6492–6500. [DOI] [PubMed] [Google Scholar]
- Baron R.; Onopriyenko A.; Katz E.; Lioubashevski O.; Willner I.; Wang S.; Tian H. Chem. Commun. 2006, 2147–2149. [DOI] [PubMed] [Google Scholar]
- Elbaz J.; Moshe M.; Willner I. Angew. Chem., Int. Ed. 2009, 48, 3834–3837. [DOI] [PubMed] [Google Scholar]
- Periyasamy G.; Collin J.-P.; Sauvage J.-P.; Levine R. D.; Remacle F. Chem.—Eur. J. 2009, 15, 1310–1313. [DOI] [PubMed] [Google Scholar]
- Pita M.; Strack G.; MacVittie K.; Zhou J.; Katz E. J. Phys. Chem. B 2009, 113, 16071–16076. [DOI] [PubMed] [Google Scholar]
- de Ruiter G.; Motiei L.; Choudhury J.; Oded N.; van der Boom M. E. Angew. Chem., Int. Ed. 2010, 49, 4780–4783. [DOI] [PubMed] [Google Scholar]
- de Ruiter G.; Tartakovsky E.; Oded N.; van der Boom M. E. Angew. Chem., Int. Ed. 2010, 49, 169–172. [DOI] [PubMed] [Google Scholar]
- Pischel U.; Andréasson J. New J. Chem. 2010, 34, 2701–2703. [Google Scholar]
- de Silva A. P.; Gunaratne H. Q. N.; Gunnlaugsson T.; Huxley A. J. M.; McCoy C. P.; Rademacher J. T.; Rice T. E. Chem. Rev. 1997, 97, 1515–1566. [DOI] [PubMed] [Google Scholar]
- de Silva A. P.; Vance T. P.; West M. E. S.; Wright G. D. Org. Biomol. Chem. 2008, 6, 2468–2480. [DOI] [PubMed] [Google Scholar]
- Duke R. M.; Veale E. B.; Pfeffer F. M.; Kruger P. E.; Gunnlaugsson T. Chem. Soc. Rev. 2010, 39, 3936–3953. [DOI] [PubMed] [Google Scholar]
- The pKa data were taken from http://research.chem.psu.edu/brpgroup/pKa_compilation.pdf.
- The protonation of the piperazinyl residue leads to a blue shift (by 24 nm) of the long wavelength aminonaphthalimide absorption band (for 2 and 5), which is indicative of the destabilization of the charge transfer state through the repulsive interaction between the protonated distant methyl-substituted N and the positive pole of the charge transfer state at the aromatic N (cf. ref (47)). The protonation of the quinolinyl moiety has stabilizing effects on the charge transfer state, which is expressed by a red shift (by 6–8 nm) of the long wavelength absorption band (for 2 and 3).
- de Silva A. P.; Gunaratne H. Q. N.; Habib-Jiwan J.-L.; McCoy C. P.; Rice T. E.; Soumillion J.-P. Angew. Chem., Int. Ed. Engl. 1995, 34, 1728–1731. [Google Scholar]
- de Silva A. P.; Rice T. E. Chem. Commun. 1999, 163–164. [Google Scholar]
- Tian H.; Xu T.; Zhao Y.; Chen K. J. Chem. Soc., Perkin Trans. 2 1999, 545–549. [Google Scholar]
- Greenfield S. R.; Svec W. A.; Gosztola D.; Wasielewski M. R. J. Am. Chem. Soc. 1996, 118, 6767–6777. [Google Scholar]
- Ferreira R.; Remón P.; Pischel U. J. Phys. Chem. C 2009, 113, 5805–5811. [Google Scholar]
- Gao Y. Q.; Marcus R. A. J. Phys. Chem. A 2002, 106, 1956–1960. [Google Scholar]
- de Silva A. P.; Goligher A.; Gunaratne H. Q. N.; Rice T. E. ARKIVOC 2003, (vii), 229–243. [Google Scholar]
- Veale E. B.; Gunnlaugsson T. J. Org. Chem. 2008, 73, 8073–8076. [DOI] [PubMed] [Google Scholar]
- Remón P.; Ferreira R.; Montenegro J.-M.; Suau R.; Pérez-Inestrosa E.; Pischel U. ChemPhysChem 2009, 10, 2004–2007. [DOI] [PubMed] [Google Scholar]
- Semeraro M.; Credi A. J. Phys. Chem. C 2010, 114, 3209–3214. [Google Scholar]
- Schulman S. G.; Capomacchia A. C. J. Am. Chem. Soc. 1973, 95, 2763–2766. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.