

Whether enzymatic or structural, the three-dimensional shape into which a protein folds is a critical determinant of all aspects of its function. Proteins fold from an initial chain of amino acids into complex helices and beta-pleated sheets based on intramolecular interactions that favor exposed hydrophilic residues and buried greasy hydrophobic side chains. The 2011 Albert Lasker Basic Medical Research Award recognizes the work of two scientists, Franz-Ulrich Hartl (Max Planck Institute of Biochemistry) and Arthur L. Horwich (Yale University) (Figure 1), whose collaborative and independent work led to a new understanding of how cells can control this complex process. Their findings not only reveal secrets of basic protein chemistry, but could also have a dramatic impact on the treatment of the many diseases in which protein folding goes awry.
Figure 1. Franz-Ulrich Hartl (left) and Arthur Horwich (right) are the recipients of the 2011 Albert Lasker Basic Medical Research Award.

Insight into the mystery of how protein folding occurs was in part revealed in the early 1960s, when Christian Anfinsen, working at the NIH, demonstrated that a denatured protein called ribonuclease could spontaneously refold in a test tube. In doing so, he showed that the amino acid sequence of a protein provided the necessary information to allow it to fold properly, dictating its three-dimensional shape and thus its activity (1–3). Following the establishment of this “Anfinsen dogma,” most biologists assumed that all proteins folded spontaneously as they were synthesized on the ribosome. As Hartl recalled in a recent interview with the JCI, “For many years, cell biologists just were not interested in protein folding; it was the domain of biophysicists and biochemists who studied the spontaneous reactions in the test tube.”
Biologists returning to the fold: the making of two careers
A decade after his discovery of spontaneous refolding of ribonuclease, Anfinsen received the Nobel Prize in Chemistry. Though he was just a budding bench scientist at the time, the event made a distinct impression on Arthur Horwich. He recalls, “We talked about that in the lab for days, and it just blew me away, because it was the most amazing experiment. I mean, how did he have the chutzpa to expect that a denatured protein would refold and become active again?”
After pediatric residency training at Yale, Horwich went to the Salk Institute to study transforming viruses with some of the leaders in the field, Walter Eckhart and Tony Hunter. His experiences there exposed him to the rapidly developing technology that was revolutionizing the field of molecular biology; as he told the JCI, “I actually watched Tony Hunter discover tyrosine phosphorylation firsthand!” In the early 1980s, Horwich returned to Yale and joined the lab of Leon Rosenberg, hoping to apply these new tools to answer lingering questions he’d encountered during his medical training. “I thought I could go after one of my old pediatric problems — pick a lethal disease and really get to a molecular understanding of it with recombinant DNA tools. It was good timing, because suddenly we could do things like isolate coding sequences and identify human mutations.”
The family of diseases he focused on were related to the urea cycle: specifically, deficiency of an enzyme called ornithine carbamoyltransferase (OTC), which results in a toxic and potentially lethal buildup of ammonia. Horwich’s research revealed that OTC was synthesized in the cytosol but required a special sequence to be “ticketed” to its functional destination, the mitochondria. Furthermore, that targeting sequence needed to be cleaved in order for OTC to become functional (Figure 2 and ref. 4).
Figure 2. Protein import into the mitochondria.

Specific N-terminal sequences are required to tag proteins for mitochondrial import. Unfolded polypeptide chains are transported through the outer and inner membranes. The targeting sequence is cleaved, and HSP60 then facilitates refolding of the imported polypeptide within the matrix. Figure adapted with permission from Sinauer Associates (25).
In Germany, Hartl was also starting to get interested in understanding the process of protein transport within the cell. After completing his own medical and scientific training at the University of Heidelberg in the mid-1980s, he joined the laboratory of Walter Neupert at the University of Munich. The group was using biochemical tools to study how proteins are imported into the mitochondria from the cytosol. In the course of this work, Hartl became aware of research demonstrating that proteins had to be unfolded for membrane translocation (5–7). As he relates, “It occurred to me then that we could then ask how these proteins refold after membrane translocation, once they’re inside the mitochondria. It seemed like we had a situation where we could really study how proteins fold in a cellular context.”
With the growing understanding that proteins must unfold to enter mitochondria and then refold to become active, researchers on both sides of the Atlantic now questioned whether that refolding could occur spontaneously. An almost heretical idea was brewing, and as Horwich describes, “We thought, what if there’s such a thing as a ‘folding machine’ on the inside of mitochondria?” Horwich expressed human OTC in yeast, and his team began a genetic screen for mutations that blocked mitochondrial import of this protein. He realized that these mutant yeast strains might allow him to identify a folding enzyme. If a strain was deficient in a factor required for refolding, it would be able to import the OTC precursor into the mitochondria and cleave the targeting sequence, but once inside the organelle, the enzyme would not fold or become functional. Screening for this combination of characteristics yielded a mutant they dubbed mitochondrial import factor 4 (Mif4).
A fortuitous phone call in 1988 altered the course of Horwich’s and Hartl’s careers. Hartl’s mentor Neupert, aware that Horwich was using yeast genetic tools to study mitochondrial protein import, reached out to him and suggested a meeting to discuss collaboration. Horwich jumped at the opportunity; as he described to the JCI, “I said, ‘I’ll be right there!’ And really, as soon as I could get a plane ticket to Munich, I went.”
In Munich, Horwich presented his work on the Mif4 mutant to Neupert and his group. They were surprised but skeptical of the findings, suggesting that the results might not necessarily demonstrate the existence of a folding enzyme. More simply, proteins might become physically stuck in the membrane while being imported into the mitochondria of mutant yeast. In that situation, the targeting peptide could be cleaved and the protein would not become enzymatically active. Hartl volunteered to do the biochemistry experiments that could determine whether proteins actually entered the mitochondria in the Mif4 mutant. As Hartl explains, “Our skill sets were very complimentary, because I was focused and trained in the biochemical aspects of a complex reaction like mitochondrial import and the subsequent folding and processing of the protein.”
Within just a few weeks, the results were in: proteins were indeed imported into the mitochondrial matrix in the mutant yeast, but then failed to refold. Hartl followed up this discovery by investigating the mitochondrial import of additional proteins, and the cumulative results supported the idea that this collaborative team had discovered a true “foldase” (8).
The next step was to identify the gene that encoded this new protein-folding enzyme. Horwich and his team screened a yeast genetic library and identified a gene that rescued the Mif4 phenotype. The gene turned out to be identical to a heat shock protein (HSP) (8) that had previously been discovered by Richard Hallberg and Thomas McMullin called HSP60 (9) and an ortholog of the bacterial factor known as GroEL. These proteins defined a subfamily of molecular chaperones called chaperonins, and Horwich, Hartl and their teams had now demonstrated that chaperonins were critical for protein folding after mitochondrial import. As Hartl explains, “Anfinsen was right that the three-dimensional structure of a protein is encoded in its sequence, and our discoveries don’t change that. What the chaperonins really do is allow a protein chain to realize its potential to fold under cellular conditions, because those are critically different from the test tube situation, and side reactions like protein aggregation are strongly favored.” In addition, chaperones allow the folding of large proteins, for which there are kinetic barriers to reaching the properly folded state (10).
In the years that followed their initial discovery, Hartl and Horwich continued to work out the details of this protein folding system. Importantly, they discovered that the folding reaction required the use of cellular energy (11), and they developed a system that allowed the reconstitution of the chaperonin-assisted protein folding process in vitro (12). They established that their foldase, HSP60, could also act to hold proteins in an unfolded state and control their localization within the mitochondria (13). Horwich’s group, in collaboration with the lab of Paul Sigler, used X-ray crystallography to reveal that chaperonins had a characteristic double-ring structure; the interior surface of these rings bound to hydrophobic surfaces to help prevent protein aggregation (Figure 3 and refs. 14, 15). Hartl and his colleagues found that chaperone-mediated folding actually required a series of steps and the action of multiple chaperone proteins that function almost as a relay team, passing off partially folded proteins (16).
Figure 3. Chaperonin structure allows protein folding within mitochondria.

(A) The bacterial chaperonin complex, based on X-ray crystallography. The GroEL rings (gold) are capped by another complex component, GroES (white). The open ring of the bottom cavity exposes at its terminal hydrophobic amino acid side chains — these capture non-native protein through their own exposed hydrophobic side chains. Such binding prevents non-native protein from aggregating. The top cavity is the site of protein folding. A protein released after initial binding in an open ring can fold in this space, which has hydrophilic walls, in solitary confinement, without the possibility of aggregation. This cage-like structure has been termed the “Anfinsen cage.” Figure reproduced with permission from Nature (15). (B) Schematic of protein folding within the GroEL-GroES complex. Image courtesy of F.-U. Hartl.
Protein folding in disease
When proteins misfold, they lose their ability to perform their normal function. This loss of function can result from single amino acid changes that disrupt normal intramolecular interactions, as is the case in cystic fibrosis. An additional level of disorder occurs because misfolded proteins tend to aggregate, in part because they expose hydrophobic residues (17). Those aggregates can form highly ordered structures called amyloid (Figure 4), which are implicated in many neurological diseases, including Alzheimer disease and Parkinson disease, and also in type 2 diabetes (18). Misfolding is also implicated in the pathogenesis of prion diseases (reviewed in ref. 10). The aggregates that form can be insoluble, and cells sequester and deposit them in specific subcellular compartments (19). It is not yet completely understood why these amyloid aggregates are toxic, though recent evidence suggests that it may be because they interact with and disrupt the function of other normal proteins (20).
Figure 4. Amyloid deposition in the brain of an Alzheimer disease patient.
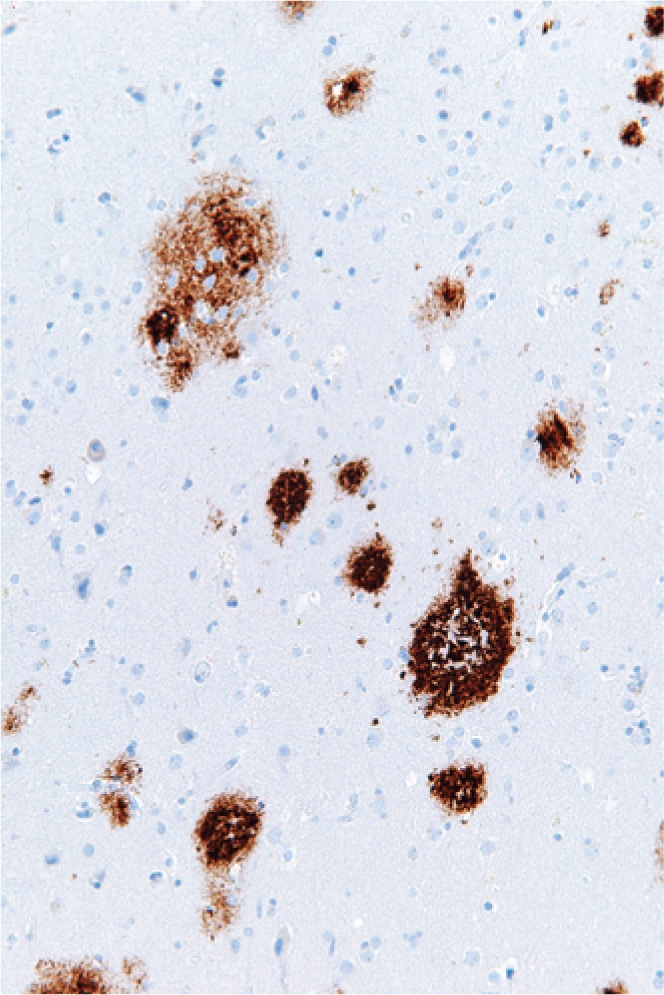
Immunohistochemistry for amyloid (brown) in cortex. Image source: Wikimedia Commons.
The folding machinery discovered by Horwich and Hartl is efficient, but as Hartl explains, research (21) suggests that “As our cells age, the normal capacity of the chaperone system declines, and this is probably one reason why many of these diseases are age dependent — because the chaperones are no longer as active as they were when were young”. Although the timing of this decline is not understood, the delay in onset may also be related to the proteins themselves; according to Horwich, “One thing we know is that most molecular chaperones function by recognizing greasy, exposed hydrophobic surfaces — it could be that most amyloidogenic proteins don’t expose enough hydrophobic residues to attract chaperones, so they misfold and aggregate without being corrected.”
The buildup of aggregates can be remedied by endogenous systems that disaggregate and refold proteins (22). Both Horwich and Hartl are now interested in how an improved understanding of protein folding could be applied to treat diseases that result from protein misfolding. One straightforward approach is to increase the level of chaperone action; chaperones are regulated by cellular stress responses, and Hartl suggests that tapping into this system might be clinically useful. He explains, “if we could find a way to mimic a cellular stress response in the absence of actual stress, chaperones might be upregulated that could resolve disease protein aggregates. Chaperones bind promiscuously to misfolded proteins, so one attractive aspect of this therapeutic strategy — if it works — is that one could potentially interfere with a number of these diseases based on the fact that the essential aggregation phenomenon is very similar between them.” Indeed, some evidence suggests that activating a cellular stress response is effective in preventing neural degeneration in cell and animal models of protein-folding diseases (23, 24). An alternative might be to use small molecules as chemical chaperones that stabilize the normal folding conformations, or to better understand and manipulate cellular protein clearance mechanisms (25).
Although progress has been made in this area, a great deal of work remains before it might be translated to clinical benefit. According to Horwich, “I’m really hopeful that we’ll get there, but we still have to develop the tools to bring our science to a point where we can treat patients. There’s nothing so humbling as being at a patient’s bedside and realizing you don’t know what is wrong. I remember when I was in residency and we had a patient with amyloidosis, and when I asked what that was, my attending described amyloid merely as ‘sticky stuff.’ Now we really know what it is, but we still don’t really have a way of treating the disease.”
Science from two perspectives
Although Hartl and Horwich utilized biochemistry and genetic studies in single-celled organisms, their findings set the stage for a new understanding of human physiology and disease. Their remarkably productive collaboration was perhaps surprising, given that these two — at least initially — approached their work in very different ways. Horwich set out with the intention of applying molecular biology tools to a medical question, but Hartl admitted, “At the time that I began these studies, I didn’t think of any medical potential applications of it at all. It was purely curiosity driven.” Thus the applicability of his work to medicine — and its recognition by the Lasker award committee — has been particularly gratifying. Said Hartl, “I think it’s important that scientists are given the chance to find out what they think is interesting — to add a new piece to the puzzle of how nature and biology work.”
Both Hartl and Horwich maintain active laboratories and continue to investigate the mechanism of protein folding and its impact on human disease. They expressed surprise and humility at the honor of receiving the Albert Lasker Basic Medical Research Award. Said Horwich, “At the lab bench, I’m basically in my sandbox. To be recognized for that work is just incredible.”
References
- 1.Haber E, Anfinsen CB. Regeneration of enzyme activity by air oxidation of reduced subtilisin-modified ribonuclease. J Biol Chem. 1961;236(2):422–424. [PubMed] [Google Scholar]
- 2.Potts JT, Jr, Young DM, Anfinsen CB. Reconstitution of fully active RNase by carboxypeptidase degraded RNase S peptide. J Biol Chem. 1963;238:2593–2594. [PubMed] [Google Scholar]
- 3.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181(96):223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 4.Horwich AL, Kalousek F, Fenton WA, Furtak K, Pollock RA, Rosenberg LE. The ornithine transcarbamylase leader peptide directs mitochondrial import through both its midportion structure and net positive charge. J Cell Biol. 1987;105(2):669–677. doi: 10.1083/jcb.105.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen WJ, Douglas MG. The role of protein structure in the mitochondrial import pathway. Unfolding of mitochondrially bound precursors is required for membrane translocation. J Biol Chem. 1987;262(32):15605–15609. [PubMed] [Google Scholar]
- 6.Pfanner N, Tropschug M, Neupert W. Mitochondrial protein import: nucleoside triphosphates are involved in conferring import-competence to precursors. Cell. 1987;49(6):815–823. doi: 10.1016/0092-8674(87)90619-2. [DOI] [PubMed] [Google Scholar]
- 7.Eilers M, Hwang S, Schatz G. Unfolding and refolding of a purified precursor protein during import into isolated mitochondria. EMBO J. 1988;7(4):1139–1145. doi: 10.1002/j.1460-2075.1988.tb02923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng MY, et al. Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature. 1989;337(6208):620–625. doi: 10.1038/337620a0. [DOI] [PubMed] [Google Scholar]
- 9.McMullin TW, Hallberg RL. A highly evolutionarily conserved mitochondrial protein is structurally related to the protein encoded by the Escherichia coli groEL gene. Mol Cell Biol. 1988;8(1):371–380. doi: 10.1128/mcb.8.1.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horwich A. Protein aggregation in disease: a role for folding intermediates forming specific multimeric interactions. J Clin Invest. 2002;110(9):1221–1232. doi: 10.1172/JCI16781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ostermann J, Horwich AL, Neupert W, Hartl FU. Protein folding in mitochondria requires complex formation with hsp60 and ATP hydrolysis. Nature. 1989;341(6238):125–130. doi: 10.1038/341125a0. [DOI] [PubMed] [Google Scholar]
- 12.Martin J, Langer T, Boteva R, Schramel A, Horwich AL, Hartl FU. Chaperonin–mediated protein folding at the surface of groEL through a ‘molten globule’-like intermediate. Nature. 1991;352(6330):36–42. doi: 10.1038/352036a0. [DOI] [PubMed] [Google Scholar]
- 13.Koll H, et al. Antifolding activity of hsp60 couples protein import into the mitochondrial matrix with export to the intermembrane space. Cell. 1992;68(6):1163–1175. doi: 10.1016/0092-8674(92)90086-r. [DOI] [PubMed] [Google Scholar]
- 14.Braig K, et al. The crystal structure of the bacterial chaperonin GroEL at 2.8 A. Nature. 1994;371(6498):578–586. doi: 10.1038/371578a0. [DOI] [PubMed] [Google Scholar]
- 15.Xu A, Horwich AL, Sigler PB. The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex. Nature. 1997;388(6644):741–750. doi: 10.1038/41944. [DOI] [PubMed] [Google Scholar]
- 16.Langer T, Lu C, Echols H, Flanagan J, Hayer MK, Hartl FU. Successive action of DnaK, DnaJ and GroEL along the pathway of chaperone-mediated protein folding. Nature. 1992;356(6371):683–689. doi: 10.1038/356683a0. [DOI] [PubMed] [Google Scholar]
- 17.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16(6):574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 18.Broadley SA, Hartl FU. The role of molecular chaperones in human misfolding diseases. FEBS Lett. 2009;583(16):2647–2653. doi: 10.1016/j.febslet.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 19.Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11(11):777–788. doi: 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- 20.Vabulas RM, Hartl FU. Aberrant protein interactions in amyloid disease. Cell Cycle. 2011;10(10):1512–1513. doi: 10.4161/cc.10.10.15432. [DOI] [PubMed] [Google Scholar]
- 21.Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311(5766):1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- 22.Sittler A, et al. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum Mol Genet. 2001;10(12):1307–1315. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- 23.Fujikake N, Nagai Y, Popiel HA, Okamoto Y, Yamaguchi M, Toda T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem. 2008;283(38):26188–26197. doi: 10.1074/jbc.M710521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rochet JC. Novel therapeutic strategies for the treatment of protein misfolding diseases. Expert Rev Mol Med. 2007;9(17):1–34. doi: 10.1017/S1462399407000385. [DOI] [PubMed] [Google Scholar]
- 25. Cooper, GM.The Cell: A Molecular Approach . 2nd edition. Sunderland, Massachusetts, USA: Sinauer Associates; 2000. [Google Scholar]