

Abstract
Metabolic syndrome has deleterious effects on the CNS, and recent evidence suggests that obesity rates are higher at presentation in children who develop epilepsy. Adiponectin is secreted by adipose tissue and acts in the brain and peripheral organs to regulate glucose and lipid metabolism. Adiponectin deficiency predisposes toward metabolic syndrome, characterized by obesity, insulin resistance, impaired glucose tolerance, hyperlipidemia, and cardiovascular morbidity. To investigate the relationship between metabolic syndrome and seizures, wild-type C57BL/6J and adiponectin knock-out mice were fed a high-fat diet, followed by treatment with low doses of kainic acid to induce seizures. Adiponectin deficiency in mice fed a high-fat diet resulted in greater fat accumulation, impaired glucose tolerance, hyperlipidemia, increased seizure severity, and increased hippocampal pathology. In contrast, there were no adverse effects of adiponectin deficiency on metabolic phenotype or seizure activity in mice fed a normal (low-fat) chow diet. These findings demonstrate that metabolic syndrome modulates the outcome of seizures and brain injury.
Introduction
Metabolic syndrome is a constellation of metabolic and cardiovascular abnormalities including obesity, impaired glucose tolerance, dyslipidemia, and cardiovascular morbidity (Grundy et al., 2005). The adverse consequences of metabolic syndrome are typically linked to vascular disease, but associations with nonvascular diseases have been noted. Interestingly, children with epilepsy have a high rate of obesity at initial presentation (Daniels et al., 2009). It is unknown whether this association indicates a causal relationship between metabolic disease and seizure susceptibility.
Adiponectin is secreted by adipocytes and improves insulin sensitivity and fat oxidation (Ahima, 2006). Adiponectin is inversely correlated with adiposity, hence metabolic syndrome is associated with low plasma adiponectin (Ahima, 2006). While adiponectin deficiency has no apparent metabolic effects in lean mice, adiponectin deficiency in mice fed a high-fat diet (HFD) results in insulin resistance, hyperlipidemia, inflammation, and vascular injury (Kubota et al., 2002; Ma et al., 2002; Maeda et al., 2002; Nawrocki et al., 2006). Low adiponectin levels are found in CSF, and both adiponectin receptors, AdipoR1 and AdipoR2, are widely expressed in the brain (Yamauchi et al., 2003, 2007; Kusminski et al., 2007). Adiponectin modulates hypothalamic and brainstem neuronal activity, and acts centrally to control peripheral metabolism (Qi et al., 2004; Fry et al., 2006; Hoyda et al., 2007; Kubota et al., 2007).
Adiponectin is protective against ischemic brain injury by modulating inflammatory pathways and endothelial function (Nishimura et al., 2008; Chen et al., 2009). Interestingly, PPARγ agonists, which are known to increase adiponectin expression, protect against seizure-related pathology (Maurois et al., 2008; Sun et al., 2008; Yu et al., 2008; Abdallah, 2010). Furthermore, the anti-epileptic drug valproic acid modulates PPARγ signaling, and alters adipoR1 and adiponectin expression (Qiao et al., 2006; Lan et al., 2008; Rauchenzauner et al., 2008). Adiponectin injected intracerebrally has also been shown to reduce kainic acid (KA)-induced excitotoxicity (Jeon et al., 2009). Thus, we hypothesized that adiponectin deficiency would enhance seizure sensitivity in the setting of metabolic syndrome. We fed C57BL/6J (wild type) and ADP-KO mice HFD or normal chow and compared body composition, glucose tolerance, lipids, KA-induced seizure, and hippocampal pathology. As predicted, adiponectin deficiency resulted in an increase in body fat, impaired glucose tolerance and increased lipids, and these changes were associated with increased seizure severity and hippocampal pathology.
Materials and Methods
Animals, diet and metabolic studies.
C57BL/6J mice and ADP-KO mice bred on the same genetic background (n = 17 per genotype), were fed HFD (Research Diets, #D12451; 45% fat, 35% carbohydrate, 20% protein; 4.7 kcal/g) for 8–12 weeks (Takahashi et al., 2002). Control WT and adiponectin-deficient mice (n = 10 per genotype), were fed normal chow (5% fat, 49% carbohydrate, 24% protein; 4 kcal/g; LabDiet). Body composition was analyzed by nuclear magnetic resonance (Echo Medical Systems) (Varela et al., 2008). To determine glucose tolerance, mice were fasted overnight (16 h), tail glucose was measured (OneTouch Ultra glucometer, Johnson & Johnson), 2 g/kg glucose was injected intraperitoneally, and tail glucose was measured at 15, 30, 60, and 120 min. Serum collected when the animals were killed was used to measure triglycerides, cholesterol, and nonesterified fatty acids (NEFAs) by enzymatic assay (Imai et al., 2007; Varela et al., 2008).
KA-induced seizure.
KA in saline was administered subcutaneously (20 mg/kg) or stereotaxically into the hippocampus (−1.8 mm, −1.8 mm, −1.8 mm relative to bregma, 100 ng). Subcutaneous saline was used as a control. Seizure activity was scored every 15 min for 4 h using a modified Racine scale (0, normal; 1, hypoactivity; 2, rigidity; 3, rearing with repetitive head/forepaw movements; 4, rearing and falling; 5, continuous rearing/falling; 6, generalized convulsions) (McKhann et al., 2003).
Brain histology and immunohistochemistry.
Two days after peripheral KA, food was removed for 4 h in the morning before the mice were killed. Serum was obtained via cardiac puncture. Mice were perfused with PBS followed by neutral buffered formalin. Brains were postfixed overnight, embedded in paraffin, and sectioned coronally (6 μm) for cresyl violet stain. Adjacent sections were subject to immunohistochemistry using the following antibodies: rat anti-GFAP (clone 2.2B10), rabbit anti-Iba1 (Wako Chemicals USA), rat anti-phospho-neurofilament (clone TA51), mouse anti-neurofilament (clone RMD020), and mouse anti-synaptophysin (clone SY38, Abcam). The slides were scored by a neuropathologist on a scale of 1–4 (1, normal; 2, mild; 3, moderate; and 4, severe).
Statistical analysis.
The effects of genotype and diet were assessed by unpaired t test or ANOVA, and pair wise comparisons were analyzed with Fisher's least significant difference test. For correlation analysis, seizure scores over time were used to calculate an area under the curve (AUC) followed by linear regressions between seizure AUC and metabolic parameters.
Results
Adiponectin deficiency increases body fat, glucose, and lipids
ADP-KO and WT mice were fed HFD to induce features of the metabolic syndrome, and assess its impact on KA-induced seizures. After 2 months on HFD, ADP-KO and WT mice had similar body weight (30.8 ± 0.8 g vs 30.6 ± 0.5 g; p = 0.842, Fig. 1A). However, ADP-KO mice had significantly greater fat mass (7.67 ± 0.58 g vs 4.63 ± 0.27 g; p = 0.0002) and less lean tissue mass (21.67 ± 0.69 g vs 24.66 ± 0.35 g; p = 0.0011) compared with WT mice (Fig. 1A). We performed intraperitoneal glucose tolerance tests as a measure of glucose homeostasis. After overnight fasting, ADP-KO mice were hyperglycemic compared with WT mice (148.5 ± 10.3 mg/dL vs 115.6 ± 11.7 mg/dL, p = 0.049). After intraperitoneal injection of glucose, blood glucose was higher in ADP-KO mice than WT (genotype p = 0.0164, time p < 0.0001, interaction p = 0.2814; Fig. 1B). At the time they were killed, ADP-KO mice had higher serum levels of triglycerides, NEFAs, and cholesterol than WT mice (Table 1). KA treatment decreased serum cholesterol levels, but this change was less compared with the effect of genotype (Table 1).
Figure 1.

Effects of HFD on body composition and glucose tolerance. A, ADP-KO and WT mice fed HFD were assessed for body weight, fat content and lean mass, shown as mean ± SE (n = 10 per group). **p < 0.01; ***p < 0.001. B, Blood glucose measurements before and after a glucose challenge are shown as mean ± SE (n = 9–10 per group; genotype p = 0.0164, time p < 0.0001, interaction p = 0.281). Post hoc analysis revealed significantly differences at times 0 (p = 0.049), 60 min (p = 0.0213) and 120 min (p = 0.0135).
Table 1.
Effects of adiponectin deficiency and kainic acid treatment on serum lipids
| Triglyceride (mg/dl) | NEFA (mEq/L) | Cholesterol (mg/dl) | |
|---|---|---|---|
| WT saline | 27.0 ± 1.4 | 0.787 ± 0.022 | 134.0 ± 8.3 |
| WT kA | 35.5 ± 2.7 | 0.762 ± 0.043 | 121.3 ± 4.7 |
| KO saline | 42.9 ± 3.4 | 0.926 ± 0.016 | 173.5 ± 8.6 |
| KO KA | 49.7 ± 8.5 | 0.866 ± 0.076 | 146.9 ± 8.4 |
| Two-way ANOVA | |||
| Genotype | 0.019 | 0.047 | 0.0006 |
| Treatment | 0.202 | 0.462 | 0.020 |
| Interaction | 0.892 | 0.764 | 0.375 |
Data are shown as mean ± SE (n = 4–6 per group). Two-way ANOVA results are shown with significant p values in bold.
Adiponectin deficiency increases seizure severity
To determine whether features of metabolic syndrome resulting from adiponectin deficiency increased seizure severity, ADP-KO and WT mice were treated with a low dose of KA (20 mg/kg) and seizures were scored from 0 (no seizure) to 6 (tonic-clonic). ADP-KO mice were more sensitive to KA-induced seizure activity than WT, with peak seizure scores of 2.7 and 1.2, respectively (p = 0.0079). Indeed, half of the ADP-KO mice had peak seizure scores of 3–4 while no WT mice scored higher than 2. Analysis of seizures over 4 h showed that the mean score was higher for ADP-KO mice at all times (genotype p = 0.0127, time p < 0.0001, interaction p < 0.0001; Fig. 2A). The duration of seizures was longer in ADP-KO mice up to 120 min, compared with 30 min in WT mice. Thus, metabolic syndrome due to adiponectin deficiency resulted in more intense and prolonged seizure activity.
Figure 2.

Kainic acid seizure in mice fed HFD. A, Data are mean ± SE; n = 6. ADP-KO mice exhibited higher seizure scores (genotype p = 0.0127, time p < 0.0001, interaction p < 0.0001). Post hoc analysis with Bonferroni's correction revealed higher seizure scores from 60 min to 105 min (p < 0.01 to p < 0.001). B, Brain sections were stained with cresyl violet (top), or for GFAP (middle), and Iba1 (bottom). Representative images of hippocampus are shown, with higher-power images of the dentate gyrus endplate shown in the insets. Hippocampal regions are labeled for reference. Scale bars: Top and middle, 500 μm; bottom, 100 μm; inset, 50 μm. C, Semiquantitative pathology scores, shown as mean ± SE; n = 6. Dashed line denotes baseline normal score of 1 (**p < 0.01, ***p < 0.001). D, Neurodegeneration and gliosis in a kainic acid-treated ADP-KO mouse. Cresyl violet (left and middle)- and Iba1 (right)-stained sections are shown. Boxed region is CA1. Arrowhead points to a pyknotic neuron in contrast with viable neuron (asterisk). Scale bars: left, 500 μm; middle, 50 μm; right, 100 μm.
Adiponectin deficiency increases post-seizure hippocampal pathology
The downstream sequelae of seizures include gliosis, neurodegeneration, and neuronal reorganization (McKhann et al., 2003). The KA dose of 20 mg/kg is low for C57BL/6J mice (Ferraro et al., 1995; McKhann et al., 2003), thus we predicted minimal gliosis and neurodegeneration in WT mice versus ADP-KO mice. Cresyl violet stained sections of brain and hippocampus showed no neurodegeneration in either WT or ADP-KO (Fig. 3A), with the exception of one KA-treated ADP-KO mouse which showed severe loss of CA1 neurons (Fig. 2D). Immunohistochemistry for glial fibrillary acidic protein (GFAP) showed mild astrocytosis in KA-treated WT mice relative to saline-treated mice, and considerably more astrocytosis in KA-treated ADP-KO mice (Fig. 2B). Immunohistochemistry for Iba1 showed no microglial activation in WT mice and saline-treated ADP-KO mice. However, mild to moderate microglial activation was noted in ADP-KO mice, including microglial hypertrophy and clustering (Fig. 2B). The single ADP-KO mouse with neurodegeneration showed profound glial activation (Fig. 2D; data not shown). Semiquantitative image analysis demonstrated that KA-treated ADP-KO mice showed significantly more astrocytic and microglial activation relative to WT mice (Fig. 2C). Neurodegeneration was not statistically different between the two groups. Even when removing the one potential outlier with severe neurodegeneration, repeat analysis still indicated that ADP-KO mice showed significantly more glial pathology compared with WT mice (data not shown). Immunohistochemistry for synaptophysin or phosphorylated neurofilament did not show any evidence of synaptic sprouting or other structural changes (data not shown). These findings indicate that the worsening of seizure severity was accompanied by increased brain injury.
Figure 3.
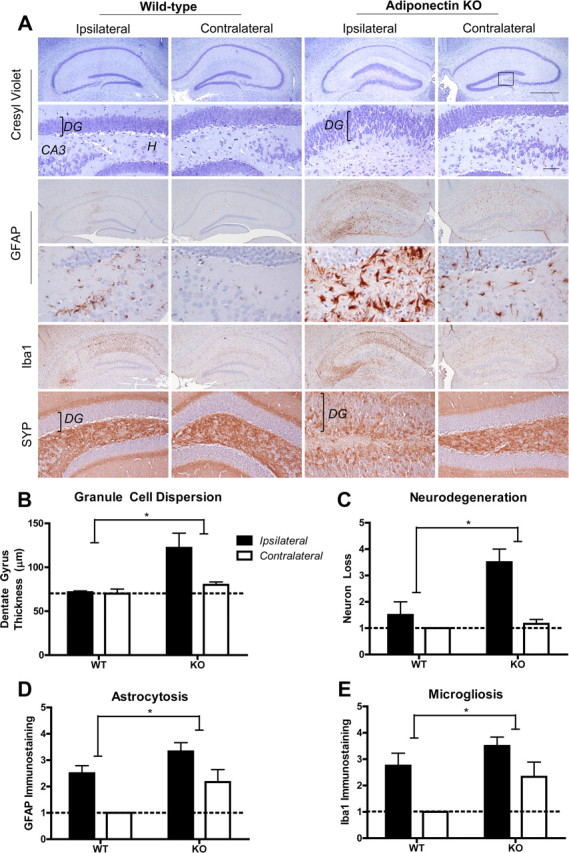
Intrahippocampal kainic acid and seizure-related pathology. A, Hippocampal sections were stained with cresyl violet (top, at low and high magnification), or for GFAP (middle, at low and high magnification), Iba1 (middle, at low magnification), and synaptophysin (SYN; bottom, at high magnification). Scale bars: lower-magnification panels, 500 μm; higher-magnification panels, 50 μm. DG, Dentate gyrus; CA3, cornu ammonis 3; H, hilum. B, Dentate gyrus thickness measurements shown as mean ± SE with dashed line denoting normal thickness. C–E, Semiquantitative pathology scores, shown as mean ± SE; n = 4–6. Dashed line denotes normal baseline score of 1. *p < 0.05 for genotype by two-way ANOVA).
Adiponectin deficiency increases chronic seizure related pathology
It is possible that altered body composition may change peripheral KA metabolism. To circumvent this issue, HFD-fed WT and ADP-KO mice were injected with a low KA dose (100 ng) directly into the hippocampus and examined after 2 weeks for chronic seizure related pathology. Severe neurodegeneration was evident in the hilum, CA3 and CA1 of ADP-KO, whereas neurodegeneration was mild or absent in WT (Fig. 3A). Intrahippocampal KA also resulted in neuronal dispersion of the dentate gyrus, and enhanced synaptophysin immunostaining in ADP-KO mice, suggesting extensive synaptic sprouting (Fig. 3A). GFAP and Iba1 staining were increased indicative of reactive astrocytosis and microgliosis in ADP-KO mice (Fig. 3A). Image analysis showed that ADP-KO displayed significantly increased granule cell dispersion (genotype p = 0.0138, laterality p = 0.0642, interaction p = 0.0827), neurodegeneration (genotype p = 0.0123, laterality p = 0.0020, interaction p = 0.0297), astrocytosis (genotype p = 0.0185, laterality p = 0.0030, interaction p = 0.6679) and microgliosis (genotype p = 0.0344, laterality p = 0.0052, interaction p = 0.5265) compared with WT (Fig. 3B–E). Thus, adiponectin deficiency enhances seizure related pathology in response to peripheral or central KA treatment.
Adiponectin deficiency in the absence of metabolic syndrome does not alter seizure activity
We hypothesized that an interaction between metabolic syndrome and adiponectin deficiency resulted in enhanced seizure activity. However, it was possible that adiponectin deficiency alone in the absence of metabolic changes may be sufficient to enhance seizure sensitivity. Thus, we examined the effects of KA in WT and ADP-KO mice fed normal chow diet. ADP-KO and WT mice had similar body weight, fat and lean mass, glucose tolerance and serum lipids (data not shown). Seizure activity was similar between WT and ADP-KO mice, peak seizure scores ranging from 0 to 1 (ADP-KO average peak score 0.6, WT average peak score 0.5, p = 0.77). Temporal analysis of seizure scores showed no significant effect of genotype (genotype p = 0.312, time p = 0.121, interaction p = 0.608).
Adiponectin deficiency is associated with increased adiposity in HFD mice, thus it is possible that seizure activity is associated with changes in metabolic parameters. We found a strong positive correlation between seizure severity and glucose intolerance (R2 = 0.5509, p = 0.0057), cholesterol (R2 = 0.5341, p = 0.0069), fat mass (R2 = 0.4391, p = 0.0189) and NEFAs (R2 = 0.4310, p = 0.0282), and a negative correlation with lean mass (R2 = 0.4706, p = 0.0138). In contrast, seizure severity was not associated with serum triglyceride (R2 = 0.1782, p = 0.1717) or body weight (R2 = 0.0756, p = 0.3869).
Discussion
It is estimated that 5–10% of individuals develop nonfebrile seizures or epilepsy in their lifetime (Hauser et al., 1993; Cockerell et al., 1995). Current efforts are underway to understand the comorbid conditions associated with epilepsy. Associations between epilepsy and obesity are confounded by the known effects of anti-epileptic drugs on peripheral metabolism (Isojärvi et al., 1996). However, a recent study has shown an increase in obesity rates in children at initial presentation before the use of anti-epileptic agents (Daniels et al., 2009).
Adiponectin deficiency results in insulin resistance, glucose intolerance, dyslipidemia and vascular injury, characteristic of metabolic syndrome. These features are reversible by adiponectin treatment (Kubota et al., 2002; Ma et al., 2002; Maeda et al., 2002; Nawrocki et al., 2006). Moreover, adiponectin has been shown to ameliorate cerebrovascular injury in mice (Nishimura et al., 2008; Chen et al., 2009). We tested whether metabolic syndrome due to adiponectin deficiency would exacerbate seizure related brain injury. The KA dose in our study was below that required to induce seizures in C57BL/6J mice (Ferraro et al., 1995; McKhann et al., 2003), hence it is remarkable that clonic seizures (seizure score of 3+) occurred in 50% of HFD-fed ADP-KO mice. ADP-KO and WT mice on HFD had similar body weight and thus received the same KA doses. Furthermore, a low dose of intrahippocampal KA which normally causes mild brain pathology (Schauwecker, 2002), resulted in severe neuronal damage and gliosis in ADP-KO mice. Thus, the worsening of pathology in ADP-KO mice cannot be attributed to altered KA pharmacokinetics in ADP-KO mice.
Adiponectin deficiency in normal chow-fed mice did not increase seizure sensitivity. Rather, there was a trend toward higher seizure activity in HFD WT and ADP-KO mice relative to chow-fed counterparts. Thus, it appears that metabolic changes, rather than adiponectin, are the major determinant of seizure severity and hippocampal pathology. It is difficult to compare the effects of HFD versus normal chow diet on subcutaneous KA-induced seizure activity in HFD WT or ADP-KO mice, because the HFD mice were heavier and received a higher KA dose than chow-fed mice. Additional studies are needed to understand how adiponectin specifically alters metabolic parameters and seizures. Adiponectin receptors are distributed widely in the brain (Guillod-Maximin et al., 2009), and central adiponectin has potent electrophysiological effects (Fry et al., 2006; Hoyda et al., 2007), raising the possibility that adiponectin directly modifies seizure activity and brain pathology. Anti-epileptic drugs modulate various metabolic pathways (Isojärvi et al., 1996). For example, valproic acid regulates adiponectin and adipoR1 expression (Qiao et al., 2006; Rauchenzauner et al., 2008). PPARγ agonists including insulin sensitizing thiazolidinediones are protective in animal seizure models (Chen et al., 2009; Jeon et al., 2009), and also increase adiponectin levels (Nawrocki et al., 2006).
Other hormones associated with energy homeostasis influence seizures. Leptin and ghrelin inhibit seizures and protect against seizure-related neuropathology (Shanley et al., 2002; Obay et al., 2007; Erbayat-Altay et al., 2008; Guo et al., 2008; Obay et al., 2008; Xu et al., 2008; Lee et al., 2010; Obeid et al., 2010). These studies demonstrate that peripheral endocrine and metabolic factors are capable of modulating seizure threshold and seizure-related pathology by acting on CNS neurons to trigger intracellular signaling pathways or modulating neuronal activity. The results of the current study indicate that changes in metabolic parameters associated with adiponectin deficiency influence seizure activity and brain pathology. Understanding of the underlying mechanisms would provide a framework for prevention and treatment of epilepsy associated with metabolic syndrome.
Footnotes
This study was supported by NIH Grants RO1-DK-062348 and PO1-DK-049210 (R.S.A.) and T32-AG00255 and K08-AG039510 (E.B.L.). We thank Drs. John Q. Trojanowski and Virginia M.-Y. Lee for providing antibody reagents, and the University of Pennsylvania Diabetes and Endocrinology Research Center (DERC) Mouse Phenotyping, Physiology and Metabolism Core for body composition analysis (NIH Grant P30-DK19525).
The authors declare no competing financial interests.
References
- Abdallah DM. Anticonvulsant potential of the peroxisome proliferator-activated receptor gamma agonist pioglitazone in pentylenetetrazole-induced acute seizures and kindling in mice. Brain Res. 2010;1351:246–253. doi: 10.1016/j.brainres.2010.06.034. [DOI] [PubMed] [Google Scholar]
- Ahima RS. Metabolic actions of adipocyte hormones: focus on adiponectin. Obesity (Silver Spring) 2006;14(Suppl 1):9S–15S. doi: 10.1038/oby.2006.276. [DOI] [PubMed] [Google Scholar]
- Chen B, Liao WQ, Xu N, Xu H, Wen JY, Yu CA, Liu XY, Li CL, Zhao SM, Campbell W. Adiponectin protects against cerebral ischemia-reperfusion injury through anti-inflammatory action. Brain Res. 2009;1273:129–137. doi: 10.1016/j.brainres.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Cockerell OC, Eckle I, Goodridge DM, Sander JW, Shorvon SD. Epilepsy in a population of 6000 re-examined: secular trends in first attendance rates, prevalence, and prognosis. J Neurol Neurosurg Psychiatry. 1995;58:570–576. doi: 10.1136/jnnp.58.5.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels ZS, Nick TG, Liu C, Cassedy A, Glauser TA. Obesity is a common comorbidity for pediatric patients with untreated, newly diagnosed epilepsy. Neurology. 2009;73:658–664. doi: 10.1212/WNL.0b013e3181ab2b11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbayat-Altay E, Yamada KA, Wong M, Thio LL. Increased severity of pentylenetetrazol induced seizures in leptin deficient ob/ob mice. Neurosci Lett. 2008;433:82–86. doi: 10.1016/j.neulet.2007.12.051. [DOI] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, Smith GG, Berrettini WH. Differential susceptibility to seizures induced by systemic kainic acid treatment in mature DBA/2J and C57BL/6J mice. Epilepsia. 1995;36:301–307. doi: 10.1111/j.1528-1157.1995.tb00999.x. [DOI] [PubMed] [Google Scholar]
- Fry M, Smith PM, Hoyda TD, Duncan M, Ahima RS, Sharkey KA, Ferguson AV. Area postrema neurons are modulated by the adipocyte hormone adiponectin. J Neurosci. 2006;26:9695–9702. doi: 10.1523/JNEUROSCI.2014-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC, Jr, Spertus JA, Costa F. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112:2735–2752. doi: 10.1161/CIRCULATIONAHA.105.169404. [DOI] [PubMed] [Google Scholar]
- Guillod-Maximin E, Roy AF, Vacher CM, Aubourg A, Bailleux V, Lorsignol A, Pénicaud L, Parquet M, Taouis M. Adiponectin receptors are expressed in hypothalamus and colocalized with proopiomelanocortin and neuropeptide Y in rodent arcuate neurons. J Endocrinol. 2009;200:93–105. doi: 10.1677/JOE-08-0348. [DOI] [PubMed] [Google Scholar]
- Guo Z, Jiang H, Xu X, Duan W, Mattson MP. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J Biol Chem. 2008;283:1754–1763. doi: 10.1074/jbc.M703753200. [DOI] [PubMed] [Google Scholar]
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia. 1993;34:453–468. doi: 10.1111/j.1528-1157.1993.tb02586.x. [DOI] [PubMed] [Google Scholar]
- Hoyda TD, Fry M, Ahima RS, Ferguson AV. Adiponectin selectively inhibits oxytocin neurons of the paraventricular nucleus of the hypothalamus. J Physiol. 2007;585:805–816. doi: 10.1113/jphysiol.2007.144519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Varela GM, Jackson MB, Graham MJ, Crooke RM, Ahima RS. Reduction of hepatosteatosis and lipid levels by an adipose differentiation-related protein antisense oligonucleotide. Gastroenterology. 2007;132:1947–1954. doi: 10.1053/j.gastro.2007.02.046. [DOI] [PubMed] [Google Scholar]
- Isojärvi JI, Laatikainen TJ, Knip M, Pakarinen AJ, Juntunen KT, Myllylä VV. Obesity and endocrine disorders in women taking valproate for epilepsy. Ann Neurol. 1996;39:579–584. doi: 10.1002/ana.410390506. [DOI] [PubMed] [Google Scholar]
- Jeon BT, Shin HJ, Kim JB, Kim YK, Lee DH, Kim KH, Kim HJ, Kang SS, Cho GJ, Choi WS, Roh GS. Adiponectin protects hippocampal neurons against kainic acid-induced excitotoxicity. Brain Res Rev. 2009;61:81–88. doi: 10.1016/j.brainresrev.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, Eto K, Yamashita T, Kamon J, Satoh H, Yano W, Froguel P, Nagai R, Kimura S, Kadowaki T, Noda T. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem. 2002;277:25863–25866. doi: 10.1074/jbc.C200251200. [DOI] [PubMed] [Google Scholar]
- Kubota N, Yano W, Kubota T, Yamauchi T, Itoh S, Kumagai H, Kozono H, Takamoto I, Okamoto S, Shiuchi T, Suzuki R, Satoh H, Tsuchida A, Moroi M, Sugi K, Noda T, Ebinuma H, Ueta Y, Kondo T, Araki E, et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 2007;6:55–68. doi: 10.1016/j.cmet.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Kusminski CM, McTernan PG, Schraw T, Kos K, O'Hare JP, Ahima R, Kumar S, Scherer PE. Adiponectin complexes in human cerebrospinal fluid: distinct complex distribution from serum. Diabetologia. 2007;50:634–642. doi: 10.1007/s00125-006-0577-9. [DOI] [PubMed] [Google Scholar]
- Lan MJ, Yuan P, Chen G, Manji HK. Neuronal peroxisome proliferator-activated receptor gamma signaling: regulation by mood-stabilizer valproate. J Mol Neurosci. 2008;35:225–234. doi: 10.1007/s12031-008-9056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Lim E, Kim Y, Li E, Park S. Ghrelin attenuates kainic acid-induced neuronal cell death in the mouse hippocampus. J Endocrinol. 2010;205:263–270. doi: 10.1677/JOE-10-0040. [DOI] [PubMed] [Google Scholar]
- Ma K, Cabrero A, Saha PK, Kojima H, Li L, Chang BH, Paul A, Chan L. Increased beta-oxidation but no insulin resistance or glucose intolerance in mice lacking adiponectin. J Biol Chem. 2002;277:34658–34661. doi: 10.1074/jbc.C200362200. [DOI] [PubMed] [Google Scholar]
- Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, Komuro R, Ouchi N, Kihara S, Tochino Y, Okutomi K, Horie M, Takeda S, Aoyama T, Funahashi T, Matsuzawa Y. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8:731–737. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- Maurois P, Rocchi S, Pages N, Bac P, Stables JP, Gressens P, Vamecq J. The PPARgamma agonist FMOC-l-leucine protects both mature and immature brain. Biomed Pharmacother. 2008;62:259–263. doi: 10.1016/j.biopha.2007.10.014. [DOI] [PubMed] [Google Scholar]
- McKhann GM, 2nd, Wenzel HJ, Robbins CA, Sosunov AA, Schwartzkroin PA. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience. 2003;122:551–561. doi: 10.1016/s0306-4522(03)00562-1. [DOI] [PubMed] [Google Scholar]
- Nawrocki AR, Rajala MW, Tomas E, Pajvani UB, Saha AK, Trumbauer ME, Pang Z, Chen AS, Ruderman NB, Chen H, Rossetti L, Scherer PE. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2006;281:2654–2660. doi: 10.1074/jbc.M505311200. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Izumiya Y, Higuchi A, Shibata R, Qiu J, Kudo C, Shin HK, Moskowitz MA, Ouchi N. Adiponectin prevents cerebral ischemic injury through endothelial nitric oxide synthase dependent mechanisms. Circulation. 2008;117:216–223. doi: 10.1161/CIRCULATIONAHA.107.725044. [DOI] [PubMed] [Google Scholar]
- Obay BD, Tasdemir E, Tümer C, Bilgin HM, Sermet A. Antiepileptic effects of ghrelin on pentylenetetrazole-induced seizures in rats. Peptides. 2007;28:1214–1219. doi: 10.1016/j.peptides.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Obay BD, Taşdemir E, Tümer C, Bilgin HM, Atmaca M. Dose dependent effects of ghrelin on pentylenetetrazole-induced oxidative stress in a rat seizure model. Peptides. 2008;29:448–455. doi: 10.1016/j.peptides.2007.11.020. [DOI] [PubMed] [Google Scholar]
- Obeid M, Frank J, Medina M, Finckbone V, Bliss R, Bista B, Majmudar S, Hurst D, Strahlendorf H, Strahlendorf J. Neuroprotective effects of leptin following kainic acid-induced status epilepticus. Epilepsy Behav. 2010;19:278–283. doi: 10.1016/j.yebeh.2010.07.023. [DOI] [PubMed] [Google Scholar]
- Qi Y, Takahashi N, Hileman SM, Patel HR, Berg AH, Pajvani UB, Scherer PE, Ahima RS. Adiponectin acts in the brain to decrease body weight. Nat Med. 2004;10:524–529. doi: 10.1038/nm1029. [DOI] [PubMed] [Google Scholar]
- Qiao L, Schaack J, Shao J. Suppression of adiponectin gene expression by histone deacetylase inhibitor valproic acid. Endocrinology. 2006;147:865–874. doi: 10.1210/en.2005-1030. [DOI] [PubMed] [Google Scholar]
- Rauchenzauner M, Laimer M, Luef G, Kaser S, Engl J, Tatarczyk T, Ciardi C, Tschoner A, Lechleitner M, Patsch J, Ebenbichler CF. Adiponectin receptor R1 is upregulated by valproic acid but not by topiramate in human hepatoma cell line, HepG2. Seizure. 2008;17:723–726. doi: 10.1016/j.seizure.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE. Modulation of cell death by mouse genotype: differential vulnerability to excitatory amino acid-induced lesions. Exp Neurol. 2002;178:219–235. doi: 10.1006/exnr.2002.8038. [DOI] [PubMed] [Google Scholar]
- Shanley LJ, O'Malley D, Irving AJ, Ashford ML, Harvey J. Leptin inhibits epileptiform-like activity in rat hippocampal neurones via PI 3-kinase-driven activation of BK channels. J Physiol. 2002;545:933–944. doi: 10.1113/jphysiol.2002.029488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Huang Y, Yu X, Li Y, Yang J, Li R, Deng Y, Zhao G. Peroxisome proliferator-activated receptor gamma agonist, rosiglitazone, suppresses CD40 expression and attenuates inflammatory responses after lithium pilocarpine-induced status epilepticus in rats. Int J Dev Neurosci. 2008;26:505–515. doi: 10.1016/j.ijdevneu.2008.01.009. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Patel HR, Qi Y, Dushay J, Ahima RS. Divergent effects of leptin in mice susceptible or resistant to obesity. Horm Metab Res. 2002;34:691–697. doi: 10.1055/s-2002-38251. [DOI] [PubMed] [Google Scholar]
- Varela GM, Antwi DA, Dhir R, Yin X, Singhal NS, Graham MJ, Crooke RM, Ahima RS. Inhibition of ADRP prevents diet-induced insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2008;295:G621–628. doi: 10.1152/ajpgi.90204.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Rensing N, Yang XF, Zhang HX, Thio LL, Rothman SM, Weisenfeld AE, Wong M, Yamada KA. Leptin inhibits 4-aminopyridine- and pentylenetetrazole-induced seizures and AMPAR-mediated synaptic transmission in rodents. J Clin Invest. 2008;118:272–280. doi: 10.1172/JCI33009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, Okada-Iwabu M, Kawamoto S, Kubota N, Kubota T, Ito Y, Kamon J, Tsuchida A, Kumagai K, Kozono H, Hada Y, Ogata H, Tokuyama K, Tsunoda M, Ide T, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007;13:332–339. doi: 10.1038/nm1557. [DOI] [PubMed] [Google Scholar]
- Yu X, Shao XG, Sun H, Li YN, Yang J, Deng YC, Huang YG. Activation of cerebral peroxisome proliferator-activated receptors gamma exerts neuroprotection by inhibiting oxidative stress following pilocarpine-induced status epilepticus. Brain Res. 2008;1200:146–158. doi: 10.1016/j.brainres.2008.01.047. [DOI] [PubMed] [Google Scholar]