

Abstract
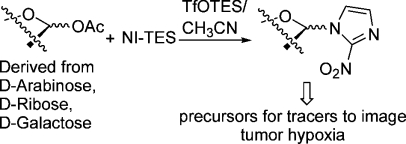
2-Nitroimidazole was silylated using hexaethyldisilazane and then reacted with 1-O-acetyl derivatives of d-arabinose, d-ribose, and d-galactose in acetonitrile at mild temperatures (−20 °C to rt), catalyzed by triethylsilyl triflate (Vorbrüggen conditions). The α-anomer was formed in the former case and the β-anomers in the latter two cases (highly) selectively. When d-arabinose and d-ribose were silylated with tert-butyldiphenylsilyl chloride in pyridine at the hydroxyl groups at C-5 and acetylated at the other ones in a one-pot reaction, mixtures of anomeric 1-O-acetyl derivatives were obtained. These were coupled by the Vorbrüggen method and then deblocked at C-5 and tosylated to give precursors for tracers to image hypoxia in four steps without using Hg(CN)2 necessary for other methods. The Vorbrüggen conditions enable a shorter route to azomycin nucleoside analogues than the previous coupling procedures.
Introduction
A large body of evidence indicates that tumor hypoxia has a negative prognosis predictive value for solid tumor progression, likeliness of metastasis and tumor prognosis, likeliness of metastasis, and overall survival.1−3 In addition, tumor cell hypoxia has a negative effect on anticancer treatment because of resistance to radiation.4,5 Accordingly, it is important to identify and localize tumor hypoxia in the management and treatment of cancer patients. Contrary to invasive methods based on the oxygen electrode system(6) to determine the oxygenation distribution in the tumor, noninvasive ones7,8 are based on imaging using radiolabeled (123I, 124I, 125I, 18F, 60Cu, 62Cu, 64Cu) tracers 1–6 (Figure 1), preferably 18F-labeled 2-nitroimidazoles. The tracer's primary cellular uptake results from diffusion and partition-based retention in lipophilic tissue.
Figure 1.

Radiolabeled tracers for imaging hypoxia.
The 2-nitroimidazole (azomycin)-based tracers are thought to be reduced in hypoxic cells to reactive intermediates, which bind to macromolecular components. Thus, they tend to be accumulated in sites of hypoxia and can be used for imaging purposes with single-photon-emission tomography (SPECT) and positron emission tomography (PET), the latter being the method of choice. Indeed, a number of radiotracers for viable hypoxic cells in solid tumors are being evaluated clinically.(9) The initial imaging studies of tumor hypoxia used iodine-123-labeled iodoazomycin arabinoside (α-IAZA, 4) with SPECT(10) and fluorine-18-labeled fluoromisonidazole (FMISO, 1) with PET.11,12 Recently, [18F]fluoroerythronitroimidazole ([18F]FETNIM, 2) was studied as a hypoxia imaging agent.(13) The copper complex copper(II) diacetylbis(N4-methylthiosemicarbazone) (Cu-ATMS, 3) labeled with the positron emitter 60Cu, 62Cu, or 64Cu is another promising hypoxic imaging agent under evaluation.(14)
More recently, the fluoroazomycin arabinoside α-[18F]FAZA (5) has been studied extensively alone and relative to FMISO.(15) It was found that α-[18F]FAZA and [18F]FMISO are taken up selelctively in vitro and in vivo in hypoxic cells.(16) Machulla et al. observed that α-[18F]FAZA shows biokinetics superior to that of [18F]FMISO and is, thus, a promising PET tracer for visualization of solid tumor hypoxia. The 125I- and 124I-labeled 2-nitroimidazole β-d-galactosides 6 were used to image tumor hypoxia in mice.(17) As the uptake of α-[18F]FAZA is diffusion controlled and its renal clearance is rapid, it is still attractive to prepare other 2-nitroimidazole nucleosides and evaluate them as precursors for tracers to image hypoxia. To improve the tumor/background ratio of tracers, their active transport by cellular transport systems such as nucleoside transporters could be exploited. Emami et al. have established a yeast system for testing the transport of fluoroazomycin nucleosides by four nucleoside transporters, which might help to find better tracers than α-[18F]FAZA.(18)
Tracer 5 most favored at present for imaging hypoxia is prepared from precursor α-7 by a substitution reaction (SN2) with cyclotron-generated 18F– in combination with Kryptofix 2.2.2, followed by base-catalyzed removal of acetyl groups and HPLC purification (Scheme 1). The 2-nitroimidazole nucleosides underlying tracers 4, 5, and 6 are formed by a Königs–Knorr reaction of 2-nitroimidazole or in some cases its trimethylsilylated species in acetonitrile at 50–60 °C with a benzoyl- or an acetyl-protected d-1-bromofuranose or d-1-bromopyranose derivatives of d-arabinose or d-galactose, respectively. Normally, excess Hg(CN)2 (2.2 equiv), sometimes in combination with SnCl4, was used as a catalyst.19−21 This method was used by Schneider et al. to prepare a number of 2-nitroimidazole nucleosides, two of which were converted to radioiodinated tracers. When we started this project to prepare known and new precursors for tracers to image hypoxia, we decided to search for an alternative to the Königs–Knorr procedure not involving metal salts, especially toxic Hg(CN)2. Ideally, the method should use easily accessible starting materials with an adequately silylated hydroxyl group at C-5 and acetyl groups for the other hydroxyls of furanoses derived from d-arabinose and d-ribose. The 2-nitroimidazole nucleosides then formed would yield products which could be deprotected and tosylated at C-5 to give the desired precursors. Additionally, the desilylated products could be transformed to the fluorides and deblocked to give the cold fluoro standards.
Scheme 1. Synthesis of α-[18F]FAZA.

Results and Discussion
Preparation of Triethylsilylated 2-Nitroimidazole and 2-Nitroimidazole Nucleosides Derived from d-Arabinose
We envisaged to test the Vorbrüggen method for the coupling first.(22) It is based on the coupling of trimethylsilylated heterocyclic bases with protected sugar derivatives with an acetoxy or methoxy group at C-1 in CH3CN, CH2Cl2, or 1,2-C2H4Cl2, catalyzed by trimethylsilyl triflate. Surprisingly, it has never been used for the synthesis of azomycin nucleosides. The solubility of 2-nitroimidazole is very low in all common solvents, especially at rt or below, and even trimethylsilylation with hexamethyldisilazane/(NH4)2SO4 in pyridine had only a marginal effect on its solubility. Therefore, we decided to use triethylsilylation to increase the solubility further, which proved to be critical for our success.
We accessed hexaethyldisilazane, a known(23) but difficult to prepare reagent, easily from (triethylsilyl)amine(24) and chlorotriethylsilane in the presence of a catalytic amount of triethylsilyl triflate (TfOTES) in dry toluene in 79% yield (Scheme 2). Initially, we refluxed a mixture of 2-nitroimidazole (NI) and 4–6 equiv of hexaethyldisilazane and a catalytic amount of (NH4)2SO4 in pyridine until the reaction mixture was homogeneous. However, later we found that 2 equiv was sufficient for silylation within 30 min and that the catalyst could be omitted. Removal of volatile components left NI-TES (2-nitro-1-(triethylsilyl)imidazole) as a crystalline product, which was used directly. Surprisingly, (triethylsilyl)amine did not silylate NI in refluxing pyridine.
Scheme 2. Synthesis of Hexaethyldisilazane and Its Use for the Triethylsilylation of 2-Nitroimidazole.

In the first place, we wanted to couple NI-TES with a suitable d-arabinofuranose derivative containing a silyl-proteced C-5 hydroxyl group. Thus, a mixture of d-arabinose and tert-butyldiphenylsilyl chloride (TBDPSCl) in pyridine was allowed to warm in the cooling bath from −20 °C to rt for 18 h. Stirring was continued for 6 h after the addition of acetic anhydride. Aqueous workup and flash chromatography furnished a mixture of anomeric acetates (α:β = 60:40) in an admixture with impurities as a syrup in 71% yield (Scheme 3). The predominating α-anomer crystallized and was fully characterized. Recently, Chatterjee et al. isolated the 5-O-TBDPS-protected d-arabinose and then acetylated it in an overall yield of 46%.(25) The coupling experiments were performed with the anomeric mixture 14. Under optimized conditions, a mixture of NI-TES and 14 in CH3CN was cooled to −20 °C, and 1 equiv of a 1 M solution of TfOTES in 1,2-C2H4Cl2 was added dropwise. Stirring was continued while the reaction mixture was allowed to warm to −8 °C within 3 h. Extractive workup and flash chromatography furnished the α-nucleoside(26) α-15 in 72% yield beside some starting material and a trace of the β-anomer detected by TLC, which was not isolated (Scheme 3). Anticipating later results, α-15 was converted in two steps into precursor α-7. As the β-nucleoside β-15 was also needed, CH3CN was replaced by 1,2-C2H4Cl2, hoping to increase the amount of the β-anomer. When NI-TMS and the mixture of α- and β-14 were coupled in 1,2-C2H4Cl2 with 0.5 equiv of TfOTMS as a catalyst for 4 h at rt, the yield for β-15 was 13% and that for α-15 16%. The yield of β-15 increased to 31%, when the coupling was modified (NI-TES, 50 °C, 1.2 equiv of TfOTES, 2 h). The low and irreproducible yields in this halogenated solvent were attributed to the unsatisfactory solubility of NI-TES. Although the β-nucleoside was formed preferentially, the method was not suitable for the preparation of relevant amounts of β-7. The preferred formation of the α-anomer is attributed to the neighboring group effect by the 2-acetoxy group, which is β-orientated.(22) However, the formation of the β-anomer results very likely from the intermediacy of a nitronate ester as suggested by others.(19)
Scheme 3. Preparation of d-Arabinofuranose Derivative 14 and Its Coupling with NI-TES by the Vorbrüggen Method.

Preparation of 2-Nitroimidazole Nucleosides Derived from d-Ribose
To broaden the scope of the method, we studied the reaction of NI-TES with d-ribose and d-galactose derivatives catalyzed by silyl triflates as well. First, d-ribose was silylated and acetylated in a one-pot reaction in the same way as d-arabinose, except that the acetylation was performed at 50 °C, to give a mixture (54:46) of α- and β-triacetate 17 which could be separated by flash chromatography (Scheme 4). Each anomer was reacted with NI-TES in CH3CN catalyzed by 1 equiv of TfOTES. When the reaction mixture was allowed to warm from −20 to −8 °C within 2 h, triacetate β-17 furnished a 72% yield of β-nucleoside β-18 and a trace of the α-nucleoside as detected by TLC. Similarly, α-triacetate yielded 87% of the β- and a trace of the α-nucleoside (20 °C to rt within 2 h). As the two anomers reacted similarly, their separation was not worthwhile. In the latter case, the influence of the temperature on the outcome was studied. Below −20 °C, coupling did not take place; between −10 and −5 °C at a reaction time of 4 h, a mixture of nucleosides resulted (β-18:α-18 = 2:1, combined yield 60%). No nucleoside was formed in 1,2-C2H4Cl2 under a variety of conditions. An alternative for the preparation of azomycin nucleosides derived from d-ribose is the coupling of commercially available acetate 19 (Scheme 5). It was found that TfOTMS was a better catalyst than TfOTES with this substrate. When the Vorbrüggen coupling was effected in CH3CN (−20 to −10 °C, 1.2 equiv of TfOTMS), a 72% yield of nucleoside β-20 resulted. However, when the same reaction was performed for 30 min at rt, a separable mixture (α-20, 42%; β-20, 31%) of nucleosides was obtained. These results underlined the necessity to control the reaction temperature carefully and use CH3CN as the solvent of choice for a good yield. For comparison, Prisbe et al. found that NI and 19 gave a 61% yield of α-20, when treated with 2 equiv of SnCl4 and Hg(CN)2 in CH3CN at 60 °C.(19) To obtain β-20 in 68% yield, acetate 19 had to be first converted to the corresponding bromide by bubbling HBr through a solution of it and then reacting it analogously to before with 2 equiv of Hg(CN)2 without SnCl4. The predominant formation of the β-anomer here is again attributed to the neighboring group effect (2-acyloxy group is α) and the α-anomer to the possible involvement of a nitronate ester.
Scheme 4. Preparation and Coupling of Protected d-Riboses 17.

Scheme 5. Coupling of 1-O-Acetyl-2,3,5-tri-O-benzoyl-α-d-ribofuranose with NI-TES by the Vorbrüggen Method.

Preparation of 2-Nitroimidazole Nucleoside Derived from d-Galactose
At last, commercially available β-d-galactopyranose pentaacetate was converted to the 2-nitroimidazole nucleoside β-22 in high yield (92%) in CH3CN under very mild conditions, when the reaction mixture was stirred for 1 h at 0 °C and 1 h at rt (Scheme 6). For comparison, the coupling of acetobromo-α-d-galactose with 2-nitroimidazole catalyzed by 2.5 equiv of Hg(CN)2 for 23 h at 40 °C in a large volume of CH3CN yielded 88% of the desired β-nucleoside.(21)
Scheme 6. Coupling of 1,2,3,4,6-Penta-O-acetyl-β-d-galactopyranose with NI-TES by the Vorbrüggen Method.

Conversion of 2-Nitroimidazole Nucleosides Derived from α- and β-d-Arabinose and d-Ribose to (Putative) Precursors for Tracers To Image Hypoxia
The precursor α-7 for the preparation of α-[18F]FAZA can be accessed easily from silylated nucleoside α-15. It was deblocked with HF generated in situ from KF/benzoic acid(27) in refluxing CH3CN in 90% yield and tosylated to give precursor α-7(28) in 94% yield, first prepared by Wiebe et al. (Scheme 7). This sequence comprises only four steps and is the shortest synthesis of precursor α-7, starting from d-arabinose.
Scheme 7. Transformation of Azomycin Nucleoside α-15 into Precursor α-7.

The synthesis of β-7 was first accomplished also by Wiebe et al., starting from 1-β-(d-ribofuranosyl)-2-nitroimidazole obtained by debenzoylation of d-ribose derivative β-20.21d,27 The hydroxyl groups at C-5′ and C-3′ were protected with the 3′,5′-O,O-(1,1,3,3-tetraisopropyldisiloxanylidene) group, and the configuration of the free hydroxyl at C-2′ was inverted by substituting the triflate with tetrabutylammonium acetate.(27) Then the disiloxanylidene group was removed, and the diol was monotosylated and acetylated to give β-7. We thought that we could shorten the lengthy synthesis by starting from a β-d-ribose derivative already containing the disiloxanylidene group (Scheme 8). Thus, d-ribose was silylated and acetylated in a one-pot reaction in the same way as the TBDPS-protected analogue 17 was prepared. The crude anomeric mixture containing impurities (47% yield) of α- and β-24 (74:19) was coupled with NI-TES in CH3CN with 1 equiv of TfOTES at 0 °C for 45 min to give separable nucleosides (β-25, 30%; α-25, 32%). Unfortunately, lowering the reaction temperature to −10 °C favored the α-anomer even more (β-25, 16%; α-25, 32%). However, the yield of the desired β-anomer was too low to pursue this approach any further. The putative precursor derived from β-d-ribose was accessed from silyl-protected β-18 (Scheme 9). It was selectively deblocked at C-5′ and tosylated to furnish precursor β-27. Analogously, the α-anomer was prepared from α-18, which was either directly accessible by coupling by the Vorbrüggen method or accessible from tribenzoate α-20 (Scheme 10). Transesterification with MeOH/MeONa gave triol nucleoside α-28 in an admixture with an unknown impurity, possibly of structure 29, already reported(19) by Prisbe et al. The two compounds could not be separated by flash chromatography, but easily after monosilylation with TBDPSCl. The silylated diol α-30 formed was acetylated to give nucleoside α-18. The seemingly obvious direct conversion of α-28 to the precursor α-27 by monotosylation followed by acetylation did not work as tosylation could not be conducted selectively, but produced a ditosylate preferentially.
Scheme 8. Preparation of 3,5-O,O-(1,1,3,3-Tetraisopropyldisiloxanylidene)-Protected d-Riboses 24 and Their Coupling with NI-TES by the Vorbrüggen Method.

Scheme 9. Preparation of Putative Precursors α- and β-27.

Scheme 10. Conversion of Tribenzoate α-20 to the Silylated Diacetate α-18.

Conclusions
Our results unambiguously establish that 1-O-acetyl-d-pentoses and d-galactose pentaacetate and triethylsilylated 2-nitroimidazole can be coupled in acetonitrile using TESOTf (Vorbrüggen method) to give 2-nitroimidazole nucleosides. The anomeric ratio of the products is delicately influenced by the substrate structure and the reaction temperature. This approach is an attractive alternative to protocols based on excess Hg(CN)2 alone or in combination with SnCl4 for coupling. 5′-O-TBDPS nucleosides can be desilylated and tosylated easily to furnish precursors for tracers to image hypoxia.
Experimental Section
1H and 13C (J-modulated; additionally, non-J-modulated spectra were recorded of compounds containing 2-nitroimidazole) NMR spectra were measured at 300 K at 400.13 and 100.61 MHz, respectively. All chemical shifts (δ) are given in parts per million. They were referenced either to residual CHCl3 (δH 7.24)/toluene-d8 (CHD2, δH 2.09)/CD3OD (CHD2, δH 3.31)/DMSO-d6 (CHD2, δH 2.50) or CDCl3 (δC 77.00)/toluene-d8 (CD3, δC 21.04)/CD3OD (CD3, δC 49.00)/DMSO-d6 (CD3, δC 39.50). IR spectra of films on a silicon disk were recorded on an FT-IR spectrometer.(29) Optical rotations were measured at 20 °C on a polarimeter in a 1 dm cell. Melting points are uncorrected.
Flash (column) chromatography was performed with silica gel 60 (230–400 mesh) and monitored by TLC, carried out on 0.25 mm thick plates, silica gel 60 F254. Spots were visualized by UV and/or by dipping the plate into a solution of (NH4)6Mo7O24·4H2O (23.0 g) and Ce(SO4)2·4H2O (1.0 g) in 10% aqueous H2SO4 (500 mL), followed by heating with a heat gun.
General Procedure A: Triethylsilylation of 2-Nitroimidazole
A mixture of 2-nitroimidazole (0.113 g, 1 mmol), hexaethyldisilazane (0.491 g, 2 mmol, 2 equiv), and dry pyridine (2 mL) was refluxed for 30 min. The solution was cooled (silylated compound crystallized), and volatile components were removed by bulb to bulb distillation (70–75 °C, 0.4 mbar) to leave brown crystalline 1-(triethylsilyl)-2-nitroimidazole, which was used immediately without further purification.
General Procedure B: Desilylation of 2-Nitroimidazole Nucleosides with a TBDPSO Group at C-5′
A mixture of the nucleoside (0.50 mmol), KF (0.407 g, 3.50 mmol, 7 equiv), and benzoic acid (0.427 g, 3.50 mmol, 7 equiv) in dry CH3CN (11.5 mL) was heated at 75 °C for 7 h (monitored by TLC).(27) The mixture was then cooled, filtered, and concentrated under reduced pressure.
General Procedure C: Tosylation of 2-Nitroimidazole Nucleosides Derived from d-Pentoses with a Free 5′-OH Group
A solution of p-toluenesulfonyl chloride (0.521 g, 2.73 mmol, 3 equiv) and nucleoside with a free 5′-OH in dry pyridine (5 mL) was kept for 18 h at 4 °C (TLC, hexanes/EtOAc, 1:2). Then water (10 mL) and 2 M HCl (10 mL) were added, and the mixture was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with saturated aq NaHCO3 (10 mL) and water (10 mL), dried (Na2SO4), and concentrated under reduced pressure.
Hexaethyldisilazane (10)
Triethylsilyl triflate (0.2 mL) was added to a stirred solution of (triethylsilyl)amine (8)(24) [9.66 g, 73.6 mmol, 3.2 equiv; 70–80 °C, 80 mmHg; 75% yield; nD20 1.4250 (lit.(24) bp 134 °C, nD 1.4267)] and chlorotriethylsilane (3.47 g, 3.86 mL, 23 mmol) in dry toluene (40 mL) at 0 °C. Precipitation of NH4Cl started immediately. After being stirred for 2 h at rt, the mixture was filtered through Celite. The filtrate was concentrated under reduced pressure, and the residue was bulb to bulb distilled (70–75 °C, 0.5 mbar; lit.(23b) bp 100 °C, 1 mmHg) to give 10 (8.92 g, 79%) as a colorless liquid: nD27 1.4502; 1H NMR (400.1 MHz, toluene-d8) δ 0.98 (t, J = 8.0 Hz, 18H), 0.53 (q, J = 8.0 Hz, 12H), −0.29 (br s, 1H, NH); 13C NMR (100.6 MHz, toluene-d8) δ 8.4, 8.0.
1,2,3-Tri-O-acetyl-5-O-(tert-butyldiphenylsilyl)-α- and 1,2,3-Tri-O-acetyl-5-O-(tert-butyldiphenylsilyl)-β-d-arabinofuranose (α- and β-14)
A mixture of d-arabinose (0.751 g, 5 mmol) in dry pyridine (10 mL) was cooled to −20 °C under argon, and TBDPSCl (t-BuPhe2SiCl) (1.374 g, 1.30 mL, 5 mmol) was added. The reaction mixture was allowed to slowly warm to rt in the cooling bath overnight (TLC, hexanes/EtOAc, 2:1). After addition of Ac2O (4 mL) the solution was stirred at rt for 6 h. Volatile components were removed under reduced pressure and the residue taken up with water (10 mL) and 2 M HCl (10 mL). The mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with saturated aq NaHCO3 (10 mL) and water (10 mL), dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified by flash chromatography (hexanes/EtOAc, 2:1, Rf 0.63) to give the mixture of anomeric acetates (1.83 g, 71%) as an oil,(25) α:β = 60:40 (by 1H NMR), [α]D20 +19.3 (c 3.0, acetone). After a few months crystals of the α-anomer formed in the oil. They were collected after the oil was dissolved in i-Pr2O/hexanes and recrystallized (i-Pr2O/hexanes) to give the homogeneous α-anomer: mp 92–93 °C; [α]D +52.1 (c 1.1, acetone). Data for α- and β-14: IR (Si) νmax 3072, 2933, 2859, 1750, 1428, 1371, 1221, 1113, 1065, 1008 cm–1; 1H NMR (400.1 MHz, CDCl3) δ(α-14) 7.67 (m, 6H), 7.38 (m, 9H), 6.19 (s, 1H), 5.37 (dd, J = 4.8, 1.0 Hz, 1H), 5.20 (d, J = 1.0 Hz, 1H), 4.24 (td, J = 4.8, 4.0 Hz, 1H), 3.84 (AB system, JAB = 11.1 Hz, JAX = JBX = 4.0 Hz, 2H), 2.10 (s, 3H), 2.09 (s, 3H), 2.03 (s, 3H), 1.06 (s, 9H); (β-14) 7.67 (m, 6H), 7.38 (m, 9H), 6.36 (d, J = 4.8 Hz, 1H), 5.62 (dd, J = 7.1, 6.0 Hz, 1H), 5.34 (dd, J = 7.1, 4.8 Hz, 1H), 4.12 (td, J = 6.0, 5.3 Hz, 1H), 3.81 (AB system, JAB = 11.0 Hz, JAX = JBX = 5.3 Hz, 2H), 2.06 (s, 3H), 2.05 (s, 3H), 1.88 (s, 3H), 1.06 (s, 9H); 13C NMR (100.6 MHz, CDCl3) δ (α-14) 169.8, 169.6, 169.3, 135.61 (2C), 135.59 (2C), 133.1, 133.0, 129.7 (2C), 127.7 (4C), 99.6, 84.8, 81.4, 76.7, 62.7, 26.7 (3C), 21.0, 20.7, 20.6, 19.3; (β-14) 169.9, 169.8, 169.4, 135.6 (2C), 135.5 (2C), 133.2, 132.9, 129.8 (2C), 127.7 (4C), 93.6, 81.6, 75.6, 74.1, 64.3, 26.7 (3C), 20.9, 20.8, 20.4, 19.2. Anal. Calcd for C27H34O8Si: C, 63.01; H, 6.66. Found for crystalline α-14: C, 62.36; H, 6.84.
1-α-[2′,3′-Di-O-acetyl-5′-O-(tert-butyldiphenylsilyl)-d-arabinofuranosyl]-2-nitroimidazole (α-15)
A solution of α- and β-14 (0.548 g, 1.06 mmol) in dry CH3CN (3 mL) was added to 2-nitroimidazole (0.113 g, 1 mmol) triethylsilylated according to general procedure A under argon in dry CH3CN (5 mL). After the stirred mixture was cooled to −20 °C, TfOTES (1 mL, 1 M in dry 1,2-C2H4Cl2) was added, and stirring was continued for 3 h at −8 °C (TLC, hexanes/EtOAc, 2:1). A saturated aq solution of NaHCO3 (10 mL) was added, and the mixture was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with water (10 mL), dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by flash chromatography (hexanes/EtOAc, 2:1, Rf 0.21) to give protected nucleoside α-15 (0.435 g, 72%) as a yellow oil (lit.(25) mp 53 °C): [α]D20 +21.3 (c 1.1, acetone); the 1H NMR data agree with those of the literature;(25) IR (Si) νmax 2932, 2859, 1755, 1540, 1475, 1428, 1369, 1230, 1106 cm–1; 13C NMR (100.6 MHz, CDCl3) δ 169.2, 168.9, 144.2, 135.64 (2C), 135.56 (2C), 132.8, 132.7, 130.0 (2C), 128.5, 127.8 (2C), 127.5 (2C), 121.9, 92.6, 87.4, 81.8, 76.8, 63.6, 26.7 (3C), 20.6, 20.4, 19.2.
Preparation of α- and β-15 in 1,2-C2H4Cl2
A solution α- and β-14 (0.680 g, 1.32 mmol) in dry 1,2-C2H4Cl2 (4 mL) was added under argon at rt to 2-nitroimidazole (0.150 g, 1.32 mmol) trimethylsilylated according to general procedure A, followed by TfOTMS (1.32 mL, 0.5 M in dry 1,2-C2H4Cl2). After the mixture was stirred for 4 h at rt, CH2Cl2 (20 mL) and a saturated aq solution of NaHCO3 (20 mL) were added. The organic phase was separated, and the aqueous one was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (hexanes/EtOAc, 3:1 and then 1:2; TLC, 2:1, Rf 0.21 for α-15 and 0.43 for β-15) to give recovered starting material (0.306 g, 45%), β-15 (0.098 g, 13%), and α-15 (0.122 g, 16%).
1,2,3-Tri-O-acetyl-5-O-(tert-butyldiphenylsilyl)-α- and Tri-O-acetyl-5-O-(tert-butyldiphenylsilyl)-β-d-ribofuranose (α- and β-17)
d-Ribose (0.751 g, 5 mmol) was silylated, acetylated, and worked up in the same way as d-arabinose, except that the acetylation was performed for 6 h at 60 °C. The crude product (α:β = 1:0.9) was purified by flash chromatography (hexanes/EtOAc, 3:1, Rf 0.73 for β-anomer and 0.58 for α-anomer, hexanes/EtOAc, 2:1) to give β-triacetate β-17 (0.720 g, 28%; 10–15% of it was unknown compounds) and α-triacetate α-17 (0.849 g, 33%; 6% of it was an impurity) as colorless, very viscous oils. Data for β-17: [α]D20 +2.17 (c 1.15, acetone); IR (Si) νmax 3050, 2933, 2859, 1 754, 1429, 1371, 1221, 1113, 1029 cm–1; 1H NMR (400.1 MHz, CDCl3) δ 7.67–7.63 (m, 4H), 7.41–7.35 (m, 6H), 6.17 (d, J = 1.5 Hz, 1H), 5.55 (dd, J = 6.3, 4.8 Hz, 1H), 5.43 (dd, J = 4.8, 1.5 Hz, 1H), 4.24 (dt, J = 6.3, 3.5 Hz, 1H), 3.76 (AB system, JAX = JBX = 3.5 Hz, JAB = 11.3 Hz, 2H), 2.10 (s, 3H), 2.03 (s, 3H), 1.93 (s, 3H), 1.05 (s, 9H); 13C NMR (100.6 MHz, CDCl3) δ 169.7, 169.48, 169.46, 135.60 (2C), 135.5 (2C), 133.0, 132.80, 129.84, 129.8, 127.8 (2C), 127.7 (2C), 98.2, 82.3, 74.5, 70.6, 63.2, 26.7 (3C), 20.9, 20.5, 20.5, 19.2. Anal. Calcd for C27H34O8Si: C, 63.01; H, 6.66. Found: C, 63.29; H, 6.78. Data for α-17: [α]D +46.3 (c 1.2, acetone); IR (Si) νmax 3072, 2933, 2859, 1749, 1472, 1428, 1370, 1222, 1113, 1074, 1051, 1011 cm–1; 1H NMR (400.1 MHz, CDCl3) δ 7.68–7.63 (m, 4H), 7.45–7.35 (m, 6H), 6.46 (d, J = 4.5 Hz, 1H), 5.50 (dd, J = 6.3, 2.0 Hz, 1H), 5.41 (dd, J = 6.3, 4.5 Hz, 1H), 4.29 (∼q, J ≈ 2.5 Hz, 1H), 3.79 (AB system, J = 11.5, 3.0, 2.3 Hz, 2H), 2.11 (s, 3H), 2.10 (s, 3H), 2.07 (s, 3H), 1.05 (s, 9H); 13C NMR 100.6 MHz, CDCl3) δ 170.2, 169.9, 169.3, 135.6 (2C), 135.6 (2C), 132.8, 132.6, 130.3, 130.2, 127.8 (2C), 127.8 (2C), 94.5, 85.0, 70.6, 70.5, 63.4, 26.7 (3C), 21.1, 20.7, 20.3, 19.2.
1-β-[2′,3′-Di-O-acetyl-5′-O-(tert-butyldiphenylsilyl)-d-ribofuranosyl]-2-nitroimidazole (β-18)
A solution of β-17 (0.395 g, 0.77 mmol) in dry CH3CN (3 mL) was added to 2-nitroimidazole (0.087 g, 0.77 mmol) triethylsilylated according to general procedure A in dry CH3CN (2 mL) under argon. After the stirred mixture was cooled to −20 °C, TESOTf (0.77 mL, 1 M in dry 1,2-C2H4Cl2) was added, and stirring was continued for 2 h while the temperature of the reaction mixture was allowed to slowly rise to −8 °C in the cooling bath (TLC, hexanes/EtOAc, 2:1). Saturated aq NaHCO3 (10 mL) was added, and the mixture was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with water (10 mL), dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by flash chromatography (hexanes/EtOAc, 3:1, Rf 0.47 for 2:1) to give nucleoside β-18(18) (0.315 g, 72%) as yellow crystals; a trace of possibly α-anomer was detected by TLC: [α]D20 +13.31 (c 1.3, acetone); mp 130–131 °C (CHCl3/i-Pr2O) (lit.(18) mp 52–53 °C).
Similarly, triacetate α-17 (0.395 g, 0.77 mmol) was reacted in the same way, except that the temperature was allowed to warm to rt within 2 h, and yielded (0.381 g, 87%) β-18; a trace of possibly α-18 could be detected in the crude product by TLC: IR (Si) νmax 2932, 1755, 1589, 1542, 1477, 1428, 1369, 1238, 1114, 1081, 1011 cm–1; the 1H and 13C NMR data agree with those of the literature.(18) Anal. Calcd for C28H33N3O8Si: C, 59.24; H, 5.86; N, 7.40. Found: C, 58.98; H, 5.56; N, 7.14.
1-β- and 1-α-(2′,3′,5-Tri-O-benzoyl-d-ribofuranosyl)-2-nitroimidazole (β- and α-20)
A solution of 1-O-acetyl-2,3,5-tri-O-benzoyl-β-d-ribofuranose (0.605 g, 1.2 mmol, 1.2 equiv) in dry CH3CN (4.6 mL) was added to the stirred mixture of 2-nitroimidazole (0.113 g, 1.0 mmol; triethylsilylated according to general procedure A) in dry CH3CN (5.0 mL) under argon. TMSOTf (0.5 M, 2.4 mL, in dry CH2Cl2) was added, and stirring was continued at rt (23 °C) for 1 h (TLC, hexanes/EtOAc, 2:1). A saturated aq solution of NaHCO3 (10 mL) was added, and the mixture was extracted with EtOAc (3 × 10 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (hexanes/EtOAc, 2:1; β-20, Rf 0.38; α-20, Rf 0.26) to yield β-20 (0.173 g, 31%) as a yellow oil and α-20 (0.235 g, 42%) as yellow crystals: mp 136–138 °C (CHCl3/i-Pr2O) [lit.(19) mp 138.5–140 °C (CH2Cl2/MeOH)]. Data for β-20: [α]D20 +27.47 (c 0.91, acetone); IR (Si) νmax 1733, 1541, 1479, 1452, 1366, 1268, 1118, 1094, 1071 cm–1; 1H NMR (400.1 MHz, CDCl3) δ 8.09–8.05 (m, 2H), 7.99–7.95 (m, 2H), δ 7.91–7.87 (m, 2H), 7.64–7.51 (m, 3H), 7.57 (d, J = 1.3 Hz, 1H), 7.50–7.44 (m, 2H), 7.43–7.38 (m, 2H), 7.37–7.32 (m, 2H), 7.12 (d, J = 1.3 Hz, 1H), 6.97 (d, J = 4.0 Hz, 1H), 5.86 (t, J = 5.6 Hz, 1H), 5.82 (dd, J = 5.6, 4.0 Hz, 1H), 4.86 (dd, J = 12.4, 2.7 Hz, 1H), 4.83 (∼td, J = 5.6, 3.5, 2.7 Hz, 1H), 4.70 (dd, J = 12.4, 3.5 Hz, 1H); 13C NMR (100.6 MHz, CDCl3) δ 166.0, 165.2, 164.8, 144.4 (br), 133.9, 133.9, 133.7, 129.9 (2C), 129.8 (2C), 129.7 (2C), 129.1, 129.1, 128.8 (2C), 128.6 (2C), 128.6 (2C), 128.3, 128.3, 121.5, 89.6, 80.7, 76.1, 70.2, 63.0. Data for α-20: [α]D −56.8 (c 1.07, acetone); IR (Si) νmax 1732, 1539, 1479, 1452, 1365, 1267, 1095, 1070, 1026 cm–1; 1H NMR (400.1 MHz, CDCl3) δ 8.14–8.09 (m, 2H), 7.71–7.67 (m, 2H), 7.64–7.56 (m, 3H), 7.54–7.46 (m, 5H), 7.34–7.27 (m, 4H), 7.23 (d, J = 1.3 Hz, 1H), 7.16 (d, J = 5.6 Hz, 1H), 6.30 (t, J = 5.6 Hz, 1H), 5.92 (dd, J = 5.6, 3.2 Hz, 1H), 5.06 (ddd ≈ q, J = 3.4 Hz, 1H), 4.79 (dd, J = 12.4, 3.4 Hz, 1H), 4.63 (dd, J = 12.4, 3.4 Hz, 1H); 13C NMR (100.6 MHz, CDCl3) δ 165.9, 165.0, 164.0, 144.6, 133.9, 133.8, 133.6, 129.7 (2C), 129.6 (2C), 129.4 (2C), 129.0, 128.7 (2C), 128.6 (2C), 128.5 (2C), 128.2, 128.2, 127.7, 122.5, 88.1, 82.7, 71.8, 71.3, 63.9.
1-β-(2′,3′,4′,6′-Tetra-O-acetyl-d-galactopyranosyl)-2-nitroimidazole (β-22)
A solution of 1,2,3,4,6-penta-O-acetyl-β-d-galactopyranose (1.171 g, 3 mmol) in dry CH3CN (20 mL) was added to the mixture of 2-nitroimidazole (0.339 g, 3.0 mmol; triethylsilylated according to general procedure A) in dry CH3CN (4 mL, sonicated for 2 min) under argon. After the stirred mixture was cooled to 0 °C, TfOTES (3.6 mL, 1.2 equiv, 1 M in dry 1,2-C2H4Cl2) was added, and stirring was continued for 1 h at 0 °C and 1 h at rt. A saturated aq solution of NaHCO3 (20 mL) was added ,and the mixture was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with water (20 mL), dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by flash column chromatography (hexanes/EtOAc, 2:1; TLC, hexanes/EtOAc, 1:1, Rf 0.45) to give nucleoside β-22(21a) (1.219 g, 92%) as a gum: [α]D20 +62.50 (c 1.48, acetone); IR (Si) νmax 1753, 1370, 1231,1094, 1064 cm–1; 1H NMR (400.1 MHz, CDCl3) δ 7.41 (d, J = 1.0 Hz, 1H), 7.16 (d, J = 1.0 Hz, 1H), 6.38 (d, J = 9.1 Hz, 1H), 5.51 (dd, J = 3.3, 1.0 Hz, 1H), 5.44 (dd, J = 10.1, 9.1 Hz, 1H), 5.33 (dd, J = 10.1, 3.3 Hz, 1H), 4.25 (ddd, J = 7.1, 5.8, 1.0 Hz, 1H), 4.14 (AB system, J = 11.4, 7.1, 5.8 Hz, 2H), 2.17 (s, 3H), 2.01 (s, 3H), 1.96 (s, 3H), 1.88 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ 170.3, 169.8, 169.6, 169.1, 144.5, 128.9, 122.1, 83.5, 74.2, 70.9, 68.7, 67.0, 61.1, 20.5 (2C), 20.4, 20.2.
1-α-(2′,3′-Di-O-acetyl-d-arabinofuranosyl)-2-nitroimidazole (α-23)
Nucleoside α-15 (0.231 g, 0.407 mmol) was desilylated by general procedure B. The crude product was purified by flash chromatography (at first hexanes/EtOAc, 1:1, Rf 0.08, and then 1:5) to give α-23 (0.113 g, 84%) as a crystalline solid: mp 115–116 °C (C2H4Cl2/i-Pr2O) (lit.(26) semisolid mass); [α]D20 −5.6 (c 1.1, acetone); IR (Si) νmax 3387, 2939, 1748, 1539, 1479, 1371, 1234, 1161, 1063 cm–1; 1H and 13C NMR data agree with those of the literature.(25)
1-α-[2′,3′-Di-O-acetyl-5′-O-(4-toluenesulfonyl)-d-arabinofuranosyl]-2-nitroimidazole (α-7)
α-23 (0.300 g, 0.91 mmol) was tosylated by general procedure C. The crude product was purified by flash chromatography (hexanes/EtOAc, 1:1, Rf 0.26) to give tosylate α-7 (0.420 g, 95%) as colorless plates: mp 102–103 °C (CHCl3/i-Pr2O); [α]D20 +39.3 (c 0.9, acetone);(28) IR (Si) νmax 1750, 1540, 1477, 1368, 1232, 1191, 1178, 1096, 1052 cm–1; the 1H and 13C NMR data agree with those of the literature, except that a signal in the 13C NMR spectrum at 20.4 is replaced by one at 17.3.(28)
1-β- and 1-α-[1,2-Di-O-acetyl-3,5-O,O-(1,1,3,3-tetraisopropyldisiloxanylidene)-d-ribofuranose (β- and α-24)
d-Ribose (0.45 g, 3 mmol) was dissolved in dry pyridine (15 mL) under argon and cooled to −35 °C. 1,3-Dichloro-1,1,3,3-tetraisopropyldisiloxane (0.946, 0.96 mL) was added, and the mixture was stirred and allowed to slowly warm in the cooling bath to rt (18 h). Ac2O (1.5 mL) was added, and stirring was continued for another 6 h, before water (2 mL) was added. After 10 min the mixture was concentrated under reduced pressure. The residue was treated with water (20 mL) and EtOAc (20 mL). The organic phase was separated, and the aq one was extraceted with EtOAc (15 mL). The combined organic layers were washed (20 mL each) with water, 2 N HCl, water, and a saturated aq solution of NaHCO3, dried (Na2SO4), and concetrated under reduced pressure. The residue was purified by flash column chromatography (hexanes/EtOAc, 10:1; TLC, Rf 0.31 for 7:1) to yield a seemingly homogeneous oily product [0.670 g, 47%; by 1H NMR a mixture of α-anomer (δ 5.93, J = 7.9 Hz):β-anomer (δ 6.03, J = 0 Hz):unknown compound (δ 6.09, J = 4.1 Hz) = 74:19:7], [α]D20 −35.81 (c 1.55, acetone), in which gradually crystals of the α-anomer formed. Hexanes were added, and the mixture was stored at −18 °C. The colorless needles were collected and recrystallized (hexanes, −18 °C). Data for α-24: mp 69–70 °C (hexanes, −18 °C); [α]D −39.75 (c 1.2, acetone); IR (Si) νmax 2946, 2868, 1747, 1464, 1373, 1216, 1169, 1125, 1078, 1010 cm–1; 1H NMR (400.1 MHz, CDCl3) δ 5.93 (d, J = 7.9 Hz, 1H), 4.80 (dd, J = 7.9, 2.8 Hz, 1H), 4.57 (dd ≈ t, J = 2.8, 2.3 Hz, 1H), 4.10 (ddd, J = 9.6, 5.2, 2.3 Hz, 1H), 3.79 (AB part of ABX system, J = 11.0, 9.6, 5.2 Hz, 2H), 2.07 (s, 3H), 2.06 (s, 3H), 1.09–0.82 (m, 28H); 13C NMR (100.6 MHz, CDCl3) δ 169.9, 169.4, 90.3, 71.1, 70.5, 70.5, 64.6, 21.0, 20.9, 17.4 (2C), 17.2 (2C), 17.2 (2C), 17.14, 17.09, 14.0, 13.6, 13.0, 12.6. Anal. Calcd for C21H40O8Si2: C, 52.91; H, 8.46. Found: C, 52.64; H, 8.38.
1-β- and 1-α-[2′-O-Acetyl-3′,5′-O,O-(1,1,3,3-tetraisopropyldisiloxanylidene)-d-ribofuranosyl]-2-nitroimidazole (β- and α-25)
A solution of α- and β-24 (0.634 g, 1.33 mmol) in dry CH3CN (5 mL) was added to 2-nitroimidazole (0.107 g, 0.95 mmol) triethylsilylated according to general procedure A in dry CH3CN (2 mL) under argon. After the stirred mixture was cooled to 0 °C, TfOTES (1.14 mL, 1 M solution in dry 1,2-C2H4Cl2) was added dropwise, and stirring was continued for 45 min at that temperature. Then a saturated aq solution of NaHCO3 (10 mL) was added, and the mixture was extracted with EtOAc (3 × 15 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The residue was flash chromatographed (hexanes/EtOAc, 5:1; β-25, Rf 0.36; α-25, Rf 0.21) to yield nucleosides β-25 (0.152 g, 30%) and α-25 (0.158 g, 32%), both colorless needles. When the reaction mixture was allowed to warm from −10 °C to rt, the β- and α-anomers were formed in yields of 16% and 32%, respectively. Data for β-25: mp 143–144 °C (i-Pr2O/hexanes); [α]D20 +4.06 (c 0.91, acetone); IR (Si) νmax 2946, 2868, 1764, 1543, 1474, 1370, 1231, 1116, 1073, 1040 cm–1; 1H NMR (400.1 MHz, CDCl3) δ 7.77 (d, J = 1.0 Hz, 1H), 7.14 (d, J = 1.0 Hz, 1H), 6.34 (s, 1H), 5.46 (d, J = 4.8 Hz, 1H), 4.43 (dd, J = 9.4, 4.8 Hz, 1H), 4.28 (d, J = 13.6 Hz, 1H), 4.10 (dd, J = 9.4, 2.5 Hz, 1H), 4.00 (dd, J = 13.6, 2.5 Hz, 1H), 2.17 (s, 3H), 1.11–0.83 (m, 28H); 13C NMR (100.6 MHz, CDCl3) δ 168.5, 143.7, 128.5, 122.3, 91.0, 82.6, 72.3, 66.8, 59.3, 20.5, 17.4, 17.3, 17.2, 17.2, 16.9, 16.8, 16.70, 16.66, 13.4, 12.9, 12.8, 12.6. Anal. Calcd for C22H39N3O8Si2: C, 49.88; H, 7.42; N, 7.93. Found: C, 49.48; H, 7.61; N, 7.83. Data for α-25: mp 106–107 °C (i-Pr2O/hexanes); [α]D +43.78 (c 0.95, acetone); IR (Si) νmax 2946, 2868, 1748, 1545, 1484, 1374, 1225, 1169, 1117, 1053, 1033, 1009 cm–1; 1H NMR (400.1 MHz, CDCl3) δ 7.26 (d, J = 1.3 Hz, 1H), 7.11 (d, J = 1.3 Hz, 1H), 6.70 (d, J = 9.1 Hz, 1H), 4.86 (dd, J = 9.1, 2.3 Hz, 1H), 4.65 (t, J = 2.3 Hz, 1H), 4.22 (td, J = 8.3, 2.3 Hz, 1H), 3.92 (d, J = 8.3 Hz, 2H), 1.88 (s, 3H), 1.15–0.82 (m, 28H); 13C NMR (100.6 MHz, CDCl3) δ 169.5, 144.9, 128.7, 121.3, 80.7, 72.4, 71.2, 70.6, 66.5, 20.4, 17.41, 17.37, 17.22, 17.16, 17.1 (2C), 17.06, 17.02, 14.1, 13.7, 12.9, 12.8. Anal. Calcd for C22H39N3O8Si2: C, 49.88; H, 7.42; N, 7.93. Found: C, 49.74; H, 7.40; N, 7.88.
1-β-[2′,3′-Di-O-acetyl-d-ribofuranosyl]-2-nitroimidazole (β-26)
Nucleoside β-18 (0.381 g, 0.67 mmol) was desilylated by general procedure B. The crude product was purified by flash chromatography (starting with hexanes/EtOAc, 1:1, Rf 0.18, and then hexanes/EtOAc, 1:5) to give alcohol β-26(18) (0.158 g, 71%) as yellow crystals: mp 125–126 °C (1,2-C2H4Cl2/i-Pr2O) (lit.(18) mp 122–123 °C); [α]D20 +29.82 (c 1.1, acetone); IR (Si) νmax 3374, 3121, 2937, 1750, 1541, 1483, 1430, 1367, 1243, 1161, 1101, 1079, 1013 cm–1; the 1H and 13C NMR data are identical to those of the literature.(18) Anal. Calcd for C12H15N3O8: C, 43.77; H, 4.59; N, 12.76. Found: C, 43.86; H, 4.42; N, 12.73.
1-β-[2′,3′-Di-O-acetyl-5′-O-(4-toluenesulfonyl)-d-ribofuranosyl]-2-nitroimidazole (β-27)
A solution of 4-toluenesulfonyl chloride (0.173 g, 0.91 mmol, 3 equiv) and alcohol β-26 (0.100 g, 0.304 mmol) in dry pyridine (2 mL) was kept for 18 h at 4 °C. When the starting alcohol was consumed (TLC, hexanes/EtOAc, 1:1), water (10 mL) and 2 M HCl (10 mL) were added. The mixture was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with saturated aq NaHCO3 (10 mL) and water (10 mL), dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by flash chromatography (hexanes/EtOAc, 1:1, Rf = 0.41) to give tosylate β-27 (0.102 g, 69%) as a yellow foam: [α]D20 +44.2 (c 1.0, acetone); IR (Si) νmax 1755, 1542, 1478, 1368, 1239, 1191, 1178, 1096 cm–1; 1H NMR (400.1 MHz, CDCl3) δ 7.79 (m, 2H), 7.49 (d, J = 1.1 Hz, 1H), 7.36 (m, 2H), 7.12 (d, J = 1.1 Hz, 1H), 6.59 (d, J = 3.8 Hz, 1H), 5.38 (dd, J = 5.6, 3.8 Hz, 1H), 5.23 (dd ≈ t, J = 6.1, 5.6 Hz, 1H), 4.44 (dd, J = 11.4, 2.4 Hz, 1H), 4.38 (dt, J = 6.1, 2.3 Hz, 1H), 4.22 (dd, J = 11.4, 2.3 Hz, 1H), 2.44 (s, 3H), 2.10 (s, 3H), 2.03 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ 169.2, 168.9, 145.7, 144.2, 132.0, 130.2 (2C), 129.1, 128.0 (2C), 121.7, 89.4, 79.6, 74.8, 68.7, 66.5, 21.7, 20.3, 20.2. Anal. Calcd for C19H21N3O10S: C, 47.20; H, 4.38; N, 8.69. Found: C, 47.48; H, 4.48; N, 8.48.
1-α-[5′-O-(tert-Butyldiphenylsilyl)-d-ribofuranosyl]-2-nitroimidazole (α-30)
A mixture of tribenzoate α-20 (0.398 g, 0.714 mmol) and MeONa/MeOH (3.0 mL, 0.05 M) was stirred at 0 °C. When the transesterification (TLC, hexanes/EtOAc, 1:2) was finished (2 h), AcOH (3 drops) was added immediately, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography (CHCl3/MeOH, 4:1, Rf 0.39) to yield an inseparable mixture of α-28 and a side product (10–20%) with the tentatively assigned structure 29 (0.132 g): 1H NMR (400.1 MHz, CD3OD) δ (α-28) 7.62 (s, 1H), 7.13 (s, 1H), 6.71 (d, J = 4.8 Hz, 1H), 4.61 (dd ≈ t, J = 5.3, 4.8 Hz, 1H), 4.31 (m, 1H), 3.84 (dd, J = 12.2, 2.8 Hz, 1H), 3.67 (dd, J = 12.2, 4.2 Hz, 1H); (29) 6.83 (s, 1H), 6.62 (s, 1H), 6.25 (d, J = 5.3 Hz, 1H), 5.56 (t, J = 5.3 Hz, 1H). TBDPSCl (0.412 g, 0.39 mL, 1.50 mmol) was added to a solution of the mixture of α-28 and 29 (0.132 g) in dry pyridine (2.5 mL), and the mixture was stirred at rt until the starting material was consumed (2 h; TLC, CHCl3/MeOH, 4:1, for starting material, hexanes/EtOAc, 1:1, for product α-30). The reaction was quenched with water (10 mL) and 2 M HCl (10 mL), and the mixture was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with saturated aq NaHCO3 (10 mL) and water (10 mL), dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified by flash chromatography (hexanes/EtOAc, 1:1, Rf 0.38) to give α-30 (0.179 g, 52%, starting from tribenzoate α-20): mp 150–151 °C (EtOAc); [α]D20 −33.6 (c 1.22, EtOH); IR (ATR) νmax 3413, 3200, 2927, 1537, 1354, 1487, 1354, 1111, 1090, 1048, 1036 cm–1; 1H NMR (400.1 MHz, DMSO-d6) δ 7.68–7.62 (m, 4H), 7.59 (s, 1H), 7.51–7.40 (m, 6H), 7.17 (s, 1H), 6.62 (d, J = 4.8 Hz, 1H), 5.49 (d, J = 5.8 Hz, 1H), 5.03 (d, J = 5.1 Hz, 1H), 4.55–4.49 (m, 1H), 4.34 (br s, 1H), 4.20–4.15 (m, 1H), 3.82 (AB system, J = 11.5, 3.7, 1.7 Hz, 2H), 1.03 (s, 9H); 13C NMR (100.61, DMSO-d6) δ 144.5, 135.1 (4C), 132.8, 132.7, 129.9 (2C), 128.0 (2C), 127.9 (2C), 127.1, 124.3, 89.9, 84.9, 71.0, 70.6, 63.8, 26.6 (3C), 18.8. Anal. Calcd for C24H29N3O6Si: C, 59.61; H, 6.04; N, 8.69. Found: C, 59.62; H, 5.98; N, 8.68.
1-α-[2′,3′-Di-O-acetyl-5′-O-(tert-butyldiphenylsilyl)-d-ribofuranosyl]-2-nitroimidazole (α-18)
Ac2O (0.3 mL) was added to a solution of diol α-30 (0.070 g, 0.145 mmol) in dry pyridine (0.3 mL) and dry CHCl3 (2 mL). The mixture was heated and stirred for 30 min at 50 °C (TLC, hexanes/EtOAc, 1:1). After cooling, water (5 mL) and 2 M HCl (5 mL) were added. The resulting mixture was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with saturated aq NaHCO3 (10 mL) and water (10 mL), dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified by flash chromatography (hexanes/EtOAc, 2:1, Rf 0.56 for hexanes/EtOAc, 1:1) to give diacetate α-18 (0.074 g, 90%): mp 134–135 °C (CHCl3/i-Pr2O); [α]D20 −41.49 (c 0.4, acetone); IR (Si) νmax 2932, 2859, 1753, 1539, 1478, 1367, 1233, 1105 cm–1; 1H NMR (400.1 MHz, CDCl3) δ 7.70–7.65 (m, 4H), 7.46–7.36 (m, 7H), 7.16 (d, J = 1.3 Hz, 1H), 6.88 (d, J = 5.8 Hz, 1H), 6.05 (dd ≈ t, J = 5.7 Hz, 1H), 5.53 (dd, J = 5.3, 1.8 Hz, 1H), 4.54 (ddd ≈ q, J = 2.1 Hz, 1H), 3.84 (AB system, J = 11.6, 2.6, 2.3 Hz, 2H), 1.89 (s, 3H), 1.84 (s, 3H), 1.10 (s, 9H); 13C NMR (100.6 MHz, CDCl3) δ 169.3, 168.2, 144.7, 135.5 (2C), 135.5 (2C), 132.2 (2C), 130.1, 130.0, 127.91 (2C), 127.90 (2C), 127.5, 122.4, 87.5, 85.6, 71.8, 71.1, 63.9, 26.7 (3C), 20.3, 19.7, 19.0. Anal. Calcd for C28H33N3O8Si: C, 59.24; H, 5.86; N, 7.40. Found: C, 59.33; H, 5.59; N, 7.42.
1-α-(2′,3′-Di-O-acetyl-d-ribofuranosyl)-2-nitroimidazole (α-26)
Nucleoside α-18 (0.194 g, 0.342 mmol) was desilylated by general procedure B. The residue was purified by flash chromatography (hexanes/EtOAc, 1:1, Rf 0.10) to give α-26 (0.102 g, 91%) as an oil: [α]D20 −47.54 (c 1.3, acetone); IR (Si) νmax 3385, 1751, 1541, 1482, 1367, 1240, 1161, 1104 cm–1; 1H NMR (400.1 MHz, CDCl3) δ 7.39 (d, J = 1.1 Hz, 1H), 7.17 (d, J = 1.1 Hz, 1H), 6.90 (d, J = 5.5 Hz, 1H), 5.87 (t, J = 5.5 Hz, 1H), 5.26 (dd, J = 5.5, 2.8 Hz, 1H), 4.56 (q, J = 2.8 Hz, 1H), 3.87 (AB system, J = 12.1, 2 × 2.8 Hz, 2H), 2.55 (br s, 1H), 1.92 (s, 3H), 1.83 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ 169.6, 168.5, 144.6, 127.6, 122.5, 87.6, 85.2, 71.2, 71.0, 62.0, 20.3, 19.8. Anal. Calcd for C12H15N3O8: C, 43.77; H, 4.59; N, 12.76. Found: C, 43.43; H, 4.26; N, 12.17.
1-α-[2′,3′-Di-O-acetyl-5′-O-(4-toluenesulfonyl)-d-ribofuranosyl]-2-nitroimidazole (α-27)
4-Toluenesulfonyl chloride (0.136 g, 0.693 mmol, 3 equiv) was dissolved in a solution of alcohol α-26 (0.076 g, 0.231 mmol) in dry pyridine (1.26 mL). The reaction mixture was kept for 18 h at 4 °C (no starting material; TLC, hexanes/EtOAc, 1:2) and then quenched by adding water (10 mL) and 2 M HCl (10 mL). The mixture was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with saturated aq NaHCO3 (10 mL) and water (10 mL), dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by flash chromatography (hexanes/EtOAc, 1:1, Rf 0.31 for hexanes/EtOAc, 1:2) to give tosylate α-27 (0.084 g, 75%): mp 119–120 °C (CHCl3/i-Pr2O); [α]D20 −18.87 (c 0.8, acetone); IR (Si) νmax 1756, 1539 1480, 1367, 1234, 1178 cm–1; 1H NMR (400.1 MHz, CDCl3) δ 7.81 (d, J = 8.3 Hz, 2H), 7.35 (d, J = 8.3 Hz, 2H), 7.29 (s, 1H), 7.14 (s, 1H), 6.62 (d, J = 5.3 Hz, 1H), 5.68 (t, J = 5.3 Hz, 1H), 5.28 (dd, J = 5.3, 3.7 Hz, 1H), 4.62 (ddd ≈ q, J = 3.5 Hz, 1H), 4.30 (AB system, J = 11.6, 3.5, 3.0 Hz, 2H), 2.42 (s, 3H), 1.90 (s, 3H), 1.82 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ 169.1, 168.0, 145.7, 144.5, 132.3, 130.1 (2C), 127.9 (2C), 127.8, 122.2, 87.5, 81.6, 70.5, 70.4, 68.0, 21.6, 20.2, 19.7. Anal. Calcd. for C19H21N3O10S: C, 47.20; H, 4.38; N, 8.69. Found: C, 46.93; H, 4.14; N, 8.50.
Acknowledgments
This work was supported by Grant L420-N19 from Der Wissenschaftsfonds (FWF). We are very much indebted to Prof. Dr. H.-J. Machulla for bringing this topic to our attention. We thank S. Felsinger for recording the NMR spectra.
Supporting Information Available
NMR spectra of selected compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Fyles A. W.; Milosevic M.; Wong R.; Kavanagh M. C.; Pintilie M.; Sun A.; Chapman W.; Levin W.; Manchul L.; Keane T. J.; Hill R. P. Radiother. Oncol. 1998, 48, 149–156. [DOI] [PubMed] [Google Scholar]
- Nordsmark M.; Hoyer M.; Keller J.; Nielsen O. S.; Jensen O. M.; Overgaard J. Int. J. Radiat. Oncol. Biol. Phys. 1996, 35, 701–708. [DOI] [PubMed] [Google Scholar]
- Brizel D. M.; Dodge R. K.; Clough R. W.; Dewhirst M. W. Radiother. Oncol. 1999, 53, 113–117. [DOI] [PubMed] [Google Scholar]
- Teicher B. A. Crit. Rev. Oncol. Hematol. 1995, 20, 9–39. [DOI] [PubMed] [Google Scholar]
- Harrison L. B.; Chandha M.; Hill R. J.; Hu K.; Shasha D. Oncologist 2002, 7, 492–508. [DOI] [PubMed] [Google Scholar]
- Raleigh J. A.; Dewhirst M. W.; Thrall D. E. Semin. Radiat. Oncol. 1996, 6, 37–45. [DOI] [PubMed] [Google Scholar]
- a Chapman J. D. N. Engl. J. Med. 1979, 301, 1429–1432. [DOI] [PubMed] [Google Scholar]; b Review:Hodgkiss R. J. Anti- Cancer Drug Des. 1998, 13, 687–702. [PubMed] [Google Scholar]
- Machulla H.-J., Ed. Imaging of Hypoxia—Tracer Developments. Kluwer Academic Publishers: Dordrecht, The Netherlands, 1999. [Google Scholar]
- Ballinger J. R. Semin. Nucl. Med. 2001, 31, 321–329. [DOI] [PubMed] [Google Scholar]
- Urtasun R. C.; Parliament M. B.; McEwan A. J.; Mercer J. R.; Mannan R. H.; Wiebe L. I.; Morin C.; Chapman J. D. Br. J. Cancer 1996, 27 (Suppl. 74), S209–S212. [PMC free article] [PubMed] [Google Scholar]
- Rasey J. S.; Koh W. J.; Evans M. L.; Peterson L. M.; Lewellen T. K.; Graham M. M.; Krohn K. A. Int. J. Radiat. Oncol. Biol. Phys. 1996, 36, 417–428. [DOI] [PubMed] [Google Scholar]
- Eschmann S.-M.; Paulsen F.; Reimold M.; Dittmann H.; Welz S.; Reischl G.; Machulla H.- J.; Bares R. J. Nucl. Med. 2005, 46, 253–260. [PubMed] [Google Scholar]
- Lehtio K.; Oikonen V.; Gronroos T.; Eskola O.; Kalliokoski K.; Bergman J.; Solin O.; Grenman R.; Nuutila P.; Minn H. J. Nucl. Med. 2001, 42, 1643–1652. [PubMed] [Google Scholar]
- a Dehdashti F.; Mintun M. A.; Lewis J. S.; Bradley J.; Govindan R.; Laforest R.; Welch M. J.; Siegel B. A. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 844–850. [DOI] [PubMed] [Google Scholar]; b Chao K. S.; Bosch W. R.; Mutic S.; Lewis J. S.; Dehdashti F.; Mintun M. A.; Dempsey J. F.; Perez C. A.; Purdy J. A.; Welch M. J. Int. J. Radiat. Oncol. Biol. Phys. 2001, 49, 1171–1182. [DOI] [PubMed] [Google Scholar]
- a Piert M.; Machulla H.-J.; Reichel G.; Ziegler S.; Kumar P.; Schwaiger M.; Wiebe L. I. J. Nucl. Med. 2002, 43 (Suppl), 278P. [Google Scholar]; b Piert M.; Machulla H.-J.; Picchio M.; Reischl G.; Ziegler S.; Kumar P.; Wester H.-J.; Beck R.; McEwan A. J. B. J. Nucl. Med. 2005, 46, 106–113. [PubMed] [Google Scholar]
- Sorger D.; Patt M.; Kumar P.; Wiebe L. I.; Barthel H.; Seese A.; Dannenberg C.; Tannapfel A.; Sabri R. K. O. Nucl. Med. Biol. 2003, 30, 317–326. [DOI] [PubMed] [Google Scholar]
- a Iyer R. V.; Haynes P. T.; Schneider R. F.; Movsas B; Chapman J. D. J. Nucl. Med. 2001, 42, 37–44. [PubMed] [Google Scholar]; b Zanzonico P.; ÓDonoghue J.; Chapman J. D.; Schneider R.; Cai S.; Larson S.; Wen B.; Chen Y; Finn R.; Ruan S.; Gerweck L.; Humm J.; Ling C. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 117–128. [DOI] [PubMed] [Google Scholar]
- Emami S.; Kumar P.; Yang J.; Kresolic Z.; Paproski R.; Cass C; McEwan A. J. B.; Wiebe L. I. J. Pharm. Sci. 2007, 10, 237–245. [PubMed] [Google Scholar]
- Prisbe E. J.; Verheyden P. H.; Moffatt J. G. J. Org. Chem. 1978, 43, 4784–4794. [Google Scholar]
- Sakaguchi M.; Larroquette C. A.; Agrawal K. C. J. Med. Chem. 1983, 26, 20–24. [DOI] [PubMed] [Google Scholar]
- a Schneider R. F.; Engelhardt E. L.; Stobbe C. C.; Fenning M. C.; Chapman J. D. J. Labelled Compd. Radiopharm. 1997, 39, 541–557. [Google Scholar]; b Kumar P.; Wiebe L. I.; Atrazheva E.; Tandon M. Tetrahedron Lett. 2001, 42, 2077–2078. [Google Scholar]; c Patt M.; Sorger D.; Scheunemann M.; Stöcklin G. Appl. Radiat. Isot. 2002, 57, 705–712. [DOI] [PubMed] [Google Scholar]; d Naimi E.; Kumar P.; McEwan A. J. B.; Wiebe L. I. Nucleosides, Nucleotides Nucleic Acids 2005, 24, 173–178. [DOI] [PubMed] [Google Scholar]
- Review:Vorbrüggen H.; Ruh-Pohlenz C. Org. React. 2000, 55, 1–109. [Google Scholar]
- a Fessenden R. J. Org. Chem. 1960, 25, 2191–2193. [Google Scholar]; b Kraus C. A.; Nelson W. K. J. Am. Chem. Soc. 1934, 56, 195–202. [Google Scholar]
- Bailey D. L.; Sommer L. H.; Whitmore F. C. J. Am. Chem. Soc. 1948, 70, 435–436. [Google Scholar]; See also: Sauer R. O.; Hasek R. H. J. Am. Chem. Soc. 1946, 68, 241–244. [Google Scholar]
- Khasnobis S.; Zhang J.; Angala S. K.; Amin A. G.; McNeil M. R.; Crick D. C.; Chatterjee D. Chem. Biol. 2006, 13, 787–795. [DOI] [PubMed] [Google Scholar]
- Kumar P.; Stypinski D.; Xia H.; McEwan A. J. B.; Machulla H.-J.; Wiebe L. I. J. Labelled Compd. Radiopharm. 1999, 42, 3–16. [Google Scholar]
- Kumar P.; Okhura K.; Beiki D.; Wiebe L. I.; Seki K.-i. Chem. Pharm. Bull. 2003, 51, 399–403. [DOI] [PubMed] [Google Scholar]
- Kumar P.; Wiebe L. I.; Asikoglu M.; Tandon M.; McEwan A. J. B. Appl. Radiat. Isot. 2002, 57, 697–703. [DOI] [PubMed] [Google Scholar]
- Mikenda W. Vib. Spectrosc. 1992, 3, 327–330. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.