

Abstract
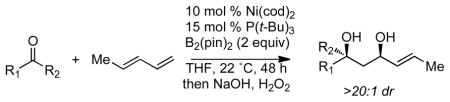
In the presence of catalytic Ni(cod)2 and P(t-Bu)3, ketones, dienes, and B2(pin)2 undergo a stereoselective multicomponent coupling reaction. Upon oxidation, the reaction furnishes 1,3-diols as the major reaction product.
Tertiary alcohols are common motifs in natural products and in medicinally active agents. Indeed, roughly 20% of the top fifty pharmaceuticals bear tertiary alcohols or their derivatives.1 While construction of these functional groups is readily accomplished by nucleophilic additions to ketones, such reactions can be challenging, particularly when stereocontrol is a required element.2 Recent studies in our laboratory have focused on the development of stereoselective borylative aldehyde-diene coupling reactions (Scheme 1).3,4,5,6 These reactions occur with exceptionally high levels of diastereoinduction when aldehydes are employed as substrates and it was of interest to ascertain whether these processes might also apply to ketone electrophiles and to determine whether high levels of stereocontrol could be maintained.7 In this report, we show that borylative ketone-diene coupling reactions can be accomplished in high yields and with excellent levels of diastereocontrol. Importantly, this reaction extends to aryl alkyl ketones as well as to dialkyl ketones. Also of note, this reaction occurs in a predictable fashion, yet with regioselection that is distinct from related aldehyde-diene coupling reactions.
Scheme 1.

Borylative Diene-Carbonyl Coupling
To initiate studies in ketone-diene coupling reactions, acetophenone was treated with (E)-1,3-pentadiene under conditions similar to those employed for aldehyde coupling reactions (10 mol % Ni(cod)2, 15 mol % P(t-Bu)3, 2 equiv B2(pin)2) and the reaction mixture was treated to oxidative work-up. As depicted in Scheme 2 (eq. 1), this reaction occurred in good yield and >20:1 diastereoselectivity. Surprisingly, the reaction of acetophenone followed a different regiochemical course compared to the analogous reaction with benzaldehyde. As depicted in Scheme 2 (eq. 2), when benzaldehyde was subjected to similar reaction conditions, 1,5-diol 2 was produced, not the 1,3-diol 1 that predominates with acetophenone. Not only is the hydroxyl-positioning different between the two products, but the diene is also incorporated into the product with opposite connectivity in the case of the ketone relative to the aldehyde electrophile.
Scheme 2.

Borylative Coupling of Acetophenone Compared to Benzaldehyde
To learn whether the reactivity observed with acetophenone is unique to this substrate, a number of other ketone electrophiles were employed in the coupling reaction. This survey of the reaction revealed that many other ketones are processed in a similar manner as acetophenone. Importantly, a variety of functional groups and substitution patterns are tolerated in the ketone electrophile. As depicted in Scheme 3, halogen substitution in the form of chloro and fluoro groups is accommodated in the reaction (compounds 3, 4, 5, 7) as is the presence of both electron-donating (compounds 10, 11) and electron-withdrawing substituents (compound 8). In all cases surveyed, >20:1 syn:anti diastereoselectivity was observed in the reaction products.
Scheme 3.

Borylative Coupling of Methyl Ketonesa
(a) Stereoselectivity determined by 1H NMR analysis. Yields refer to isolated yield of purified material; value is an average of two or more experiments.
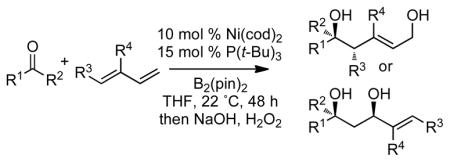
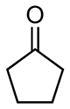
Whereas aromatic methyl ketones were found to react with good yield and selectivity providing the 1,3 diol regioselectively, reactions of aliphatic ketones delivered the regioisomeric 1,5 diol. For example, 4-phenyl-2-butanone (entry 1, Table 1) underwent smooth coupling with pentadiene but, in contrast to reactions of aromatic ketones, was converted to the derived 1,5-diol in a manner similar to that observed for reactions of benzaldehyde (Scheme 2, eq. 2). Suspecting that the steric encumbrance surrounding the reacting carbonyl may determine the regiochemical outcome, we examined constrained cyclic ketones and found that these also deliver the 1,5-diol product (entries 2–4, Table 1). It should be noted that the product of entry 4 appears to arise by preferred equatorial attachment of the diene to the carbonyl carbon of the substrate. Lastly, isoprene was found to react in a regioselective manner as well.
Table 1.
Borylative Coupling of Ketones and Dienesa
 | |||||
|---|---|---|---|---|---|
| entry | ketone | diene | product | dr | % yield |
| 1 |
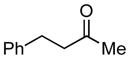
|
|
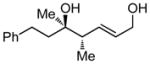 12 |
7:1 | 69 |
| 2 |
|
|
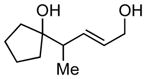 13 |
N/A | 56 |
| 3 |
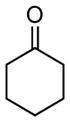
|
|
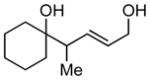 14 |
N/A | 52 |
| 4 |
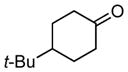
|
|
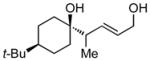 15 |
>20:1 | 53 |
| 5 |
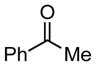
|
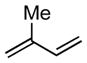
|
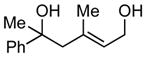 16 |
N/A | 39 |
Stereoselectivity determined by 1H NMR analysis. Yields refer to isolated yield of purified material; value is an average of two or more experiments.
Diene-carbonyl coupling reactions have been extensively studied by Tamaru and Mori, both of whom executed these transformations under reducing conditions with silane, organozinc reductants, and organoborane reductants.4,7 Related alkylative reactions are known with organoboronate reagents.7j,8 These processes are thought to occur by conversion of the diene and aldehyde to metallacyclic intermediates. The identity of this intermediate was secured by Ogoshi and Kurosawa who isolated such structures from stoichiometric reactions of Ni(cod)2, PCy3, a 1,3-diene, and benzaldehyde.9 Considering the similar reaction conditions, it is considered likely that such mechanisms also operate in the borylative carbonyl-diene coupling reactions and these can be used to understand the regiochemical course of the coupling processes with hindered and non-hindered carbonyl substrates. As depicted in Scheme 4, with non-hindered electrophiles, reaction with pentadiene may occur to give nickellacycle 17. Subsequent σ-bond metathesis with B2(pin)2 would then deliver π-allyl 18, a compound that may engage in π-σ-π isomerization prior to reductive elimination to afford 19. With more hindered carbonyls, it is tenable that steric effects retard C-C bond formation with the substituted terminus of the diene and, instead, the less hindered end of the diene adds to the carbonyl. Subsequent σ-bond metathesis delivers a π-allyl 20, which undergoes reductive elimination to furnish the 1,3-diol. It is conceivable that with a substituted π-allyl the π-σ-π isomerization is retarded relative to reductive elimination and the geometry of the initial σ-bond metathesis product dictates the regiochemical outcome of the reaction.
Scheme 4.

In conclusion, we have demonstrated that the borylative carbonyl-diene coupling reaction can extend to ketone substrates and that it occurs in a regio- and stereoselective fashion. While the outcome of the reaction is distinct from that which is observed with aldehyde substrates, it is consistent with known features of this class of transformations. Further studies on the utility of these processes are in progress.
Supplementary Material
Acknowledgments
AllyChem is acknowleged for a generous donation of B2(pin)2. This work was supported by the NIH (GM 59417). HYC is grateful for a Rodin Fellowship and an ACS Organic Division Fellowship.
Footnotes
Supporting Information Available. Complete experimental procedures and characterization data (1H and 13C NMR, IR, and mass spectrometry). This material is free of charge via the internet at http://pubs.acs.org.
References
- 1.See http://cbc.arizona.edu/njardarson/group/top-pharmaceuticals-poster
- 2.Reviews: Hatano M, Ishihara K. Synthesis. 2008:1647.Shibasaki M, Kanai M. Chem Rev. 2008;108:2853. doi: 10.1021/cr078340r.Garcia C, Martin VS. Curr Org Chem. 2006;10:1849.
- 3.(a) Cho HY, Morken JP. J Am Chem Soc. 2008;130:16140. doi: 10.1021/ja806113v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cho HY, Morken JP. J Am Chem Soc. 2010;132:7576. doi: 10.1021/ja101513d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For related reductive or alkylative Ni-catalyzed diene carbonyl couplings, see: Kimura M, Ezoe A, Shibata K, Tamaru Y. J Am Chem Soc. 1998;120:4033.Takimoto M, Hiraga Y, Sato Y, Mori M. Tetrahedron Lett. 1998;39:4543.Sato Y, Takanashi T, Hoshiba M, Mori M. Tetrahedron Lett. 1998;39:5579.Sato Y, Saito N, Mori M. Tetrahedron. 1998;54:1153.Sato Y, Saito N, Mori M. J Am Chem Soc. 2000;122:2371.Kimura M, Shibata K, Koudahashi Y, Tamaru Y. Tetrahedron Lett. 2000;41:6789.Shibata K, Kimura M, Kojima K, Tanaka S, Tamaru Y. J Organomet Chem. 2001;624:348.Sato Y, Sawaki R, Mori M. Organometallics. 2001;20:5510.Shibata K, Kimura M, Shimizu M, Tamaru Y. Org Lett. 2001;3:2181. doi: 10.1021/ol0100879.Sato Y, Saito N, Mori M. J Org Chem. 2002;67:9310. doi: 10.1021/jo020438c.Sato Y, Sawaki R, Saito N, Mori M. J Org Chem. 2002;67:656. doi: 10.1021/jo0106086.Loh TP, Song HY, Zhou Y. Org Lett. 2002;4:2715. doi: 10.1021/ol026216i.Takimoto M, Kajima Y, Sato Y, Mori M. J Org Chem. 2005;70:8605. doi: 10.1021/jo051283m.Hirashita T, Kambe S, Tsuji H, Araki S. Chem Commun. 2006:2595. doi: 10.1039/b603597c.
- 5.For reviews and selected recent examples of related nickel-catalyzed couplings, see: Montgomery J. Angew Chem Int Ed. 2004;43:3890. doi: 10.1002/anie.200300634.Kimura M, Tamaru Y. Top Curr Chem. 2007;279:173.Moslin RM, Miller-Moslin K, Jamison TF. Chem Commun. 2007:4441. doi: 10.1039/b707737h.Ho CY, Jamison TF. Angew Chem, Int Ed. 2007;46:782. doi: 10.1002/anie.200603907.Baxter RD, Montgomery J. J Am Chem Soc. 2008;130:9662. doi: 10.1021/ja803774s.Ng SS, Ho CY, Schleicher KD, Jamison TF. Pure Appl Chem. 2008;80:929. doi: 10.1351/pac200880050929.Liu P, McCarren P, Cheong PHY, Jamison TF, Houk KN. J Am Chem Soc. 2010;132:2050. doi: 10.1021/ja909562y.Malik HA, Sormunen GJ, Montgomery J. J Am Chem Soc. 2010;132:6304. doi: 10.1021/ja102262v.Baxter RD, Montgomery J. J Am Chem Soc. 2011;133:5728. doi: 10.1021/ja200867d.
- 6.For aligned studies in diene-carbonyl couplings, see: Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:6340. doi: 10.1021/ja802001b.Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:14891. doi: 10.1021/ja805722e.Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514. doi: 10.1021/ja808857w.Skucas E, Zbieg JR, Krische MJ. J Am Chem Soc. 2009;131:5054. doi: 10.1021/ja900827p.Han SB, Han H, Krische MJ. J Am Chem Soc. 2010;132:1760. doi: 10.1021/ja9097675.Han SB, Gao X, Krische MJ. J Am Chem Soc. 2010;132:9153. doi: 10.1021/ja103299f.
- 7.For diene-carbonyl couplings that employ ketones, see: Sato Y, Takimoto M, Hayashi K, Katsuhara T, Takagi K, Mori M. J Am Chem Soc. 1994;116:9771.Sato Y, Takimoto M, Mori M. Tetrahedron Lett. 1996;37:887.Kimura M, Fujimatsu H, Ezoe A, Shibata K, Shimizu M, Matsumoto S, Tamaru Y. Angew Chem, Int Ed. 1999;38:397. doi: 10.1002/(SICI)1521-3773(19990201)38:3<397::AID-ANIE397>3.0.CO;2-Y.Kimura M, Matsuo S, Shibata K, Tamaru Y. Angew Chem, Int Ed. 1999;38:3386. doi: 10.1002/(sici)1521-3773(19991115)38:22<3386::aid-anie3386>3.0.co;2-w.Sato Y, Takimoto M, Mori M. J Am Chem Soc. 2000;122:1624.Ezoe A, Kimura M, Inoue T, Mori M, Tamaru Y. Angew Chem Int Ed. 2002;41:2784. doi: 10.1002/1521-3773(20020802)41:15<2784::AID-ANIE2784>3.0.CO;2-A.Kimura M, Kojima K, Tamaru Y. Synthesis. 2004:3089.Kimura M, Ezoe A, Mori M, Tamaru Y. J Am Chem Soc. 2005;127:201. doi: 10.1021/ja0469030.Kimura M, Ezoe A, Mori M, Iwata K, Tamaru Y. J Am Chem Soc. 2006;128:8559. doi: 10.1021/ja0608904.Saito N, Yamazaki T, Sato Y. Chem Lett. 2009;38:594.
- 8.Saito N, Yamazaki T, Sato Y. Tetrahedron Lett. 2008;49:5073. [Google Scholar]
- 9.Ogoshi S, Tonomori K, Oka M, Kurosawa H. J Am Chem Soc. 2006;128:7077. doi: 10.1021/ja060580l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.