

Abstract
p73 is one of the tumor-suppressor p53 family of nuclear transcription factor. As expected from the structural similarity between p53 and p73, p73 has a tumor-suppressive function. However, p73 was rarely mutated in human primary tumors. Under normal physiological conditions, p73 is kept at an extremely low level to allow cells normal growth. In response to a certain subset of DNA damages, p73 is induced dramatically and transactivates an overlapping set of p53-target genes implicated in the promotion of cell cycle arrest and/or apoptotic cell death. Cells undergo cell cycle arrest and/or apoptotic cell death depending on the type and strength of DNA damages. p73 is regulated largely through the posttranslational modifications such as phosphorylation and acetylation. These chemical modifications are tightly linked to direct protein-protein interactions. In the present paper, the authors describe the functional significance of the protein-protein interactions in the regulation of proapoptotic p73.
1. Introduction
For a long time, p53 has been believed to be a solitary gene. This classical point of view has been challenged by a discovery of novel human p53 homologues termed p73 and p63 [1–3]. Thus, p53 family is composed of p53, p73 and p63. p73 contains an NH2-terminal transactivation domain (TA; amino acid residues 1–54), a central core sequence-specific DNA-binding domain (DB; amino acid residues 131–310), an oligomerization domain (OD; amino acid residues 345–380) and a COOH-terminal sterile α motif (SAM; amino acid residues 484–549) domain [1]. Among them, the central core sequence-specific DNA-binding domain is highly conserved (more than 60% amino acid sequence identity) across the family, and p53 lacks the COOH-terminal SAM domain which is involved in the protein-protein interaction [4]. As expected from the structural similarity between p53 and p73, p73 has an ability to transactivate an overlapping set of p53-target genes implicated in cell cycle arrest and/or apoptotic cell death such as p21WAF1, MDM2, BAX, PUMA, and NOXA [5–7]. Indeed, forced expression of p73 induced cell cycle arrest and/or apoptotic cell death in certain cancerous cells [1, 8, 9].
Since p73 was mapped at human chromosome 1p36.3 where genomic aberrations are frequently observed in a variety of tumors [10–14], it is likely that p73 could be one of the classical Knudson-type tumor-suppressor genes. To address this issue, the extensive mutation search for p73 was carried out. In a sharp contrast to p53 bearing loss of-function mutations in 50% of human tumors [15, 16], p73 was rarely mutated in human tumors [17], suggesting that p73 is not a Knudson-type tumor-suppressor gene. In support with these results, initial genetic studies revealed that p73-deficient mice do not develop spontaneous tumors [18]. p53-deficient mice underwent spontaneous tumor development, mainly sarcomas and lymphomas [19]. Of note, Flores et al. demonstrated that p73 is required for p53-dependent apoptotic cell death, indicating that p73 is one of the essential coactivators for p53, and these observations also emphasized the functional importance of p73 in the regulation of DNA damage-induced p53-dependent proapoptotic pathway [20]. Subsequent genetic studies revealed that mice harboring hemizygous p73 develop spontaneous tumors, and their spectrum is quite different from that of p53-deficient mice [21]. Based on these results, p73 has been considered to be one of the critical tumor-suppressors, although p73 was infrequently mutated in human primary tumors.
Extensive expression studies demonstrated that p73 encodes at least seven alternative splicing variants with different COOH termini (p73α, p73β, p73γ, p73δ, p73ε, p73η, and p73ξ), termed the TA variants [1, 22–24]. Since these splicing variants contain the intact NH2-terminal transactivation domain, they have the varied transcriptional potential. Until now, each TA variant-specific biological function remains unclear. In addition to COOH-terminal splicing variants, p73 produced NH2-terminally truncated forms of p73 termed the ΔNp73 variants (Δp73α, Δp73β, Δp73γ, Δp73δ, Δp73ε, Δp73η, and Δp73ξ) arising from the alternative promoter usage [25]. ΔNp73 lacked NH2-terminal transactivation domain and had an oncogenic potential [26, 27]. Indeed, higher expression levels of ΔNp73 were strongly correlated with poor prognostic outcome in neuroblastoma patients [28]. In a good agreement with these observations, ΔNp73 was frequently overexpressed in a variety of human tumor tissues as compared with their corresponding normal tissues [29]. Of note, ΔNp73 displayed a dominant-negative behavior toward TAp73 as well as wild-type p53 [25]. Furthermore, TAp73 was also inhibited by mutant forms of p53 [30]. Intriguingly, Liu et al. reported that ΔNp73β has an ability to induce cell cycle arrest and apoptotic cell death in association with the upregulation of p53-target genes [31]. Unlike ΔNp73β, ΔNp73α was inactive in suppressing cell growth. This complicated issue should be addressed further.
Under normal physiological conditions, steady-state expression of p73 is maintained at quite low level and thereby keeping this dangerous proapoptotic protein in an inactive form [7]. Initial studies showed that p73 is not induced in response to DNA damage such as actinomycin D treatment and UV exposure [1]. However, subsequent studies demonstrated that p73 is induced and activated in response to a certain subset of DNA damaging agents [32]. Accumulated evidence strongly suggests that DNA damage-mediated induction and activation of p73 is regulated by posttranslational modifications such as phosphorylation and acetylation, which is tightly linked to protein-protein interactions [5–7].
In the present paper, we describe the regulatory mechanisms of proapoptotic p73 through protein-protein interactions.
2. Negative Autoregulatory Feedback Loop
As described in [25], ΔNp73 acts as a dominant-negative inhibitor toward wild-type p73. Since ΔNp73 had an oncogenic potential [26, 27], the balance between the intracellular expression levels of proapoptotic wild-type p73 and antiapoptotic ΔNp73 plays a critical role in the regulation of cell-fate determination. We and others found that wild-type p73 has an ability to transactivate its dominant-negative inhibitor ΔNp73 [29, 33, 34], and thereby creating a feedback control loop which tightly regulates the proapoptotic function of wild-type p73 (Figure 1). Since ΔNp73 retains an intact central core sequence-specific DNA-binding domain as well as an oligomerization domain, it is likely that ΔNp73 inhibits wild-type p73 by direct protein-protein interaction between them through the oligomerization domain (defective hetero-oligomer formation) and/or the dominant-negative competition for DNA-binding sites with wild-type p73 [5, 35]. Thus, ΔNp73 contributes to a safety system preventing inappropriate cell death.
Figure 1.
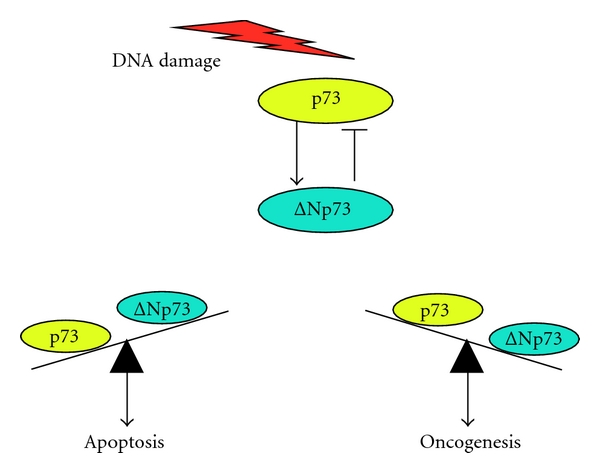
Dominant-negative effect of ΔNp73 on wild-type p73. In response to DNA damage, wild-type p73 transactivates its dominant-negative inhibitor ΔNp73. The intracellular balance between the expression levels of wild-type p73 and ΔNp73 is a critical determinant of cell fate.
Maisse et al. demonstrated that ΔNp73 is rapidly degraded in response to DNA damage [36]. Although the precise molecular mechanisms behind the selective degradation of ΔNp73 in response to DNA damage remains elusive, this preferential degradation of ΔNp73 might relieve the inhibitory effect of ΔNp73 on wild-type p73, and results in the promotion of cell cycle arrest and/or apoptotic cell death of damaged cells. Recently, Sayan et al. found that the RING finger domain ubiquitin protein ligase termed PIR2 is a direct p73-target gene product, and PIR2 preferentially promotes the proteasomal degradation of ΔNp73 [37]. Based on their results, PIR2 expression was induced upon DNA damage in association with a significant accumulation of wild-type p73. In addition, a strong interaction between ΔNp73 and PIR2 was detectable, whereas wild-type p73 bound to PIR2 with a much lower affinity. Therefore, the preferential degradation of ΔNp73 mediated by PIR2 might be due to differential binding affinity of PIR2 to ΔNp73 and wild-type p73.
Previously, Ohtsuka et al., described that p73 directly transactivates cyclin G [38]. According to their results, cyclin G bound to p73 and promoted its proteolytic degradation in a ubiquitination-independent manner. Although the precise molecular mechanisms underlying cyclin G-dependent proteolytic degradation remains unclear, there could exist a negative feedback control pathway in which cyclin G regulates the stability of p73.
3. Proteolytic Degradation of p73
Under normal physiological conditions, p73 is kept at an extremely low level. Since proteasome inhibitor treatment resulted in a significant accumulation of the endogenous p73 [39], p73 is regulated at least in part in a ubiquitin/proteasome-dependent manner. MDM2 which targets p53 for ubiquitin/proteasome-dependent degradation, bound to NH2-terminal transactivation domain of p73 but did not promote its degradation even though MDM2 inhibited p73-mediated transcriptional as well as proapoptotic activity [40–43]. It might be due to the fact that p73 lacks the degradation signal located within amino acid residues 92–112 of p53, which has been considered to confer MDM2 degradability [44]. Rossi et al. employed a phage display procedure to identify a novel p73-binding partner [45]. After the several rounds of screening, they found that HECT-type E3 ubiquitin protein ligase termed Itch is associated with p73. According to their results, their interaction was mediated by PY motif of p73 and WW domain of Itch. Itch had an ability to ubiquitinate efficiently p73 but not p53, and promoted its proteasome-dependent degradation. Itch did not interact with p53, which lacks PY motif. In contrast to p53, ΔNp73 was also the substrate of Itch. Of note, Itch was rapidly reduced in response to DNA damage through as yet unknown mechanisms, and thereby allowing p73 to increase. Asher et al. described that binding of YAP1 to PY motif of p73 prevents the interaction between Itch and p73 to stabilize p73 [46]. Consistent with these observations, Levy et al. demonstrated that YAP1 stabilizes p73 by displacing Itch binding to p73 [47]. Similarly, NEDD4-binding protein termed N4BP1 was able to interfere the complex formation of Itch with p73 [48]. N4BP1 had an undetectable effect on the stability of Itch. It appears to be important to clarify the molecular mechanisms behind DNA damage-induced downregulation of Itch.
F-box protein termed FBXO45 contains NH2-terminal F-box domain and SPRY domain. SPRY domain has been shown to be involved in protein-protein interactions [49]. Recently, Peschiaroli et al. reported that FBXO45 promotes the proteasome-dependent degradation of p73 [50]. Based on their results, FBXO45 bound to p73. Their complex formation was mediated by SAM domain of p73 and SPRY domain of FBXO45. Forced expression of FBXO45 resulted in a significant reduction in expression level of p73. Treatment of proteasome inhibitor rescued FBXO45-mediated downregulation of p73, suggesting that p73 is degraded by FBXO45 in a proteasome-dependent manner. Like Itch, FBXO45 was capable to promote the ubiquitin/proteasome-dependent degradation of wild-type p73 as well as ΔNp73. Additionally, FBXO45 was down-regulated in response to DNA damage. Zhang et al. demonstrated that one of PIAS SUMO-ligase family members termed PIASy is associated with p73 and PIASy-mediated sumoylation induces proteasomal degradation of p73 [51]. PIASy significantly reduced p73-mediated transcriptional activation. Intriguingly, we have found that HECT-type E3 ubiquitin protein ligase termed NEDL2 has an ability to polyubiquitinate p73 and increase its stability [52], indicating that polyubiquitination of p73 does not always act as a degradation signal.
It has been shown that p73 is also regulated in a proteasome-independent manner [53]. For example, Munarriz et al. revealed that calpain I is able to cleave p73 at two distinct sites including NH2-terminal transactivation domain and COOH-terminal oligomerization domain [54]. Consistent with these results, forced expression of the endogenous calpain inhibitor termed calpastatin resulted in an increase in the steady-state expression level of p73.
4. Phosphorylation-Dependent Activation of p73
Like p53, multiple phosphorylations following DNA damage regulate the stability as well as activity of p73 [5–7]. It has been shown that DNA damage activates nonreceptor tyrosine kinase c-Abl through ATM-dependent phosphorylation of c-Abl at Ser-465 [55–59]. Initial studies demonstrated that c-Abl is associated with p73 through SH3 domain of c-Abl and PY motif of p73, and directly phosphorylates p73 at Tyr-99 in response to CDDP and ionizing radiation [60–62]. Phosphorylated form of p73 underwent nuclear redistribution and became associated with the nuclear matrix [63]. c-Abl-mediated phosphorylation of p73 at Tyr-99 increased its stability and enhanced its transcriptional as well as proapoptotic activity. It has been shown that c-Abl stimulates the catalytic activity of p38 MAP kinase [64]. Sanchez-Prieto et al. found that c-Abl-mediated activation of p38 MAP kinase leads to phosphorylation of Thr residues adjacent to Pro residues of p73, and thereby increasing the protein stability of p73 [65]. Mantovani et al. described that COOH-terminal three amino acid residues including Ser-412, Thr-442, and Thr-482 are the phosphorylation sites mediated by p38 MAPK [66]. Previous studies showed that DNA damage induces c-Abl-dependent phosphorylation of PKCδ and promotes the nuclear translocation of PKCδ [67]. Ren et al. revealed that PKCδ catalytic fragment (PKCδCF) interacts with NH2-terminal transactivation domain as well as central core sequence-specific DNA-binding domain of p73, and phosphorylates p73 at Ser-289 within central core sequence-specific DNA-binding domain [68]. PKCδCF-mediated phosphorylation of p73 increased the stability of p73 and enhanced its transcriptional as well as proapoptotic activity.
Chk1 and Chk2 have been shown to be the downstream effector kinases of ATM and ATR, which play a critical role in the regulation of DNA damage response [69, 70]. Gonzalez et al. described that Chk1 interacts with p73 and phosphorylates p73 at Ser-47 in response to DNA damage [71]. According to their results, Chk1 but not Chk2 had an ability to phosphorylate p73. Chk1-mediated phosphorylation enhanced p73-dependent transactivation capacity as well as proapoptotic function. Subsequent studies demonstrated that Chk1 and Chk2 are required for DNA damage-induced accumulation of p73 [72]. Based on their results, Chk1 and Chk2 contributed to the transcriptional activation of p73 gene, which might be due to the stabilization of E2F1 in response to DNA damage. As described previously [73–75], E2F1 acts as a transcription factor for p73.
It has been shown that c-Abl activates the JNK signaling pathway in response to DNA damage, and JNK is closely involved in the apoptotic response to a variety of genotoxic stresses [56, 76, 77]. Recently, Jones et al. reported that JNK has an ability to stabilize p73 in a phosphorylation-dependent manner [78]. JNK was associated with p73 and phosphorylated its multiple Ser and Thr residues including Ser-8, Ser-97, Ser-110, Ser-333, Ser-412, Thr-442, and Thr-482. JNK-mediated phosphorylation enhanced the transcriptional as well as proapoptotic activity of p73.
Previously, we described that COOH-terminal Pro-rich domain (amino acid residues 380–513) contains transactivation ability as examined by GAL4 system [79]. Recently, Nyman et al. precisely identified the COOH-terminal transactivation domain of p73 which exists within amino acid residues 381 to 399 [80]. Intriguingly, this second COOH-terminal transactivation domain preferentially regulated the transcription of p53-target genes implicated in the induction of cell cycle arrest rather than apoptotic cell death. PKC phosphorylated p73 at Ser-388, and markedly enhanced the transcriptional activity of p73. Mutation at Ser-388 resulted in a remarkable reduction of the p73-mediated transcriptional activation. Taken together, their results indicate that PKC-mediated phosophorylation of p73 at Ser-388 contributes to the selective induction of cell cycle arrest.
It has been well known that IKK (IκB kinase) complex acts as an upstream regulator of prosurvival NF-κB signaling pathway [81]. IKK complex is composed of three functional subunits including IKK-α, IKK-β, and IKK-γ. We have found that nuclear IKK-α is induced to accumulate in response to DNA damage and associated with the central core sequence-specific DNA-binding domain of p73 in nuclear matrix [82]. DNA damage-mediated nuclear accumulation of IKK-α was dependent on phospho-ATM [83]. IKK-α had an ability to stabilize p73 by inhibiting its polyubiquitination, and enhanced p73-mediated transcriptional activation as well as proapoptotic activity. In addition, IKK-α phosphorylated the NH2-terminal portion of p73 and a kinase-deficient mutant form of IKK-α had undetectable effect on p73. Thus, IKK-α-mediated activation of p73 was regulated in a phosphorylation-dependent manner (Figure 2).
Figure 2.
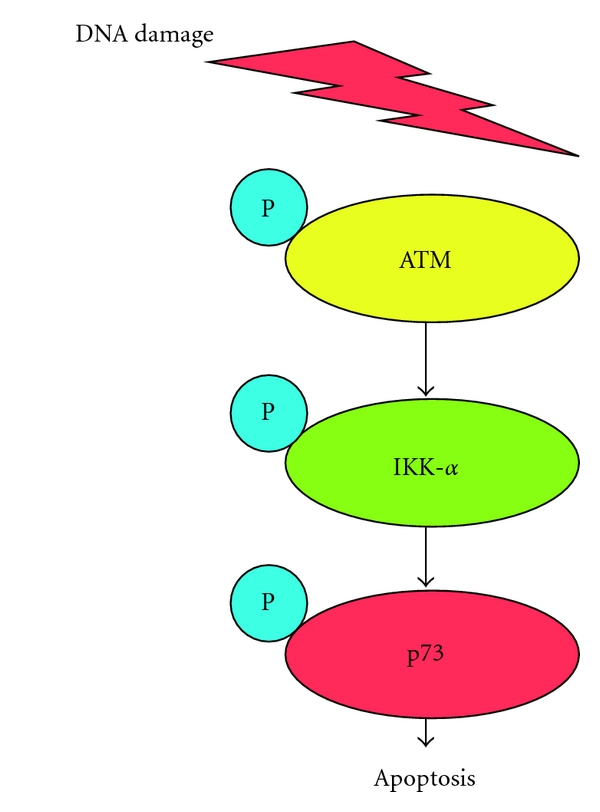
p73-dependent apoptotic cell death in response to DNA damage. Upon DNA damage, phospho-ATM phosphorylates IKK-α and promotes nuclear accumulation of IKK-α. IKK-α then enhances transcriptional as well as proapoptotic function of p73 in a p53-independent manner.
5. Phosphorylation-Dependent Inhibition of p73
Phosphorylation does not always act as an activation signal. Irwin et al. described that expression of p73 is regulated in a cell cycle-dependent manner [74]. Under their experimental conditions, p73 started to increase at the end of G1 phase, which was mediated by E2F1. Gaiddon et al. found that p73 interacts with cyclin/CDK complex through its cyclin recognition motif (CRM) [84]. p73 contains two CRM sequences located at NH2-terminal portion (149-KKL-151) and COOH-terminal region (515-RAL-517). Cyclin/CDK complex phosphorylated p73 at Thr-86 and p73 was most highly phosphorylated at Thr-86 in G2 as well as M phase. From the functional point of view, cyclin/CDK-mediated phosphorylation at Thr-86 reduced the transcriptional activity of p73. However, the biological significance of phospho-p73 in the regulation of cell cycle progression remains unclear.
During the extensive search for p73-binding partners by using a yeast-based two-hybrid screening procedure, we have identified PKA catalytic subunit β (PKA-Cβ) as a novel p73-binding protein [85]. p73 bound to PKA-Cβ in mammalian cells through its NH2-terminal portion (amino acid residues 63–130) and COOH-terminal region (amino acid residues 469–636). In vitro kinase reactions revealed that PKA-Cβ is able to phosphorylate NH2-terminal region of p73. Within this region, there exists a putative PKA recognition site (78-RAAS-82). Of note, PKA-Cβ inhibited p73-mediated transcriptional activation as well as proapoptotic activity following DNA damage in a phosphorylation-dependent manner.
Hck is one of the nonreceptor tyrosine kinase Src family [86–88]. Considering that Hck interacts with c-Abl and their interaction modulates each other's activity [89], Paliwal et al. investigated the possible role of Hck in the regulation of p73 [90]. According to their results, Hck was associated with p73 through its SH3 domain, and phosphorylated p73 at Tyr-28. Forced expression of Hck led to the stabilization of cytoplasmic p73 but not of nuclear p73 in a phosphorylation-dependent manner. Hck inhibited p73-mediated transcriptional as well as proapoptotic activity, which required the intact SH3 domain of Hck, suggesting that physical interaction between Hck and p73 plays an important role in the inhibition of p73.
Plk (Polo-like kinase) family is composed of four members including Plk1, Plk2, Plk3, and Plk4. Plk1 has been considered to be a positive cell cycle regulator [91–93], and also shown to be a negative regulator of p53 [94]. Recently, we have found for the first time that Plk1 attenuates transcriptional as well as proapoptotic function of p73 [53]. Based on our results, Plk1 was associated with p73 through NH2-terminal domain of p73 and catalytic domain of Plk1. Kinase-deficient Plk1 had a marginal effect on p73. Indeed, in vitro kinase reactions demonstrated that Plk1 phosphorylates p73 at Thr-27. Komatsu et al. revealed that Plk1 also suppresses another p53 family member, p63, through physical interaction and phosphorylation [95]. Plk1 phosphorylated p63 at Ser-52.
Plk3 has been shown to be closely involved in the regulation of DNA damage response through the direct interaction with p53 accompanied with an induction of its phosphorylation at Ser-20 [96, 97]. In contrast to Plk3/p53 interaction, we have demonstrated that Plk3 is associated with NH2-terminal region of p73 and inhibits p73-mediated transacriptional activation as well as proapoptotic activity [98]. In addition, Plk3 decreased the stability of p73. Since kinase-deficient Plk3 had an undetectable effect on p73, Plk3-mediated inhibition was regulated in a phosphorylation-dependent manner. In fact, Plk3 was able to phosphorylate certain amino acid residue(s) between 63 and 113 of p73. Thus, it is likely that Plk3 attenuates the inappropriate p73-mediated apoptotic cell death in response to DNA damage.
6. Acetylation-Dependent Regulation of p73
Like p53, p73 is also regulated by p300-mediated acetylation. Zeng et al. showed that histone acetyltransferase p300 binds to p73 through its NH2-terminal CH1 domain and NH2-terminal transactivation domain of p73 [99]. p300 stimulated p73-mediated transcriptional activation as well as proapoptotic function. Subsequent studies demonstrated that p73 is acetylated by p300 at Lys-321, Lys-327 and Lys-331 in response to DNA damage [100]. Furthermore, c-Abl was required for DNA damage-mediated acetylation of p73 and acetylated forms of p73 preferentially transactivated p53-target genes implicated in the induction of apoptotic cell death such as p53AIP1. Mantovani et al. found that prolyl isomerase Pin1 promotes the conformational change of p73, and enhances proapoptotic activity of p73 [66]. According to their results, c-Abl enhanced the phosphorylation-dependent interaction between Pin1 and p73, and thereby inducing the acetylation of p73 mediated by p300. Indeed, Pin1 greatly enhanced p73 acetylation by p300. In addition, forced expression of p300 resulted in a remarkable stabilization of p73. Strano et al. revealed that YAP1 promotes p73/p300 complex formation and potentiates p300-mediated acetylation of p73 [101]. Therefore, it is likely that p300-mediated acetylation of p73 reduces the ubiquitination levels of p73 by competition between acetylation and ubiquitination. In support with these observations, SIRT1 with an intrinsic deacetylase activity abrogated p73-mediated transacriptional activation and apoptotic cell death [102]. On the other hand, Zeng et al. demonstrated that p300 acts as a coactivator of p73 without inducing its acetylation [103]. Further studies should be required to address this issue.
7. Subcellular Localization of p73
Subcellular localization is one of the critical determinants for the activity of p73. Kim et al. identified HIPK2 as a novel p73-binding partner by using yeast-based two-hybrid screening [104]. HIPK2 has been shown to be Ser/Thr kinase which phosphorylates p53 at Ser-46 and enhance proapoptotic activity of p53 [105]. HIPK2 bound to p73 through its COOH-terminal region containing PEST sequence and the oligomerization domain of p73. Reporter assays revealed that HIPK2 enhances the transcriptional activity of p73. Intriguingly, HIPK2 colocalized with p73 in the nuclear body, suggesting that nuclear structures such as nuclear body might provide an important subnuclear locale for p73 function.
Previously, Kim et al. employed the yeast-based two-hybrid procedure to identify a novel p73-binding protein [106]. After the extensive screening, they identified amphiphysin IIb-1. Amphiphysin IIb-1 which contains NH2-terminal BAR domain and COOH-terminal SH3 domain, was one of the splicing variants of cytoplasmic amphiphysin IIb [107]. According to their results, amphiphysin IIb-1 interacted with p73 through its COOH-terminal SH3 domain and COOH-terminal region of p73 (amino acid residues 321–376). This interaction significantly inhibited p73-mediated transacriptional activation and apoptotic cell death. Subsequent studies demonstrated that amphiphysin IIb-1 promotes the cytoplasmic relocalization of p73 by masking the nuclear localization signal (NLS) of p73 (amino acid residues 338–348).
WWOX gene is located at 16q23.2-24.1, a genomic region with a high incidence of loss of heterozygosity (LOH) and homozygous deletions [108]. Ectopic expression of WWOX in breast cancer cells inhibited tumor growth in vivo [109], indicating that WWOX might be a candidate tumor-suppressor. Since WWOX contains two WW domains, Fabbri et al. performed the affinity assays to search for cellular proteins which could bind to WW domain of WWOX [110]. Finally, they found that PY motif of p73 interacts with WW domain of WWOX. In contrast to p73, WWOX failed to bind to p53 which lacks PY motif. Based on their results, WWOX caused the redistribution of p73 from nuclear compartment to the cytoplasm and repressed the transactivation function of p73. Of note, cytoplasmic p73 enhanced proapoptotic activity of WWOX in a transcription-independent manner. However, the precise molecular mechanisms behind the cytoplasmic p73-mediated activation of WWOX remains obscure.
8. Positive Regulation of p73 through Protein-Protein Interaction
Protein-protein interaction is one of the central events in a variety of cellular biological response. In this connection, p73 is regulated by protein-protein interactions.
By using the extensive pull-down assays, Strano et al. identified YAP1 as one of p73-binding proteins [111]. YAP1 was originally identified as an adaptor protein binding to SH3 domain of the Yes proto-oncogene product belonging to the Src family of protein tyrosine kinases. YAP1 bound to PY motif of p73 through its WW domain. YAP1 was not associated with p53. Introduction of the mutation into PY motif of p73 abrogated the interaction between p73 and YAP1, suggesting that structural integrity of PY motif is necessary for the interaction with YAP1. Forced expression of YAP1 enhanced transactivation ability of p73, indicating that YAP1 acts as a coactivator for p73.
As described previously [112], ASPP1 and ASPP2 belonging to ASPP family, bound to the central core sequence-specific DNA-binding domain of p53, and specifically stimulated the transcription of p53-target genes implicated in the induction of apoptotic cell death. Bergamaschi et al. examined the possible effect of ASPP1 and ASPP2 on p73 [113]. According to their results, ASPP1 and ASPP2 were able to interact with p73, and selectively enhanced p73-mediated transactivation of proapoptotic BAX, PIG3, and PUMA genes but not of p21WAF1 and MDM2 genes. Consistent with these observations, ASPP1 and ASPP2 enhanced proapoptotic function of p73 in response to DNA damage. It is likely that ASPP1 and ASPP2 are closely involved in the regulation of promoter selectivity of p73 (Figure 3).
Figure 3.
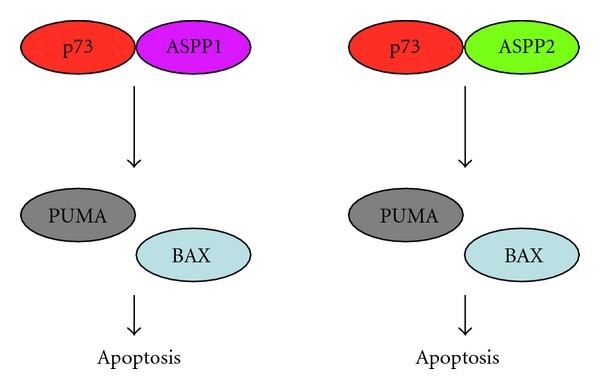
p73/ASPP complex preferentially induces the expression of proapoptotic BAX and PUMA. p73 forms a complex with ASPP1 or with ASPP2, and these transcriptional complexes selectively transactivate proapoptotic p73-target genes such as BAX and PUMA.
Jeong et al. identified p19ras as a novel p73-binding protein by using the yeast-based two-hybrid screening [114]. p19ras is an alternative splicing variant of c-H-ras, and lacks transforming potential [115]. Systematic deletion analysis revealed that p19ras interacts with p73 DNA-binding domain-containing region (amino acid residues 54–310). Of note, p19ras remarkably disrupted the complex formation of p73 with its negative regulator MDM2. As expected, p19ras blocked MDM2-mediated transcriptional repression of p73 and led to the activation of p73.
Similarly, we have employed the yeast-based two-hybrid screening to identify p73-binding proteins. After several rounds of extensive screening, we have obtained MM1 and RanBPM [116, 117]. MM1 has been shown to interact with c-Myc and suppress its transactivation function [118]. MM1 bound to COOH-terminal region of p73 (amino acid residues 551–636) in cell nucleus. In contrast to p73, MM1 failed to interact with p53. Forced expression of MM1 resulted in an enhancement of p73-mediated transcriptional as well as growth-suppressing activity. As expected, MM1 had an undetectable effect on p53. RanBPM was initially identified as a cellular protein which interacts with Ran nuclear-cytoplasmic transport protein [119, 120], and contains the putative SPRY domain which might be involved in protein-protein interactions. The region containing the canonical SPRY domain of RanBPM was associated with COOH-terminal region of p73 (amino acid residues 551–636). Like MM1, RanBPM did not bind to p53. RanBPM had an ability to stabilize p73 by inhibiting its ubiquitination. Finally, RanBPM enhanced transcriptional and proapoptotic activities of p73.
9. Negative Regulation of p73 through Protein-Protein Interactions
As described previously [121, 122], WT1 gene mapped at human chromosome 11p13 has been considered to be a candidate tumor-suppressor gene for Wilm's tumor. Subsequent studies demonstrated that WT1 contains zinc finger domain and acts as a sequence-specific transcription factor [123]. Scharnhorst et al. reported that WT1 interacts with p73 through its zinc finger domain [124]. WT1 had a negligible effect on the stability of p73 but strongly inhibited p73-mediated transcriptional activation.
Tax which is encoded by the human T-cell leukemia virus type I (HTLV-I), has been shown to be HTLV-I-associated malignant transformation [125, 126]. Indeed, Tax induced tumorigenesis and leukemogenesis in mice [127]. Although Tax had an ability to stabilize p73, p73-mediated transcriptional activation was significantly inhibited by Tax [128]. Since CBP with an intrinsic histone acetyl transferase activity acts as transcriptional coactivator for Tax and p73 [99, 129], Tax-mediated repression of p73 might be due to the competition between Tax and p73 for CBP. Thus, it is likely that inactivation of p73 by Tax contributes to HTLV-I-dependent leukemogenesis.
Hepatitis C virus (HCV) core protein has been shown to modulate p53 [130, 131]. Alisi et al. found that HCV core protein directly interacts with COOH-terminal portion of p73 (amino acid residues 321–353) [132]. Luciferase reporter assays demonstrated that HCV core protein enhances the p73-dependent luciferase activity driven by MDM2 promoter, whereas p73-dependent transactivation of BAX is significantly inhibited by HCV core protein. Consistent with these observations, HCV core protein almost completely blocked the ability of p73 to reduce cell viability.
It has been shown that adenovirus E1B, SV40 large T antigen and human papilloma virus E6 which inhibit p53, do not interact with p73 and do not affect p73 activity [133, 134]. Similarly, adenovirus E4orf6 protein with oncogenic potential bound to COOH-terminal region of p53 and inhibited transcriptional activity of p53 [135]. Higashino et al. reported that E4orf6 is also associated with COOH-terminal region of p73 and blocks p73-mediated transcriptional as well as proapoptotic activity [136]. Thus, it is possible that E4orf6-mediated inhibition of p53 and p73 is responsible for the enhanced tumorigenic potential of adenovirus. Additionally, these findings suggest that only a subset of viral oncoproteins interact with p73.
Nagatani et al. found that the CCAAT-binding transcription factor CTF2 is overexpressed in CDDP-resistant cells, suggesting that CTF2 is involved in the acquisition of drug-resistant phenotype of tumor cells [137]. Subsequent studies revealed that CTF2 interacts with central core sequence-specific DNA-binding domain of p73 (amino acid residues 228–312) [138]. According to their results, CTF2 strongly inhibited the sequence-specific DNA-binding activity of p73. Therefore, it is likely that CTF2-dependent inhibition of p73 contribute to the acquisition of drug-resistant phenotype of tumor cells.
Bcl-2 associated athanogene BAG-1 protected cells from cellular stress-mediated apoptotic cell death [139–141]. In accordance with these results, BAG-1 expression correlated with important clinical parameters of certain tumors [142]. Wang et al. reported that BAG-1 is associated with p73 through its intact BAG domain, and strongly attenuates transcriptional activity of p73 [143]. Knocking down of the endogenous BAG-1 resulted in a reactivation of p73. Additionally, BAG-1 reduced the stability of p73. Collectively, BAG-1-mediated inhibition of p73 might be one of the molecular mechanisms by which BAG-1 interfered with stress-induced apoptotic cell death.
TIP60 is a member of MYST family of histone acetyltransferase which is conserved from yeast to human [144]. It has been shown that TIP60 acts as a coactivator for p53 by inhibiting MDM2-mediated proteolytic degradation of p53 [145]. TIP60-mediated acetylation of p53 at Lys-120 had a critical role in the regulation of p53-dependent apoptotic response [146, 147]. Recently, Kim et al. described that, in contrast to p53, TIP60 strongly inhibits transcriptional as well as proapoptotic function of p73 [148]. TIP60 formed a ternary complex with p73, which was bridged by MDM2. It is likely that TIP60 recruits MDM2 onto p73 thereby inhibiting the activity of p73.
It is worth noting that human tumor-derived p53 mutants strongly inhibit p73 [30]. As described previously [149], forced expression of p53 mutant caused increased chemoresistance to anticancer drugs, suggesting that mutant p53 inactivates p73-induced apoptosis through the interaction with p73. Strano et al. found that mutant p53 interacts with p73 through its central core sequence-specific DNA-binding domain and p73 region containing central core sequence-specific DNA-binding domain and oligomerization domain [150]. Consistent with the previous observations [30], This interaction markedly suppressed transcriptional activity of p73. Since mutant p53 bound to the oligomerization domain of p73, homotetramer formation of p73 might be blocked by mutant p53. Alternatively, binding of mutant p53 to central core sequence-specific DNA-binding domain of p73 might attenuate its binding to p53/p73-responsive element. Sang et al. described that forced expression of p73 induces apoptotic cell death in human breast cancer-derived and chemo-resistant MDA-MB-436 cells bearing p53 mutation [151], indicating that the balance between the intracellular expression levels of mutant p53 and p73 is a critical determinant of chemosensitivity.
10. Future Perspective
As described in the present paper, a variety of cellular and viral proteins are involved in the positive and negative regulations of proapoptotic p73 through protein-protein interactions. From the clinical point of view, over 50% human tumors carry p53 mutations. In some cases, tumors bearing p53 mutations displayed chemoresistant phenotype. On the other hand, p73 was infrequently mutated in human tumors. Therefore, it is quite important to clarify the precise molecular mechanisms underlying p73-dependent apoptotic pathway in response to DNA damage. Furthermore, mutant p53 acts as a dominant-negative inhibitor toward p73 as well as p53 (Figure 4). In this connection, development of a novel strategy to overcome and/or eliminate the dominant-negative effect of mutant p53 on p73 and p53 is required for the effective treatment of tumors carrying p53 mutations.
Figure 4.

Dominant-negative behavior of mutant p53 toward wild-type p73 and p53. Mutant form of p53 binds to wild-type p73 as well as p53 and strongly inhibits their tumor suppressive function.
References
- 1.Kaghad M, Bonnet H, Yang A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90(4):809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 2.Yang A, Kaghad M, Wang Y, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Molecular Cell. 1998;2(3):305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 3.Osada M, Ohba M, Kawahara C, et al. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nature Medicine. 1998;4(7):839–843. doi: 10.1038/nm0798-839. [DOI] [PubMed] [Google Scholar]
- 4.Thanos CD, Bowie JU. p53 Family members p63 and p73 are SAM domain-containing proteins. Protein Science. 1999;8(8):1708–1710. doi: 10.1110/ps.8.8.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang A, McKeon F. p63 and p73: p53 mimics, menaces and more. Nature Reviews Molecular Cell Biology. 2000;1(3):199–207. doi: 10.1038/35043127. [DOI] [PubMed] [Google Scholar]
- 6.Melino G, De Laurenzi V, Vousden KH. p73: friend or foe in tumorigenesis. Nature Reviews Cancer. 2002;2(8):605–615. doi: 10.1038/nrc861. [DOI] [PubMed] [Google Scholar]
- 7.Ozaki T, Nakagawara A. p73, a sophisticated p53 family member in the cancer world. Cancer Science. 2005;96(11):729–737. doi: 10.1111/j.1349-7006.2005.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jost CA, Marin MC, Kaelin WG., Jr. p73 is a human p53-related protein that can induce apoptosis. Nature. 1997;389(6647):191–194. doi: 10.1038/38298. [DOI] [PubMed] [Google Scholar]
- 9.Rödicker F, Pützer BM. p73 is effective in p53-null pancreatic cancer cells resistant to wild-type TP53 gene replacement. Cancer Research. 2003;63(11):2737–2741. [PubMed] [Google Scholar]
- 10.Brodeur GM, Sekhon GS, Goldstein MN. Chromosomal aberrations in human neuroblastomas. Cancer. 1977;40(5):2256–2263. doi: 10.1002/1097-0142(197711)40:5<2256::aid-cncr2820400536>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 11.Balaban GB, Herlyn M, Clark WH, Jr., Nowell PC. Karyotypic evolution in human malignant melanoma. Cancer Genetics and Cytogenetics. 1986;19(1-2):113–122. doi: 10.1016/0165-4608(86)90378-x. [DOI] [PubMed] [Google Scholar]
- 12.Sozzi G, Bertoglio MG, Pilotti S, Rilke F, Pierotti MA, Della Porta G. Cytogenetic studies in primary and metastatic neuroendocrine Merkel cell carcinoma. Cancer Genetics and Cytogenetics. 1988;30(1):151–158. doi: 10.1016/0165-4608(88)90104-5. [DOI] [PubMed] [Google Scholar]
- 13.Dracopoli NC, Harnett P, Bale SJ, et al. Loss of alleles from the distal short arm of chromosome 1 occurs late in melanoma tumor progression. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(12):4614–4618. doi: 10.1073/pnas.86.12.4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ross RA, Spengler BA, Domenech C, Porubcin M, Rettig WJ, Biedler JL. Human neuroblastoma I-type cells are malignant neural crest stem cells. Cell Growth and Differentiation. 1995;6(4):449–456. [PubMed] [Google Scholar]
- 15.Hollstein M, Shomer B, Greenblatt M, et al. Somatic point mutations in the p53 gene of human tumors and cell lines: updated compilation. Nucleic Acids Research. 1996;24(1):141–146. doi: 10.1093/nar/24.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hollstein M, Hergenhahn M, Yang Q, Bartsch H, Wang Z-Q, Hainaut P. New approaches to understanding p53 gene tumor mutation spectra. Mutation Research. 1999;431(2):199–209. doi: 10.1016/s0027-5107(99)00162-1. [DOI] [PubMed] [Google Scholar]
- 17.Ikawa S, Nakagawara A, Ikawa Y. p53 family genes: structural comparison, expression and mutation. Cell Death and Differentiation. 1999;6(12):1154–1161. doi: 10.1038/sj.cdd.4400631. [DOI] [PubMed] [Google Scholar]
- 18.Yang A, Walker N, Bronson R, et al. p73-Deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. p73-Deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404(6773):99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- 19.Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356(6366):215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 20.Flores ER, Tsai KY, Crowley D, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416(6880):560–564. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- 21.Flores ER, Sengupta S, Miller JB, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7(4):363–373. doi: 10.1016/j.ccr.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 22.De Laurenzi V, Costanzo A, Barcaroli D, et al. Two new p73 splice variants, γ and δ, with different transcriptional activity. Journal of Experimental Medicine. 1998;188(9):1763–1768. doi: 10.1084/jem.188.9.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Laurenzi V, Catani MV, Terrinoni A, et al. Additional complexity in p73: induction by mitogens in lymphoid cells and identification of two new splicing variants ε and ζ . Cell Death and Differentiation. 1999;6(5):389–390. doi: 10.1038/sj.cdd.4400521. [DOI] [PubMed] [Google Scholar]
- 24.Ueda Y, Hijikata M, Takagi S, Chiba T, Shimotohno K. New p73 variants with altered C-terminal structures have varied transcriptional activities. Oncogene. 1999;18(35):4993–4998. doi: 10.1038/sj.onc.1202817. [DOI] [PubMed] [Google Scholar]
- 25.Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289(5477):304–306. doi: 10.1126/science.289.5477.304. [DOI] [PubMed] [Google Scholar]
- 26.Ishimoto O, Kawahara C, Enjo K, Obinata M, Nukiwa T, Ikawa S. Possible oncogenic potential of ΔNp73: a newly identified isoform of human p73. Cancer Research. 2002;62(3):636–641. [PubMed] [Google Scholar]
- 27.Stiewe T, Zimmermann S, Frilling A, Esche H, Pützer BM. Transactivation-deficient δTA-p73 acts as an oncogene. Cancer Research. 2002;62(13):3598–3602. [PubMed] [Google Scholar]
- 28.Casciano I, Mazzocco K, Boni L, et al. Expression of ΔNp73 is a molecular marker for adverse outcome in neuroblastoma patients. Cell Death and Differentiation. 2002;9(3):246–251. doi: 10.1038/sj.cdd.4400993. [DOI] [PubMed] [Google Scholar]
- 29.Zaika AI, Slade N, Erster SH, et al. ΔNp73, a dominant-negative inhibitor of wild-type p53 and TAp73, is up-regulated in human tumors. Journal of Experimental Medicine. 2002;196(6):765–780. doi: 10.1084/jem.20020179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Como CJ, Gaiddon C, Prives C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Molecular and Cellular Biology. 1999;19(2):1438–1449. doi: 10.1128/mcb.19.2.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu G, Nozell S, Xiao H, Chen X. ΔNp73β is active in transactivation and growth suppression. Molecular and Cellular Biology. 2004;24(2):487–501. doi: 10.1128/MCB.24.2.487-501.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG., Jr. Chemosensitivity linked to p73 function. Cancer Cell. 2003;3(4):403–410. doi: 10.1016/s1535-6108(03)00078-3. [DOI] [PubMed] [Google Scholar]
- 33.Grob TJ, Novak U, Maisse C, et al. Human ΔNp73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death and Differentiation. 2001;8(12):1213–1223. doi: 10.1038/sj.cdd.4400962. [DOI] [PubMed] [Google Scholar]
- 34.Nakagawa T, Takahashi M, Ozaki T, et al. Autoinhibitory regulation of p73 by ΔNp73 to modulate cell survival and death through a p73-specific target element within the ΔNp73 promoter. Molecular and Cellular Biology. 2002;22(8):2575–2585. doi: 10.1128/MCB.22.8.2575-2585.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stiewe T, Theseling CC, Pützer BM. Transactivation-deficient ΔTA-p73 inhibits p53 by direct competition for DNA binding. Implications for tumorigenesis. The Journal of Biological Chemistry. 2002;277(16):14177–14185. doi: 10.1074/jbc.M200480200. [DOI] [PubMed] [Google Scholar]
- 36.Maisse C, Munarriz E, Barcaroli D, Melino G, De Laurenzi V. DNA damage induces the rapid and selective degradation of the ΔNp73 isoform, allowing apoptosis to occur. Cell Death and Differentiation. 2004;11(6):685–687. doi: 10.1038/sj.cdd.4401376. [DOI] [PubMed] [Google Scholar]
- 37.Sayan BS, Yang AL, Conforti F, et al. Differential control of TAp73 and DeltaNp73 protein stability by the ring finger ubiquitin ligase PIR2. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12877–12882. doi: 10.1073/pnas.0911828107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohtsuka T, Ryu H, Minamishima YA, Ryo A, Lee SW. Modulation of p53 and p73 levels by cyclin G: implication of a negative feedback regulation. Oncogene. 2003;22(11):1678–1687. doi: 10.1038/sj.onc.1206306. [DOI] [PubMed] [Google Scholar]
- 39.Lee C-W, La Thangue NB. Promoter specificity and stability control of the p53-related protein p73. Oncogene. 1999;18(29):4171–4181. doi: 10.1038/sj.onc.1202793. [DOI] [PubMed] [Google Scholar]
- 40.Bálint E, Bates S, Vousden KH. Mdm2 binds p73α without targeting degradation. Oncogene. 1999;18(27):3923–3929. doi: 10.1038/sj.onc.1202781. [DOI] [PubMed] [Google Scholar]
- 41.Dobbelstein M, Wienzek S, König C, Roth J. Inactivation of the p53-homologue p73 by the mdm2-oncoprotein. Oncogene. 1999;18(12):2101–2106. doi: 10.1038/sj.onc.1202512. [DOI] [PubMed] [Google Scholar]
- 42.Ongkeko WM, Wang XQ, Siu WY, et al. MDM2 and MDMX bind and stabilize the p53-related protein p73. Current Biology. 1999;9(15):829–832. doi: 10.1016/s0960-9822(99)80367-4. [DOI] [PubMed] [Google Scholar]
- 43.Zeng X, Chen L, Jost CA, et al. MDM2 suppresses p73 function without promoting p73 degradation. Molecular and Cellular Biology. 1999;19(5):3257–3266. doi: 10.1128/mcb.19.5.3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gu J, Nie L, Wiederschain D, Yuan Z-M. Identification of p53 sequence elements that are required for MDM2-mediated nuclear export. Molecular and Cellular Biology. 2001;21(24):8533–8546. doi: 10.1128/MCB.21.24.8533-8546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rossi M, De Laurenzi V, Munarriz E, et al. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO Journal. 2005;24(4):836–848. doi: 10.1038/sj.emboj.7600444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asher G, Tsvetkov P, Kahana C, Shaul Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes and Development. 2005;19(3):316–321. doi: 10.1101/gad.319905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levy D, Adamovich Y, Reuven N, Shaul Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death and Differentiation. 2007;14(4):743–751. doi: 10.1038/sj.cdd.4402063. [DOI] [PubMed] [Google Scholar]
- 48.Oberst A, Malatesta M, Aqeilan RI, et al. The Nedd4-binding partner 1 (N4BP1) protein is an inhibitor of the E3 ligase Itch. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(27):11280–11285. doi: 10.1073/pnas.0701773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ponting C, Schultz J, Bork P. SPRY domains in ryanodine receptors (Ca2+-release channels) Trends in Biochemical Sciences. 1997;22(6):193–194. doi: 10.1016/s0968-0004(97)01049-9. [DOI] [PubMed] [Google Scholar]
- 50.Peschiaroli A, Scialpi F, Bernassola F, Pagano M, Melino G. The F-box protein FBXO45 promotes the proteasome-dependent degradation of p73. Oncogene. 2009;28(35):3157–3166. doi: 10.1038/onc.2009.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang C, Yuan X, Yue L, et al. PIASy interacts with p73alpha and regulates cell cycle in HEK293 cells. Cellular Immunology. 2010;263:235–240. doi: 10.1016/j.cellimm.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 52.Miyazaki K, Ozaki T, Kato C, et al. A novel HECT-type E3 ubiquitin ligase, NEDL2, stabilizes p73 and enhances its transcriptional activity. Biochemical and Biophysical Research Communications. 2003;308(1):106–113. doi: 10.1016/s0006-291x(03)01347-0. [DOI] [PubMed] [Google Scholar]
- 53.Koida N, Ozaki T, Yamamoto H, et al. Inhibitory role of Plk1 in the regulation of p73-dependent apoptosis through physical interaction and phosphorylation. The Journal of Biological Chemistry. 2008;283(13):8555–8563. doi: 10.1074/jbc.M710608200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Munarriz E, Bano D, Sayan AE, Rossi M, Melino G, Nicotera P. Calpain cleavage regulates the protein stability of p73. Biochemical and Biophysical Research Communications. 2005;333(3):954–960. doi: 10.1016/j.bbrc.2005.05.188. [DOI] [PubMed] [Google Scholar]
- 55.Yuan Z-M, Huang Y, Whang Y, et al. Role for c-Abl tyrosine kinase in growth arrest response to DNA damage. Nature. 1996;382(6588):272–274. doi: 10.1038/382272a0. [DOI] [PubMed] [Google Scholar]
- 56.Kharbanda S, Ren R, Pandey P, et al. Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature. 1995;376(6543):785–788. doi: 10.1038/376785a0. [DOI] [PubMed] [Google Scholar]
- 57.Liu Z-G, Baskaran R, Lea-Chou ET, et al. Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature. 1996;384(6606):273–276. doi: 10.1038/384273a0. [DOI] [PubMed] [Google Scholar]
- 58.Baskaran R, Wood LD, Whitaker LL, et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature. 1997;387(6632):516–519. doi: 10.1038/387516a0. [DOI] [PubMed] [Google Scholar]
- 59.Shafman T, Khanna KK, Kedar P, et al. Interaction between ATM protein and c-Abl in response to DNA damage. Nature. 1997;387(6632):520–523. doi: 10.1038/387520a0. [DOI] [PubMed] [Google Scholar]
- 60.Gong J, Costanzo A, Yang H-Q, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399(6738):806–809. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- 61.Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73α and their collaboration to induce apoptosis. Nature. 1999;399(6738):809–812. doi: 10.1038/21697. [DOI] [PubMed] [Google Scholar]
- 62.Yuan Z-M, Shioya H, Ishiko T, et al. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature. 1999;399(6738):814–817. doi: 10.1038/21704. [DOI] [PubMed] [Google Scholar]
- 63.Ben-Yehoyada M, Ben-Dor I, Shaul Y. c-Abl tyrosine kinase selectively regulates p73 nuclear matrix association. The Journal of Biological Chemistry. 2003;278(36):34475–34482. doi: 10.1074/jbc.M301051200. [DOI] [PubMed] [Google Scholar]
- 64.Pandey P, Raingeaud J, Kaneki M, et al. Activation of p38 mitogen-activated protein kinase by c-Abl-dependent and -independent mechanisms. The Journal of Biological Chemistry. 1996;271(39):23775–23779. doi: 10.1074/jbc.271.39.23775. [DOI] [PubMed] [Google Scholar]
- 65.Sanchez-Prieto R, Sanchez-Arevalo VJ, Servitja J-M, Gutkind JS. Regulation of p73 by c-Abl through the p38 MAP kinase pathway. Oncogene. 2002;21(6):974–979. doi: 10.1038/sj.onc.1205134. [DOI] [PubMed] [Google Scholar]
- 66.Mantovani F, Piazza S, Gostissa M, et al. Pin1 links the activities of c-Abl and p300 in regulating p73 function. Molecular Cell. 2004;14(5):625–636. doi: 10.1016/j.molcel.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 67.Yuan Z-M, Utsugisawa T, Ishiko T, et al. Activation of protein kinase Cδ by the c-Abl tyrosine kinase in response to ionizing radiation. Oncogene. 1998;16(13):1643–1648. doi: 10.1038/sj.onc.1201698. [DOI] [PubMed] [Google Scholar]
- 68.Ren J, Datta R, Shioya H, et al. p73β is regulated by protein kinase Cδ catalytic fragment generated in the apoptotic response to DNA damage. The Journal of Biological Chemistry. 2002;277(37):33758–33765. doi: 10.1074/jbc.M110667200. [DOI] [PubMed] [Google Scholar]
- 69.Walworth N, Davey S, Beach D. Fission yeast chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature. 1993;363(6427):368–371. doi: 10.1038/363368a0. [DOI] [PubMed] [Google Scholar]
- 70.Murakami H, Okayama H. A kinase from fission yeast responsible for blocking mitosis in S phase. Nature. 1995;374(6525):817–819. doi: 10.1038/374817a0. [DOI] [PubMed] [Google Scholar]
- 71.Gonzalez S, Prives C, Cordon-Cardo C. p73α regulation by Chk1 in Response to DNA Damage. Molecular and Cellular Biology. 2003;23(22):8161–8171. doi: 10.1128/MCB.23.22.8161-8171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72.Urist M, Tanaka T, Poyurovsky MV, Prives C. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes and Development. 2004;18(24):3041–3054. doi: 10.1101/gad.1221004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stiewe T, Putzer BM. Role of the p53-homologue p73 in E2F1-induced apoptosis. Nature Genetics. 2000;26(4):464–469. doi: 10.1038/82617. [DOI] [PubMed] [Google Scholar]
- 74.Irwin M, Marin MC, Phillips AC, et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature. 2000;407(6804):645–648. doi: 10.1038/35036614. [DOI] [PubMed] [Google Scholar]
- 75.Lissy NA, Davis PK, Irwin M, Kaelin WG, Dowdy SF. A common E2F-1 and p73 pathway mediates cell death induced by TCR activation. Nature. 2000;407(6804):642–645. doi: 10.1038/35036608. [DOI] [PubMed] [Google Scholar]
- 76.Kharbanda S, Pandey P, Ren R, Mayer B, Zon L, Kufe D. c-Abl activation regulates induction of the SEK1/stress-activated protein kinase pathway in the cellular response to 1-β-D-arabinofuranosylcytosine. The Journal of Biological Chemistry. 1995;270(51):30278–30281. doi: 10.1074/jbc.270.51.30278. [DOI] [PubMed] [Google Scholar]
- 77.Zanke BW, Boudreau K, Rubie E, et al. The stress-activated protein kinase pathway mediates cell death following injury induced by cis-platinum, UV irradiation or heat. Current Biology. 1996;6(5):606–613. doi: 10.1016/s0960-9822(02)00547-x. [DOI] [PubMed] [Google Scholar]
- 78.Jones EV, Dickman MJ, Whitmarsh AJ. Regulation of p73-mediated apoptosis by c-Jun N-terminal kinase. Biochemical Journal. 2007;405(3):617–623. doi: 10.1042/BJ20061778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takada N, Ozaki T, Ichimiya S, Todo S, Nakagawara A. Identification of a transactivation activity in the COOH-terminal region of p73 which is impaired in the naturally occurring mutants found in human neuroblastomas. Cancer Research. 1999;59(12):2810–2814. [PubMed] [Google Scholar]
- 80.Nyman U, Vlachos P, Cascante A, Hermanson O, Zhivotovsky B, Joseph B. Protein kinase C-dependent phosphorylation regulates the cell cycle-inhibitory function of the p73 carboxy terminus transactivation domain. Molecular and Cellular Biology. 2009;29(7):1814–1825. doi: 10.1128/MCB.00585-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annual Review of Immunology. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 82.Furuya K, Ozaki T, Hanamoto T, et al. Stabilization of p73 by nuclear IκB kinase-α mediates cisplatin-induced apoptosis. The Journal of Biological Chemistry. 2007;282(25):18365–18378. doi: 10.1074/jbc.M610522200. [DOI] [PubMed] [Google Scholar]
- 83.Yoshida K, Ozaki T, Furuya K, et al. ATM-dependent nuclear accumulation of IKK-α plays an important role in the regulation of p73-mediated apoptosis in response to cisplatin. Oncogene. 2008;27(8):1183–1188. doi: 10.1038/sj.onc.1210722. [DOI] [PubMed] [Google Scholar]
- 84.Gaiddon C, Lokshin M, Gross I, et al. Cyclin-dependent kinases phosphorylate p73 at Threonine 86 in a cell cycle-dependent manner and negatively regulate p73. The Journal of Biological Chemistry. 2003;278(30):27421–27431. doi: 10.1074/jbc.M300251200. [DOI] [PubMed] [Google Scholar]
- 85.Hanamoto T, Ozaki T, Furuya K, et al. Identification of protein kinase A catalytic subunit β as a novel binding partner of p73 and regulation of p73 function. The Journal of Biological Chemistry. 2005;280(17):16665–16675. doi: 10.1074/jbc.M414323200. [DOI] [PubMed] [Google Scholar]
- 86.Ernst M, Gearing DP, Dunn AR. Functional and biochemical association of Hck with the LIF/IL-6 receptor signal transducing subunit gp130 in embryonic stem cells. EMBO Journal. 1994;13(7):1574–1584. doi: 10.1002/j.1460-2075.1994.tb06420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Quintrell N, Lebo R, Varmus H, et al. Identification of a human gene (HCK) that encodes a protein-tyrosine kinase and is expressed in hemopoietic cells. Molecular and Cellular Biology. 1987;7(6):2267–2275. doi: 10.1128/mcb.7.6.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ziegler SF, Marth JD, Lewis DB, Perlmutter RM. Novel protein-tyrosine kinase gene (hck) preferentially expressed in cells of hematopoietic origin. Molecular and Cellular Biology. 1987;7(6):2276–2285. doi: 10.1128/mcb.7.6.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tanis KQ, Veach D, Duewel HS, Bornmann WG, Koleske AJ. Two distinct phosphorylation pathways have additive effects on Abl family kinase activation. Molecular and Cellular Biology. 2003;23(11):3884–3896. doi: 10.1128/MCB.23.11.3884-3896.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Paliwal P, Radha V, Swarup G. Regulation of p73 by Hck through kinase-dependent and independent mechanisms. BMC Molecular Biology. 2007;8, article 45 doi: 10.1186/1471-2199-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barr FA, Silljé HHW, Nigg EA. Polo-like kinases and the orchestration of cell division. Nature Reviews Molecular Cell Biology. 2004;5(6):429–440. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- 92.Xie S, Xie B, Lee MY, Dai W. Regulation of cell cycle checkpoints by polo-like kinases. Oncogene. 2005;24(2):277–286. doi: 10.1038/sj.onc.1208218. [DOI] [PubMed] [Google Scholar]
- 93.Dai W. Polo-like kinases, an introduction. Oncogene. 2005;24(2):214–216. doi: 10.1038/sj.onc.1208270. [DOI] [PubMed] [Google Scholar]
- 94.Ando K, Ozaki T, Yamamoto H, et al. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. The Journal of Biological Chemistry. 2004;279(24):25549–25561. doi: 10.1074/jbc.M314182200. [DOI] [PubMed] [Google Scholar]
- 95.Komatsu S, Takenobu H, Ozaki T, et al. Plk1 regulates liver tumor cell death by phosphorylation of TAp63. Oncogene. 2009;28(41):3631–3641. doi: 10.1038/onc.2009.216. [DOI] [PubMed] [Google Scholar]
- 96.Xie S, Wu H, Wang Q, et al. Plk3 functionally links DNA damage to cell cycle arrest and apoptosis at least in part via the p53 pathway. The Journal of Biological Chemistry. 2001;276(46):43305–43312. doi: 10.1074/jbc.M106050200. [DOI] [PubMed] [Google Scholar]
- 97.Bahassi EM, Conn CW, Myer DL, et al. Mammalian Polo-like kinase 3 (Plk3) is a multifunctional protein involved in stress response pathways. Oncogene. 2002;21(43):6633–6640. doi: 10.1038/sj.onc.1205850. [DOI] [PubMed] [Google Scholar]
- 98.Sang M, Ando K, Okoshi R, et al. Plk3 inhibits pro-apoptotic activity of p73 through physical interaction and phosphorylation. Genes to Cells. 2009;14(7):775–788. doi: 10.1111/j.1365-2443.2009.01309.x. [DOI] [PubMed] [Google Scholar]
- 99.Zeng X, Li X, Miller A, et al. The N-terminal domain of p73 interacts with the CH1 domain of p300/CREB binding protein and mediates transcriptional activation and apoptosis. Molecular and Cellular Biology. 2000;20(4):1299–1310. doi: 10.1128/mcb.20.4.1299-1310.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Costanzo A, Merlo P, Pediconi N, et al. DNA damage-dependent acetylation of p73 dictates the selective activation of apoptotic target genes. Molecular Cell. 2002;9(1):175–186. doi: 10.1016/s1097-2765(02)00431-8. [DOI] [PubMed] [Google Scholar]
- 101.Strano S, Monti O, Pediconi N, et al. The transcriptional coactivator yes-associated protein drives p73 gene-target specificity in response to DNA damage. Molecular Cell. 2005;18(4):447–459. doi: 10.1016/j.molcel.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 102.Dai JM, Wang ZY, Sun DC, Lin RX, Wang SQ. SIRT1 interacts with p73 and suppresses p73-dependent transcriptional activity. Journal of Cellular Physiology. 2007;210(1):161–166. doi: 10.1002/jcp.20831. [DOI] [PubMed] [Google Scholar]
- 103.Zeng X, Lee H, Zhang Q, Lu H. p300 does not require its acetylase activity to stimulate p73 function. The Journal of Biological Chemistry. 2001;276(1):48–52. doi: 10.1074/jbc.C000722200. [DOI] [PubMed] [Google Scholar]
- 104.Kim E-J, Park J-S, Um S-J. Identification and characterization of HIPK2 interacting with p73 and modulating functions of the p53 family in vivo. The Journal of Biological Chemistry. 2002;277(35):32020–32028. doi: 10.1074/jbc.M200153200. [DOI] [PubMed] [Google Scholar]
- 105.D’Orazi G, Cecchinelli B, Bruno T, et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nature Cell Biology. 2002;4(1):11–19. doi: 10.1038/ncb714. [DOI] [PubMed] [Google Scholar]
- 106.Kim K-C, Kim T-S, Kang K-H, Choi K-H. Amphiphysin IIb-1, a novel splicing variant of amphiphysin II, regulates p73β function through protein-protein interactions. Oncogene. 2001;20(46):6689–6699. doi: 10.1038/sj.onc.1204839. [DOI] [PubMed] [Google Scholar]
- 107.Butler MH, David C, Ochoa G-C, et al. Amphiphysin II (SH3p9; BIN1), a member of the amphiphysin/Rvs family, is concentrated in the cortical cytomatrix of axon initial segments and nodes of ranvier in brain and around T tubules in skeletal muscle. Journal of Cell Biology. 1997;137(6):1355–1367. doi: 10.1083/jcb.137.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ried K, Finnis M, Hobson L, et al. Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Human Molecular Genetics. 2000;9(11):1651–1663. doi: 10.1093/hmg/9.11.1651. [DOI] [PubMed] [Google Scholar]
- 109.Bednarek AK, Keck-Waggoner CL, Daniel RL, et al. WWOX, the FRA16D gene, behaves as a suppressor of tumor growth. Cancer Research. 2001;61(22):8068–8073. [PubMed] [Google Scholar]
- 110.Fabbri M, Iliopoulos D, Trapasso F, et al. WWOX gene restoration prevents lung cancer growth in vitro and in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4041–4046. doi: 10.1073/pnas.0505485102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Strano S, Munarriz E, Rossi M, et al. Physical Interaction with Yes-associated Protein Enhances p73 Transcriptional Activity. The Journal of Biological Chemistry. 2001;276(18):15164–15173. doi: 10.1074/jbc.M010484200. [DOI] [PubMed] [Google Scholar]
- 112.Samuels-Lev Y, O’Connor DJ, Bergamaschi D, et al. ASPP proteins specifically stimulate the apoptotic function of p53. Molecular Cell. 2001;8(4):781–794. doi: 10.1016/s1097-2765(01)00367-7. [DOI] [PubMed] [Google Scholar]
- 113.Bergamaschi D, Samuels Y, Jin B, Duraisingham S, Crook T, Lu X. ASPP1 and ASPP2: common activators of p53 family members. Molecular and Cellular Biology. 2004;24(3):1341–1350. doi: 10.1128/MCB.24.3.1341-1350.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jeong M-H, Bae J, Kim W-H, et al. p19ras interacts with and activates p73 by involving the MDM2 protein. The Journal of Biological Chemistry. 2006;281(13):8707–8715. doi: 10.1074/jbc.M513853200. [DOI] [PubMed] [Google Scholar]
- 115.Cohen JB, Broz SD, Levinson AD. Expression of the H-ras proto-oncogene is controlled by alternative splicing. Cell. 1989;58(3):461–472. doi: 10.1016/0092-8674(89)90427-3. [DOI] [PubMed] [Google Scholar]
- 116.Watanabe K-I, Ozaki T, Nakagawa T, et al. Physical interaction of p73 with c-Myc and MM1, a c-Myc-binding protein, and modulation of the p73 function. The Journal of Biological Chemistry. 2002;277(17):15113–15123. doi: 10.1074/jbc.M111281200. [DOI] [PubMed] [Google Scholar]
- 117.Kramer S, Ozaki T, Miyazaki K, Kato C, Hanamoto T, Nakagawara A. Protein stability and function of p73 are modulated by a physical interaction with RanBPM in mammalian cultured cells. Oncogene. 2005;24(5):938–944. doi: 10.1038/sj.onc.1208257. [DOI] [PubMed] [Google Scholar]
- 118.Mori K, Maeda Y, Kitaura H, Taira T, Iguchi-Ariga SMM, Ariga H. MM-1, a novel c-Myc-associating protein that represses transcriptional activity of c-Myc. The Journal of Biological Chemistry. 1998;273(45):29794–29800. doi: 10.1074/jbc.273.45.29794. [DOI] [PubMed] [Google Scholar]
- 119.Nakamura M, Masuda H, Horii J, et al. When overexpressed, a novel centrosomal protein, RanBPM, causes ectopic microtubule nucleation similar to γ-tubulin. Journal of Cell Biology. 1998;143(4):1041–1052. doi: 10.1083/jcb.143.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nishitani H, Hirose E, Uchimura Y, et al. Full-sized RanBPM cDNA encodes a protein possessing a long stretch of proline and glutamine within the N-terminal region, comprising a large protein complex. Gene. 2001;272(1-2):25–33. doi: 10.1016/s0378-1119(01)00553-4. [DOI] [PubMed] [Google Scholar]
- 121.Pritchard-Jones K, Fleming S, Davidson D, et al. The candidate Wilms’ tumour gene is involved in genitourinary development. Nature. 1990;346(6280):194–197. doi: 10.1038/346194a0. [DOI] [PubMed] [Google Scholar]
- 122.Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60(3):509–520. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- 123.Menke AL, Van Der Eb AJ, Jochemsen AG. The Wilms’ tumor 1 gene: oncogene or tumor suppressor gene? International Review of Cytology. 1998;181:151–212. doi: 10.1016/s0074-7696(08)60418-0. [DOI] [PubMed] [Google Scholar]
- 124.Scharnhorst V, Dekker P, Van Der Eb AJ, Jochemsen AG. Physical interaction between Wilms tumor 1 and p73 proteins modulates their functions. The Journal of Biological Chemistry. 2000;275(14):10202–10211. doi: 10.1074/jbc.275.14.10202. [DOI] [PubMed] [Google Scholar]
- 125.Grassmann R, Berchtold S, Radant I, et al. Role of human T-cell leukemia virus type 1 X region proteins in immortalization of primary human lymphocytes in culture. Journal of Virology. 1992;66(7):4570–4575. doi: 10.1128/jvi.66.7.4570-4575.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Smith MR, Greene WC. Type I human T cell leukemia virus tax protein transforms rat fibroblasts through the cyclic adenosine monophosphate response element binding protein/activating transcription factor pathway. Journal of Clinical Investigation. 1991;88(3):1038–1042. doi: 10.1172/JCI115364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Grossman WJ, Kimata JT, Wong F-H, Zutter M, Ley TJ, Ratner L. Development of leukemia in mice transgenic for the tax gene of human T- cell leukemia virus type I. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(4):1057–1061. doi: 10.1073/pnas.92.4.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lemasson I, Nyborg JK. Human T-cell leukemia virus type I tax repression of p73beta is mediated through competition for the C/H1 domain of CBP. The Journal of Biological Chemistry. 2001;276(19):15720–15727. doi: 10.1074/jbc.M100131200. [DOI] [PubMed] [Google Scholar]
- 129.Van Orden K, Giebler HA, Lemasson I, Gonzales M, Nyborg JK. Binding of p53 to the KIX domain of CREB binding protein. A potential link to human T-cell leukemia virus, type I-associated leukemogenesis. The Journal of Biological Chemistry. 1999;274(37):26321–26328. doi: 10.1074/jbc.274.37.26321. [DOI] [PubMed] [Google Scholar]
- 130.Ray RB, Meyer K, Ray R. Suppression of apoptotic cell death by hepatitis C virus core protein. Virology. 1996;226(2):176–182. doi: 10.1006/viro.1996.0644. [DOI] [PubMed] [Google Scholar]
- 131.Otsuka M, Kato N, Lan K-H, et al. Hepatitis C virus core protein enhances p53 function through augmentation of DNA binding affinity and transcriptional ability. The Journal of Biological Chemistry. 2000;275(44):34122–34130. doi: 10.1074/jbc.M000578200. [DOI] [PubMed] [Google Scholar]
- 132.Alisi A, Giambartolomei S, Cupelli F, et al. Physical and functional interaction between HCV core protein and the different p73 isoforms. Oncogene. 2003;22(17):2573–2580. doi: 10.1038/sj.onc.1206333. [DOI] [PubMed] [Google Scholar]
- 133.Roth J, König C, Wienzek S, Weigel S, Ristea S, Dobbelstein M. Inactivation of p53 but not p73 by adenovirus type 5 E1B 55-kilodalton and E4 34-kilodalton oncoproteins. Journal of Virology. 1998;72(11):8510–8516. doi: 10.1128/jvi.72.11.8510-8516.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Marin MC, Jost CA, Irwin MS, DeCaprio JA, Caput D, Kaelin WG., Jr. Viral oncoproteins discriminate between p53 and the p53 homolog p73. Molecular and Cellular Biology. 1998;18(11):6316–6324. doi: 10.1128/mcb.18.11.6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dobner T, Horikoshi N, Rubenwolf S, Shenk T. Blockage by adenovirus E4orf6 of transcriptional activation by the p53 tumor suppressor. Science. 1996;272(5267):1470–1473. doi: 10.1126/science.272.5267.1470. [DOI] [PubMed] [Google Scholar]
- 136.Higashino F, Pipas JM, Shenk T. Adenovirus E4orf6 oncoprotein modulates the function of the p53-related protein, p73. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(26):15683–15687. doi: 10.1073/pnas.95.26.15683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nagatani G, Nomoto M, Takano H, et al. Transcriptional activation of the human HMG1 gene in cisplatin-resistant human cancer cells. Cancer Research. 2001;61(4):1592–1597. [PubMed] [Google Scholar]
- 138.Uramoto H, Izumi H, Nagatani G, et al. Physical interaction of tumour suppressor p53/p73 with CCAAT-binding transcription factor 2 (CTF2) and differential regulation of human high-mobility group 1 (HMG1) gene expression. Biochemical Journal. 2003;371(2):301–310. doi: 10.1042/BJ20021646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Alberti S, Esser C, Höhfeld J. BAG-1—a nucleotide exchange factor of Hsc70 with multiple cellular functions. Cell Stress and Chaperones. 2003;8(3):225–231. doi: 10.1379/1466-1268(2003)008<0225:bnefoh>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Townsend PA, Cutress RI, Sharp A, Brimmell M, Packham G. BAG-1: a multifunctional regulator of cell growth and survival. Biochimica et Biophysica Acta. 2003;1603(2):83–98. doi: 10.1016/s0304-419x(03)00002-7. [DOI] [PubMed] [Google Scholar]
- 141.Townsend PA, Stephanou A, Packham G, Latchman DS. BAG-1: a multi-functional pro-survival molecule. International Journal of Biochemistry and Cell Biology. 2005;37(2):251–259. doi: 10.1016/j.biocel.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 142.Tang S-C, Shaheta N, Chernenko G, Khalifa M, Wang X. Expression of BAG-1 in invasive breast carcinomas. Journal of Clinical Oncology. 1999;17(6):1710–1719. doi: 10.1200/JCO.1999.17.6.1710. [DOI] [PubMed] [Google Scholar]
- 143.Wang X-H, O’Connor D, Brimmell M, Packham G. The BAG-1 cochaperone is a negative regulator of p73-dependent transcription. British Journal of Cancer. 2009;100(8):1347–1357. doi: 10.1038/sj.bjc.6604985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yang X-J. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Research. 2004;32(3):959–976. doi: 10.1093/nar/gkh252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Legube G, Linares LK, Tyteca S, et al. Role of the histone acetyl transferase Tip60 in the p53 pathway. The Journal of Biological Chemistry. 2004;279(43):44825–44833. doi: 10.1074/jbc.M407478200. [DOI] [PubMed] [Google Scholar]
- 146.Sykes SM, Mellert HS, Holbert MA, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Molecular Cell. 2006;24(6):841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Molecular Cell. 2006;24(6):827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 148.Kim J-W, Song PI, Jeong M-H, et al. TIP60 represses transcriptional activity of p73β via an MDM2-bridged ternary complex. The Journal of Biological Chemistry. 2008;283(29):20077–20086. doi: 10.1074/jbc.M800161200. [DOI] [PubMed] [Google Scholar]
- 149.Blandino G, Levine AJ, Oren M. Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene. 1999;18(2):477–485. doi: 10.1038/sj.onc.1202314. [DOI] [PubMed] [Google Scholar]
- 150.Strano S, Munarriz E, Rossi M, et al. Physical and functional interaction between p53 mutants and different isoforms of p73. The Journal of Biological Chemistry. 2000;275(38):29503–29512. doi: 10.1074/jbc.M003360200. [DOI] [PubMed] [Google Scholar]
- 151.Sang M, Li Y, Ozaki T, et al. p73-dependent induction of 14-3-3σ increases the chemo-sensitivity of drug-resistant human breast cancers. Biochemical and Biophysical Research Communications. 2006;347(1):327–333. doi: 10.1016/j.bbrc.2006.06.079. [DOI] [PubMed] [Google Scholar]