

Background: The mechanism underlying nuclear transport of RNA polymerase II (RNAPII) is unclear.
Results: Npa3 is required for nuclear localization of RNAPII and binds it in a GTP-dependent manner.
Conclusion: RNAPII nuclear import takes place via an unconventional pathway involving Npa3 and a cycle of GTP-dependent Npa3-RNAPII binding and release.
Significance: Learning the mechanism of RNAPII nuclear import is crucial for understanding the regulation of gene expression.
Keywords: Gene Expression, Gene Transcription, GTPase, Nuclear Transport, RNA Polymerase II
Abstract
We identified XAB1 in a proteomic screen for factors that interact with human RNA polymerase II (RNAPII). Because XAB1 has a conserved Saccharomyces cerevisiae homologue called Npa3, yeast genetics and biochemical analysis were used to dissect the significance of the interaction. Degron-dependent Npa3 depletion resulted in genome-wide transcription decreases, correlating with a loss of RNAPII from genes as measured by chromatin immunoprecipitation. Surprisingly, however, transcription in vitro was unaffected by Npa3, suggesting that it affects a process that is not required for transcription in yeast extracts. Indeed, Npa3 depletion in vivo affects nuclear localization of RNAPII; the polymerase accumulates in the cytoplasm. Npa3 is a member of the GPN-LOOP family of GTPases. Npa3 mutants that either cannot bind GTP or that bind but cannot hydrolyze it are inviable and unable to support nuclear transport of RNAPII. Surprisingly, we were unable to detect interactions between Npa3 and proteins in the classical importin α/β pathway for nuclear import. Interestingly, Npa3-RNAPII binding is significantly increased by the addition of GTP or its slowly hydrolyzable analogue guanosine 5′-3-O-(thio)triphosphate (GTPγS). Moreover, the Npa3 mutant that binds GTP, but cannot hydrolyze it, binds RNAPII even in the absence of added GTP, whereas the mutant that cannot bind GTP is unable to bind the polymerase. Together, our data suggest that Npa3 defines an unconventional pathway for nuclear import of RNAPII, which involves GTP-dependent binding of Npa3 to the polymerase.
Introduction
RNA polymerase II (RNAPII)2 is a multisubunit enzyme responsible for transcription of all eukaryotic protein-encoding genes. The structure of RNAPII (1, 2), as well as its function and regulation (3, 4), has been intensely studied for decades, but surprisingly little is known about its biogenesis. Furthermore, RNAPII is a nuclear protein, but exactly how it enters the nucleus is still unclear. For example, there is no obvious nuclear localization signal (NLS) on any of the polymerase subunits that might target it to the nucleus. One possibility is that NLS-containing carrier protein binds the polymerase and presents it to the nuclear import machinery. There is precedence for such a chaperone mechanism for protein transport. For example, nuclear export of the large 60 S ribosomal subunit depends on the adapter protein Nmd3, which provides the nuclear export signal (5, 6). Another question is whether RNAPII subunits enter the nucleus separately and independently or whether they are preassembled in the cytoplasm and imported as larger complexes or even as the whole RNAPII complex. Only very recently, a few studies have addressed these questions. One investigated the assembly of RNAPII from its individual subunits and concluded that it takes place in the cytoplasm and is a prerequisite for correct nuclear import (7). Three other studies, also published while our work was in progress or preparation for publication, were focused on the nuclear import of RNAPII and reported that two different proteins, namely XAB1/RPAP4/GPN1/Npa3 (8, 9) and Iwr1 (10), have roles in transporting RNAPII to the nucleus.
In our study, we focused on Saccharomyces cerevisiae Npa3. Npa3 was originally named due to its interaction with nucleolar preribosomal associated 1 (Npa1) (11), but unlike Npa1 (which is found in the nucleolus), Npa3 is primarily a cytoplasmic protein (11, 12). NPA3 is an essential gene in yeast (13). Its human orthologue, XPA binding-protein 1 (XAB1, also called RPAP4/GPN1 or MBDin), was initially identified as a xeroderma pigmentosum A (XPA)-interacting protein in a two-hybrid screen (14), but it has also been studied in the context of the methylated DNA-binding protein MBD2, where it appears to reactivate transcription from methylated promoters repressed by MBD2 (15).
We identified human XAB1 while looking for novel RNAPII-associated proteins by affinity purification coupled with mass spectrometry (MS). Indeed, when its yeast orthologue Npa3 was tagged and purified, the only protein that consistently co-purified with it was RNAPII. Here we show that Npa3 is required for transcription of a substantial fraction of yeast genes because it is needed for nuclear localization of RNAPII. We show evidence for an important role for the GTP-binding domain of Npa3 and the GTP hydrolysis cycle in the association with RNAPII. Our results also argue for a role of Npa3 in nuclear import of RNAPII via a non-classical nuclear import pathway.
EXPERIMENTAL PROCEDURES
Strains and Plasmids
Unless stated, all strains were based on w303-1a (MATa leu2-3,112 his3-11,15 ade2-1 ura3-1 trp1-1 can1-100). Npa3-HA3 (Npa3 C-terminally tagged with HA3::HIS3), Npa3-FLAG-TEV2-Myc9 (Npa3 C-terminally tagged with FLAG-TEV2-Myc9::TRP1), Npa3-FLAG-TEV2-Myc18 (Npa3 C-terminally tagged with FLAG-TEV2-Myc18::HIS3), Npa3degHA3 (Npa3deg::KanMX C-terminally tagged with HA3::URA3), Nup133-Myc9 (Nup133 C-terminally tagged with Myc9::TRP1), Nup133-Myc9 and Npa3-HA3 (as Nup133-Myc9, but Npa3 also C-terminally tagged with HA3::HIS3), Npa3degHA3 and Xpo1-HA3 (as Npa3degHA3, but Xpo1 also C-terminally tagged with HA3::HIS3), Rpb3-EGFP (Rpb3 C-terminally tagged with EGFP::HIS3), and Npa3degHA3 and Rpb3-EGFP (as Npa3degHA3, but Rpb3 also C-terminally tagged with EGFP::HIS3), where Npa3deg indicates Npa3-degron. Npa3degHA3 strain was constructed from the yjr072c-td strain (16), a kind gift from Dr. Karim Labib, Paterson Institute, Manchester, UK. First, a C-terminal HA3::URA3 tag was added to the Npa3deg (yjr072c-td), and then the strain was mated and sporulated to remove TUB1-GFP::TRP1 and ubr1delta::GAL-HA-UBR1::HIS3. For NLS-EGFP (and EGFP) tagging of endogenous Rpb3, the plasmid pYM28 (10), a kind gift from Dr. Patrick Cramer, Gene Center Munich, Germany, was used.
Degron Induction
Npa3degHA3 cells were grown at 25 °C (permissive temperature) in synthetic complete medium or synthetic dropout medium, with the addition of 10 μm CuSO4 to maintain near physiological Npa3 levels (as confirmed by Western blotting). For Npa3 degradation, cells were washed two times with medium without CuSO4 and shifted to 37 °C (restrictive temperature) for 6 or 8 h, resulting in less than 10% of cellular Npa3 remaining. Induction of Ubr1 was not required for efficient degradation.
Protein Expression and Purification
To purify Npa3 with its interacting partners from yeast, Npa3-FLAG-TEV2-Myc9 cells were harvested, resuspended in 150 mm Tris acetate (pH 7.8), 50 mm potassium acetate, 1 mm EDTA, 5 mm dithiothreitol, 20% glycerol, 0.01% Nonidet P-40, and protease inhibitors (17), and lysed using a freezer mill. The extracts were precleared at 12,000 × g for 20 min, potassium acetate was added to a final concentration of 500 mm, and the extracts were ultracentrifuged using a Beckman Ti45 rotor (40,000 rpm, 1 h, 4 °C). The soluble layer was precleared over a protein A column and then loaded onto a 9E11 affinity column at a flow rate of 0.25 ml/min. The column was washed with 40 column volumes of lysis buffer supplemented with 500 mm potassium acetate and then with 20 column volumes of lysis buffer supplemented with 50 mm potassium acetate and finally equilibrated in TEV cleavage buffer (10 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1 mm β-mercaptoethanol). TEV cleavage was carried out overnight in TEV cleavage buffer by adding 100 μg of AcTEV protease (Invitrogen), and the proteins bound to the column were eluted with TEV cleavage buffer containing 300 mm NaCl. In the next purification step, the eluates were further purified either by M2 (anti-FLAG affinity) chromatography or over a HiTrapQ FF column.
In Vitro Transcription
Promoter-specific transcription was carried out using yeast whole-cell extract (WCE) from Npa3-FLAG-TEV2-Myc18 and the guanine-less (G-less) cassette template plasmid pGCYC1-402 (18, 19). Briefly, reactions were performed in a final volume of 25 μl (40 mm HEPES (pH 7.5); 15 mm MgCl2; 8 mm DTT; 100 mm KOAc; 0.8 mm CTP, ATP, and GTP; 12 μm UTP; 10 μCi of [32P]UTP (3000 Ci/mmol; PerkinElmer Life Sciences); and 40 units of RNasin (Promega)). In each reaction, 0, 140, or 280 μg of Npa3-depleted or mock-depleted WCE was incubated with 100 ng of Gal4-VP16, and 800 ng of pGCYC1-402 DNA at room temperature for 30 min in the presence or absence of 600 ng of rNpa3. Reactions were stopped by the addition of 200 μl of stop buffer (10 mm Tris (pH 7.5), 0.3 m NaCl, and 5 mm EDTA) and 100 units of RNase T1 (Roche Applied Science) for 15 min at room temperature. Samples were then treated with proteinase K, phenol/chloroform was extracted, and samples were run on a sequencing gel as described (20). Results were visualized by autoradiography.
WCE depleted of Npa3 was generated by incubating 500 μl of WCE (89 mg/ml) with 50 μl of protein A/G (1:1)-agarose beads to which 9E11 anti-Myc antibody had been coupled at 4 mg/ml. Mock depletion was with protein A/G beads alone. Incubations were carried out on a rotator at 4 °C sequentially for 2 h, 40 min, and then 20 min, respectively, using fresh beads each time. Depletion levels were checked by Western blotting.
Immunoprecipitation and ChIP
Npa3-HA3 was immunoprecipitated with an anti-HA antibody (ab9110), and Npa3-FLAG was immunoprecipitated using M2-agarose and eluted with 3×FLAG peptide. The same antibodies were used for detection of Npa3-HA3 and Npa3-FLAG by Western blotting. Xpo1-HA3 was detected with ab9110 antibody, and Nup133-Myc9 was detected with 9E11 anti-Myc antibody. ChIP assays were carried out as described (21). For Npa3-HA3 ChIP, ab9110 antibody was used, and Rpb1 ChIP was done with a mix of 4H8 and 8WG16 antibodies.
RT-PCR and Clonal Sequencing Methods
RNA was prepared from wild type and Npa3-degron strains grown in synthetic complete medium at 37 °C in the absence of Cu2+ for 6 h using an RNeasy kit (Qiagen). Clonal sequencing of RNA was carried out on a Genome Analyzer IIx instrument, by single-end, 72-bp analysis. Reads were mapped to the June 2008 (sacCer2) version of the S. cerevisiae genome using Bowtie (22) with default settings. The Bioconductor package GenomicFeatures was used to count read overlaps with protein-coding transcripts from the Saccharomyces Genome Database (SGD). Differential expression analysis for each transcript between Npa3-degron and WT groups was assessed using the Bioconductor package edgeR (23). Library size was normalized using the “TMM” method of the “calcNormFactors” function. An estimate of common dispersion was calculated using the “estimateCommonDisp” function before implementation of the “exactTest” function.
For RT-PCR analysis, RNA was treated with Turbo DNase (Ambion), and cDNA was synthesized using the Taqman reverse transcriptase kit (Applied Biosystems). PCR oligonucleotide sequences are available on request. Real-time PCR analysis was carried out on a Bio-Rad CFX96 real-time system.
Fluorescence Microscopy
Yeast immunofluorescence was carried out according to the method of Ayscough and Drubin (24). Rpb1 was detected using a 4H8 monoclonal antibody at 1:5000 and Alexa Fluor 488 anti-mouse at 1:800. Deltavision microscopy was used to visualize yeast using an X100 UplanSApo 1.40 NA oil objective lens on an Olympus inverted microscope (IX71). Images were captured using SoftWorx computer software (Applied Precision). Three-dimensional data sets were computationally deconvolved and flattened.
RESULTS
Npa3 Interacts with RNA Polymerase II in Yeast and Human
To gain new insight into elongation by RNAPII, we used a human HEK293 cell line expressing a C-terminally FLAG-tagged Rpb3 subunit of RNAPII and purified proteins associated with the elongating form of RNAPII as described previously (25). In an attempt to identify proteins specifically associated with DNA damage-stalled RNAPII, one-half of the cells were irradiated with 30 J/m2 of ultraviolet (UV) light. Significant amounts of Cockayne syndrome B and DNA damage-binding protein 1 (DDB1) were found to interact with RNAPII in UV-irradiated cells only, as expected from previous data (26), but we failed to identify novel proteins associating with RNAPII in a UV-dependent manner using this approach (data not shown). Interestingly, however, we repeatedly (in both conditions) identified XAB1, a poorly characterized GTPase, which was initially identified through its interaction with the repair protein xeroderma pigmentosum A (14). We were intrigued by the interaction of XAB1 with RNAPII, so we decided to characterize the interaction further.
We initially constructed a stable human cell line expressing near normal levels of C-terminally FLAG-tagged XAB1 to identify any additional interacting partners, which might provide a clue to its function. Unfortunately, although we employed the same well established procedure used in the purification of Rpb3-FLAG, we were for unknown reasons unable to purify sufficient amounts of XAB1 complex for mass spectrometric analysis. Because XAB1 has a highly conserved yeast homologue, Npa3, we decided to switch to yeast and take advantage of a powerful affinity purification strategy in this system (27) (Fig. 1A). We genomically tagged Npa3 with a C-terminal FLAG-TEV2-Myc9 tag, purified it over an anti-Myc affinity column, and then eluted it by proteolytic cleavage with TEV protease. The eluate was further purified over an anti-FLAG M2-agarose affinity column and eluted with 3×FLAG peptide. The co-purifying proteins were analyzed by mass spectrometry (Fig. 1B, M2). In addition to Npa3, we identified several different heat-shock proteins, some of which are frequently observed during purification of other proteins by this procedure as well. More importantly, mass spectrometric analysis identified RNAPII subunits Rpb1, Rpb2 and Rpb5 in this Npa3 purification. The result was similar when we employed HiTrapQ ion exchange chromatography after TEV cleavage elution (Fig. 1B, Q). In this case, we identified Rbp2 and Rpb3 associated with Npa3. In general, only RNAPII subunits consistently co-purified with Npa3 in all the purifications of differing stringency that we performed (Fig. 1B and data not shown).
FIGURE 1.
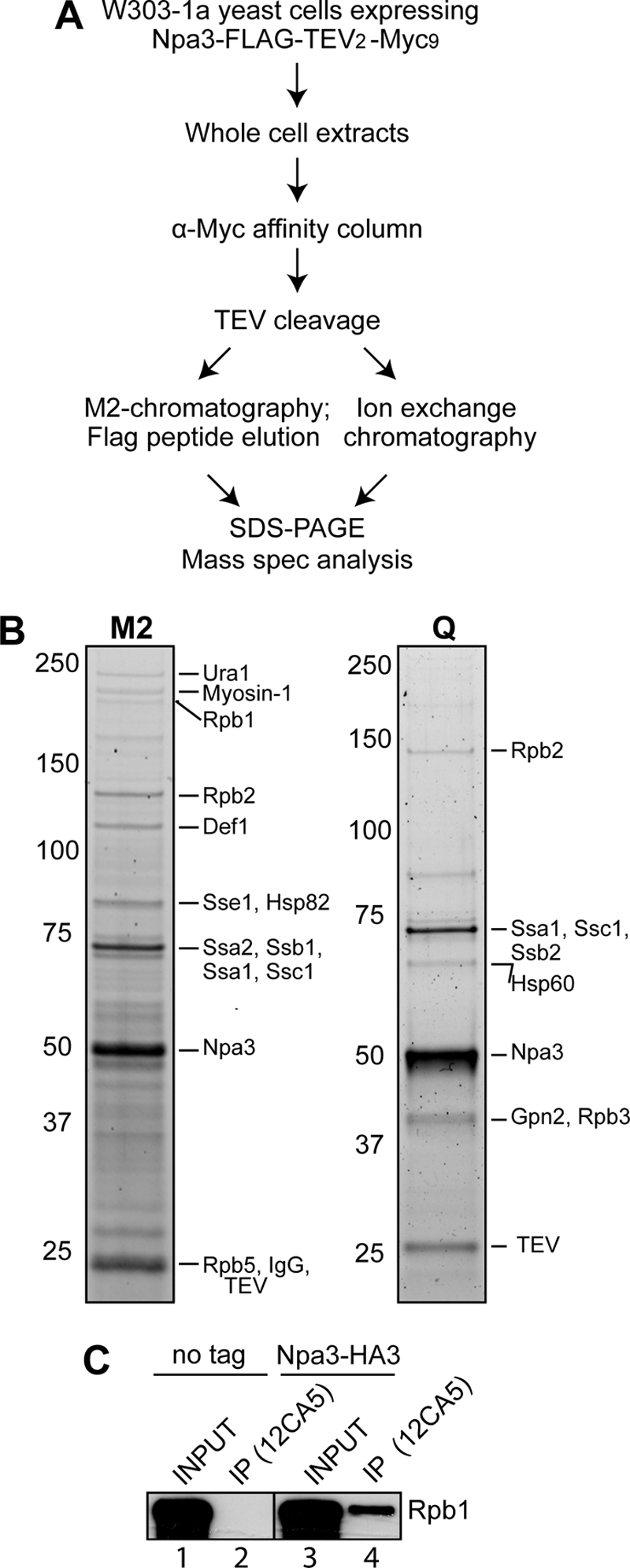
RNAPII co-purifies with Npa3. A, outline of the Npa3 purification procedure. Mass spec analysis, mass spectrometric analysis. B, eluates from M2-agarose (M2) and a HiTrapQ FF column (Q) (peak fraction) were separated by 4–12% SDS-PAGE and stained with SYPRO Ruby. Positions of proteins identified by mass spectrometric analysis are indicated on the right, and marker protein migration is indicated on the left. C, immunoprecipitation (IP) from whole cell extracts from a strain expressing endogenous Npa3 with a C-terminal 3×HA tag (Npa3-HA3) or an untagged control strain (no tag), with anti-HA (12CA5) antibody. Rpb1 was detected by Western blotting with a mixture of 4H8 and 8WG16 antibodies.
To confirm the interaction between Npa3 and RNAPII, we constructed a strain expressing 3×HA-tagged Npa3 from its native chromosomal location and pulled down Npa3 with 12CA5 (anti-HA) antibody. The non-tagged parental strain was used as a negative control. We reproducibly observed a small proportion of polymerase co-precipitating with 12CA5 antibodies from NPA3-HA cells, but not the untagged cell line, as assayed by Western blotting with anti-Rpb1 antibodies (Fig. 1C, compare lanes 2 and 4).
Npa3 Is Associated with Chromatin and Affects Transcription
The finding that Npa3 interacts with RNAPII raised the intriguing possibility that Npa3 might have a role in transcription. To investigate the effect of Npa3 on transcription, mRNA levels were compared by clonal sequencing of mRNA isolated from wild type or an Npa3-degron strain (16) at the restrictive temperature, where most Npa3 is degraded (Fig. 2A). In three independent experiments, we obtained reads for transcripts from 6109 genes. Expression levels of 1392 genes (22.8% of the genome) were changed by a factor of 2 or more in the Npa3-degron strain when compared with the control strain. The vast majority of these, 1294 genes, were down-regulated (93%), whereas only 98 genes (7%) were up-regulated. To verify the RNA-Seq results, we measured the expression of six down-regulated genes, two up-regulated genes, and two genes that were not affected in the degron strain by reverse transcription followed by quantitative PCR (RT-PCR) (Table 1). The RT-PCR results closely matched those obtained by RNA-Seq. Note that YEF3 gene was not called as a down-regulated gene in the genome-wide analysis, although the quantitative RT-PCR indicated an average expression reduction of 2.2-fold. This is because the RNA-Seq average difference only showed a 1.8-fold average reduction. It is highly likely that a much higher proportion than ∼23% of the transcriptome is affected by Npa3, also because it was not possible to degrade all the Npa3 within the time frame of the experiment.
FIGURE 2.

Npa3 depletion affects transcription in vivo, but not in vitro. A, depletion of Npa3-degron after inducing degradation by raising the temperature to 37 °C. No degradation of Npa3 is observed in a WT congenic strain (not shown). B, Rpb1 ChIP using WT or Npa3-degron cells at the non-depleting temperature (25 °C) or depletion-inducing temperature (37 °C). ChIP signals on ZPS1 and NAT4 are expressed as the percentage of input. Error bars denote S.D. of three biological replicates. C, Western blots of extracts from an Npa3-FLAG-TEV2-Myc18 strain before (titration on left) or after depletion of Npa3 by 9E10 (anti-Myc) or control beads (Mock). Note that the Npa3 signal after 9E10 depletion (lane 7) is similar to 1–5% of the signal before depletion (lanes 4 and 5), whereas the signal in the mock depletion is similar to 100% (compare lane 6 with lane 1). D, promoter-driven transcription in mock- or Npa3-depleted extract in the absence or presence of added recombinant Npa3.
TABLE 1.
Validation of RNA-Seq data by reverse transcription and real-time (quantitative) PCR
-Fold change in expression for three biological replicates, with averages shown. NA, not applicable.
| RNA-Seq 1 | RNA-Seq 2 | RNA-Seq 3 | RNA-Seq average | RT-PCR 1 | RT-PCR 2 | RT-PCR 3 | RT-PCR average | |
|---|---|---|---|---|---|---|---|---|
| Down (WT/degron) | ||||||||
| ZPS1 | 14.4 | 9.0 | 15.6 | 13.0 | 10.5 | 12.5 | 15.2 | 12.7 |
| NAT4 | 4.1 | 12.6 | NA | 8.4 | 2.4 | 2.9 | 3.9 | 3.1 |
| ADH1 | 1.3 | 3.0 | 5.9 | 3.4 | 1.4 | 2.6 | 3.4 | 2.5 |
| PMA1 | 1.4 | 2.8 | 5.0 | 3.1 | 4.0 | 3.8 | NA | 3.9 |
| FLP1 | 2.6 | 1.7 | 3.5 | 2.6 | 2.4 | 1.9 | 2.4 | 2.2 |
| YEF3 | 1.3 | 1.6 | 2.7 | 1.8 | 1.2 | 2.4 | 3.0 | 2.2 |
| No change (WT/degron) | ||||||||
| RPB3 | 0.8 | 1.6 | 1.8 | 1.4 | 0.9 | 1.5 | 1.8 | 1.4 |
| RPB1 | 0.9 | 1.2 | 1.7 | 1.3 | 0.7 | 0.8 | 1.0 | 0.8 |
| Up (degron/WT) | ||||||||
| PTR2 | 7.6 | 5.2 | 4.3 | 5.7 | 10.0 | 8.3 | 7.7 | 8.7 |
| HSP78 | 6.1 | 5.0 | 1.9 | 4.3 | 5.3 | 5.0 | 2.9 | 4.4 |
To test whether the dramatic effect on mRNA levels observed in the RNA-Seq experiment was caused by a defect in transcription, we measured RNAPII occupancy by ChIP on some of the affected genes. The ZPS1 and NAT4 genes, which were down-regulated, also had significantly less RNAPII bound in the absence of Npa3 (37 °C), indicating a transcriptional defect (Fig. 2B). Together, these results indicate that loss of Npa3 results in impaired transcription of a large number of yeast genes.
Depletion of Npa3 from Transcription-competent Cell Extracts Has No Effect on Transcription in Vitro
After observing global effects on transcription upon Npa3 depletion in vivo, we wanted to study its role in transcription in more detail in vitro. Promotor-driven transcription was assayed using yeast WCE (18) on a DNA template containing a G-less cassette. For production of WCE, we used the strain expressing FLAG-TEV2-Myc18-tagged Npa3 from its native chromosomal location, and we then removed Npa3 from the extract using anti-Myc antibody (Fig. 2C). This led to >95% depletion of Npa3 from the extract (Fig. 2C, compare lane 7 with lanes 4 and 5). Npa3-depleted and mock-depleted extracts were then used to reconstitute promoter-driven RNAPII transcription from the G-less cassette-containing DNA template, with or without the addition of Npa3-GST purified from Escherichia coli. Surprisingly, neither immunodepletion (Fig. 2D, compare lanes 1 and 2 with lanes 3 and 4) nor the addition of purified Npa3 protein to the extracts (compare lanes 6 and 7 with lanes 8 and 9) had an effect on transcription. This was in stark contrast with our in vivo data, which had suggested a general role for Npa3 in transcription. We therefore reasoned that Npa3 might be involved in a process that is not relevant in the in vitro transcription system. Two obvious possibilities were a function in chromatin transactions or nuclear localization and/or assembly of RNAPII. Because a substantial fraction of XAB1 (the human homologue of Npa3) is cytoplasmic, we now investigated whether Npa3 might play a role in assembly or nuclear transport of RNAPII.
Npa3 Is Required for Nuclear Localization of RNAPII
Initially, to investigate the effect of Npa3 on RNAPII complex assembly, we purified RNAPII from an Npa3-degron strain at the restrictive temperature and compared its subunit composition with RNAPII purified from the wild type strain. There was no significant difference in the yield or subunit composition between the two polymerases (data not shown), suggesting that Npa3 is not absolutely required for RNAPII biogenesis.
To test the hypothesis that Npa3 is needed for correct localization of RNAPII, we looked at the localization of two RNAPII subunits, Rpb1 and Rpb3, in wild type and Npa3-degron cells by immunofluorescence (Fig. 3). At the permissive temperature, Rpb1 was strictly nuclear in both strains; no staining was observed in the cytoplasm (Fig. 3A, two upper rows). In contrast, although the localization of Rpb1 did not change at the restrictive temperature in wild type cells (third row), a significant fraction became cytoplasmic in Npa3-degron cells (lower row), indicating a defect in its nuclear import in the absence of Npa3. To monitor the localization of another RNAPII subunit, Rpb3 was genomically tagged with a C-terminal EGFP tag, and its localization was monitored by live cell imaging (Fig. 3B). As observed for Rpb1, Rpb3-EGFP was found only in the nucleus of both wild type and degron cells at the permissive temperature, whereas clear cytoplasmic accumulation of Rpb3-EGFP was seen in the cells depleted for Npa3 (lower panels), but not in wild type cells, at the elevated temperature.
FIGURE 3.

RNAPII localizes to the cytoplasm in the absence of Npa3. A, localization of Rpb1 in WT or Npa3-degron (Npa3deg) cells at non-depleting (25 °C) or depletion-inducing temperature (37 °C) was assessed by immunofluorescence, using anti-Rpb1 (4H8) antibody. B, WT and Npa3-degron (Npa3deg) cells expressing an endogenous C-terminally EGFP-tagged Rpb3 were grown at 25 °C overnight (O.N.), shifted to 37 °C for 8 h, and then shifted back to 25 °C for 4 h. Localization of Rpb3 in WT and Npa3-degron cells was monitored via EGFP fluorescence.
As cells had to be kept at the restrictive temperature for several hours to achieve efficient depletion of the essential Npa3 protein, there was a possibility that altered RNAPII localization was a secondary effect of, for example, cell death. Therefore, we investigated whether cells resumed growth and normal Rpb3 localization when brought back to the permissive temperature. Indeed, when shifted back to the permissive temperature so that Npa3 function was reactivated, cells not only resumed growing (data not shown), but also, more importantly, RNAPII again became exclusively nuclear (Fig. 3B, compare the second and third column). Together, these data show that Npa3 plays an important role in the nuclear localization of RNAPII; although RNAPII is observed in the nucleus in all conditions, partial depletion of Npa3 (Fig. 2A) results in substantial accumulation of the polymerase in the cytoplasm.
Npa3 Interacts with Nup133 and Crm1/Xpo1
The data above strongly indicate that Npa3 is required for nuclear import of RNAPII, but which other transport factors are responsible for the translocation of this complex to the nucleus? We investigated interactions of Npa3 with known transport factors by co-immunoprecipitation experiments. We failed to detect an interaction of Npa3 with yeast importin α, Srp1 (data not shown). Moreover, we found no evidence for interaction between Npa3 and Kap95, a yeast importin β, or Gsp1, the yeast Ran orthologue (data not shown), which are otherwise the central components of the classical nuclear import pathway.
Because we failed to uncover evidence that Npa3 and its cargo enter the nucleus via the classical nuclear import pathway, we wondered whether there is an alternative, more direct pathway that Npa3 might utilize. One possibility would be that Npa3 binds directly to nuclear pore proteins, potentially bypassing the need for importin β to make the contact with the nuclear pore. Interestingly, we had indeed identified Nup133, one of the nuclear pore outer ring proteins, in one of our Npa3 purifications. To verify this interaction, we genomically tagged Nup133 with a 9×Myc tag in the strain already expressing genomically 3×HA-tagged Npa3. Npa3-HA3 was pulled down with an anti-HA antibody, and the interaction with Nup133-Myc9 was examined by Western blotting. Nup133 was co-immunoprecipitated with Npa3-HA3 from the strain expressing tagged Npa3, but not from the untagged control strain (Fig. 4A), confirming the Npa3-Nup133 interaction.
FIGURE 4.
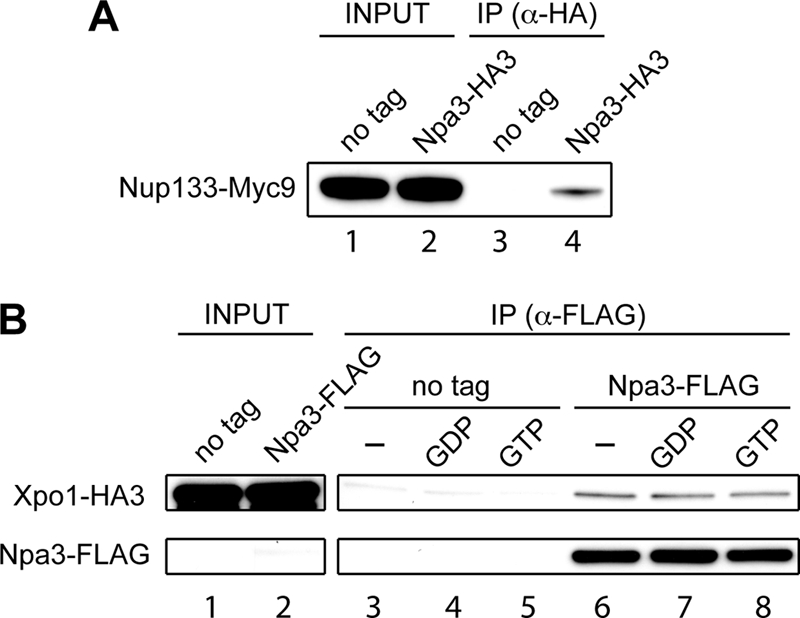
Npa3 interacts with nuclear pore protein Nup133 and nuclear export protein Crm1/Xpo1. A, Npa3 was immunoprecipitated (IP) with an anti-HA antibody from extracts of a strain expressing endogenous Npa3 with a C-terminal 3×HA tag and Nup133 with a C-terminal 9×Myc tag (Npa3-HA3). A strain expressing only Myc9-tagged Nup133 was used as a negative control (no tag). Nup133 was detected by Western blotting with a 9E11 anti-Myc antibody. Lanes 1 and 2: 0.5% of input. B, Npa3 was immunoprecipitated with an anti-FLAG antibody from extracts of cells expressing Npa3-FLAG and Xpo1-HA3. Bound proteins were eluted with a 3×FLAG peptide. A strain expressing only tagged Xpo1 was used as a negative control (no tag). Xpo1 was detected by Western blotting with an ab9110 anti-HA antibody. Lanes 1 and 2: 0.5% input. Prior to immunoprecipitation, extracts were incubated with no added GTP analogue (lanes 3 and 6), 500 μm GDP (lanes 4 and 7), or 500 μm GTP (lanes 5 and 8) to measure the effect of nucleotide on binding.
Upon unloading its cargo in the nucleus, Npa3 needs to be recycled to the cytoplasm. This prompted us to test whether Npa3 also binds the nuclear export factor Crm1/Xpo1. Indeed, 3×HA-tagged Crm1/Xpo1 was co-immunoprecipitated with anti-FLAG antibodies from a strain expressing FLAG-tagged Npa3, but not from an untagged control strain (Fig. 4B, compare lanes 3 and 6). Crm1/Xpo1 bound Npa3-FLAG regardless of the addition of GDP or GTP to the cell extracts (compare lanes 6–8).
A Functional Npa3 GTPase Domain Is Required for Cell Viability and Proper Nuclear Localization of RNAPII
Database searches revealed only one conserved domain in Npa3: a GTPase motif of the GPN family (28). To establish whether GTPase activity is essential for its function, we mutated two crucial residues in Npa3; the D106A mutation will abolish GTP binding (28), whereas the Q110L mutation corresponds to a naturally occurring mutation in the homologous Ras protein that retains the ability to bind GTP but that does not support GTP hydrolysis (29). We introduced wild type Npa3 and the two mutant forms into Npa3-degron cells and assessed cell viability at the permissive and restrictive temperature (Fig. 5A). At the permissive temperature, all cell types grew similarly well. More importantly, cells expressing the D106A and Q110L mutants were even more sensitive to elevated temperature (where Npa3-degron is degraded) than cells transformed with an empty vector, whereas wild type Npa3 rescued the temperature sensitivity of the cells, as expected. This indicates that GTPase activity is indeed essential for the function of Npa3.
FIGURE 5.

Characterization of Npa3 GTPase domain mutants. Npa3-degron cells were transformed with a yeast centromeric plasmid (pRS415) carrying Npa3-FLAG (Npa3 WT), Npa3-FLAG D106A, or Npa3-FLAG Q110L under the control of its natural promoter and terminator. A, serial dilutions of cells grown in liquid cultures at 25 °C overnight were spotted on plates containing 10 μm CuSO4 and grown for 3 days at 25 °C (no Npa3-degron depletion) or spotted on plates with no added CuSO4 and grown for 2 days at 37 °C (Npa3-degron depletion induced). B, Rpb1 localization in cells grown for 6 h at 37 °C (Npa3-degron depletion induced) assessed by immunofluorescence, using a 4H8 anti-Rpb1 antibody.
Next we wanted to test whether GTPase function of Npa3 is needed for proper nuclear localization of RNAPII. Therefore, we monitored Rpb1 localization when expressing wild type or mutant Npa3 protein in Npa3-degron cells (Fig. 5B). As expected, a significant portion of Rpb1 was cytoplasmic at the restrictive temperature in Npa3-degron cells transformed with an empty vector (Fig. 5B, upper row), whereas Rpb1 was exclusively nuclear in cells expressing wild type Npa3 protein (second row). Interestingly, cytoplasmic accumulation of Rpb1 was very pronounced in the cells expressing mutant D106A and Q110L protein (lower two rows), strongly indicating an essential function of the Npa3 GTPase activity in nuclear localization of RNAPII.
A GTP Cycle for Npa3 Association with RNAPII
Having shown that the GTPase domain of Npa3 is essential for nuclear localization of RNAPII, we wanted to know whether GTP binding by Npa3 regulates its interaction with RNAPII. Npa3-degron cells expressing undegradable WT-, D106A-, or Q110L Npa3-FLAG or an empty vector were grown at 37 °C for 6 h to degrade Npa3-degron. WCE from these cells was then incubated with M2 FLAG-agarose before Npa3-FLAG and interacting proteins were eluted with FLAG peptide, and eluates were analyzed by Western blotting (Fig. 6). As Npa3 is a GTPase, we first tested whether the addition of different versions of guanine nucleotide phosphates to the extracts affects the interaction between Npa3 and Rpb1. Strikingly, although the addition of GDP did not have much effect (Fig. 6, compare lanes 7 and 8), the addition of GTP, and its slowly hydrolyzable form GTPγS in particular, consistently resulted in significantly larger amounts of Rpb1 being pulled down with Npa3-FLAG (Fig. 6, compare lane 7 with lanes 9 and 10, respectively), indicating that GTP binding is important for the interaction between Npa3 and Rpb1.
FIGURE 6.

RNAPII-Npa3 association is regulated by GTP binding and hydrolysis. Npa3 (WT or mutant indicated on top) was immunoprecipitated from whole cell extracts (after growth for 6 h at 37 °C to degrade endogenous Npa3-degron) via a C-terminal FLAG tag on the plasmid-expressed Npa3 proteins (Empty vector is shown as control). Bound proteins were eluted with 3×FLAG peptide, and Rpb1 was detected by Western blotting with a mixture of 4H8 and 8WG16 anti-Rpb1 antibodies. Npa3-FLAG was detected with anti-FLAG antibodies. Lanes 1, 6, 11, and 16: 1% input. Prior to immunoprecipitation, extracts were incubated with no GTP analogue (−) or 500 μm GDP, GTP, or GTPγS, as indicated.
In agreement with this conclusion, the two Npa3 mutants behaved very differently in the RNAPII binding assay. Npa3 D106A, the mutant defective in nucleotide binding, was thus completely defective in Rpb1 binding (Fig. 6, lanes 12–15), strongly supporting the idea that nucleotide binding is essential for the association of RNAPII with Npa3. Indeed, the Npa3 Q110L mutant, which binds but cannot hydrolyze GTP, bound Rpb1 very well, even without the addition of exogenous GTP to the extract, to a level comparable with wild type Npa3 in the presence of exogenous GTP and GTPγS (compare lanes 9 and 10 with lanes 17–20). We conclude that an intact Npa3 GTPase domain and GTP binding are essential not only for viability and RNAPII nuclear localization, but also for proper interaction of Npa3 with RNAPII.
DISCUSSION
RNAPII is a large enzyme consisting of 12 subunits. Due to the structural studies of the polymerase in past couple of decades (1, 2), we now have a detailed model of the enzyme, clearly depicting the positions and interactions of individual subunits. However, how those subunits are assembled into the RNAPII complex and delivered to the nucleus from the cytoplasm has remained unclear. Here we show that in the absence of Npa3, a conserved and essential GTPase, RNAPII accumulates in the cytoplasm, and transcription is therefore severely affected. The nuclear transport function of Npa3 is strictly dependent on its GTPase activity as point mutations in the GTPase domain affect binding of Npa3 to RNAPII, resulting in its cytoplasmic accumulation. We found no evidence for binding of Npa3 to any of the classical nuclear import proteins, but it did bind the nuclear pore protein Nup133, opening the possibility that Npa3 might bring its RNAPII cargo to the nucleus via an interaction with nuclear pore proteins.
Our initial interest in Npa3 was triggered by our discovery that human RNA polymerase II was found associated with XAB1/RPAP4/GPN1 in chromatin. This finding might suggest that the protein functions in the nucleus, in apparent agreement with a study on the function of XAB1/RPAP4/GPN1 in reactivating transcription from methylated promoters repressed by MBD2 (15). Our subsequent data in yeast confirmed the interaction with RNAPII and also initially supported a function for Npa3 in transcription, as expression of (and RNAPII association with) numerous genes was significantly affected by Npa3 depletion. However, we were unable to uncover any effect on transcription reconstituted in crude extracts, which might indicate an indirect effect and would seem in agreement with the fact that the majority of the pool of the protein is cytoplasmic. This led us to investigate the potential role of Npa3 in RNAPII localization and the realization that it is important for normal nuclear import of the polymerase.
The data on the role of Npa3 in nuclear import of RNAPII presented here complements several studies published on RNAPII assembly and transport published recently (7–10). In one of these studies, it was found that HSP90 is involved in RNAPII assembly in the cytoplasm in human cells and that only assembled RNAPII complexes are transported (7). In support of a conserved role for heat-shock proteins in the assembly of RNAPII, the yeast homologue of HSP90, Hsc82, was also detected in our Npa3 purifications. In general, several GPNs (GPN1/2/3) were found with RNAPII in different studies in humans (7–9), and we detected both Npa3 and Gpn2 with RNAPII from yeast as well. It thus appears that the basic cellular apparatus used for assembly and transport of RNAPII has remained conserved from yeast to human.
In humans, the orthologue of Npa3, XAB1/RPAP4/GPN1, was found to interact with karyopherin α2 (8), a human importin α protein, and importin 9 (9), member of the importin β family. Somewhat surprisingly, we failed to detect an interaction between Npa3 and Kap60 (importin α), Kap95 (importin β), or Gsp1 (Ran), which are components of the classical nuclear import pathway. This might simply imply that the interaction between Npa3 and the importins is too weak or too transient to be detected in the type of assay we used (co-immunoprecipitation), but it might also mean that Npa3 is imported via an alternative pathway in yeast. In this connection, it is relevant to note that although most types of characterized NLS-containing proteins are bound by importin α, which then binds importin β, which in turn contacts the nuclear pore, some transported proteins bind directly to importin β and do not require importin α for translocation to the nucleus (30). Other proteins are even imported without Ran (31, 32), and there are even examples of proteins, such as β-catenin, whose transport seems to require neither importins nor Ran as these proteins can bind directly to the nuclear pore (33). We found that Npa3 can also bind at least one nuclear pore protein, Nup133, and although Nup133 is not one of the several phenylalanine-glycine repeat nucleoporins that have so far been implicated in the binding and translocation of import complexes, it is possible that Npa3 can directly bind nuclear pore proteins and therefore does not depend on the classical nuclear import pathway.
Another feature of Npa3 lends further credence to the idea that its mechanism of RNAPII nuclear import is via a non-conventional pathway; Npa3 itself is a GTPase. As a matter of fact, we found that Npa3 binds RNAPII much better in the active, GTP-bound form and that mutations in the GTPase domain affect binding in a meaningful and potentially telling manner. Thus, a D106A point mutation, which abolishes GTP binding (28), also renders Npa3 unable to associate with RNAPII, whereas Q110L mutation, which allows small GTPases to bind GTP, but not to hydrolyze it (29), results in very strong RNAPII binding even in conditions where nucleotide is limiting. Unsurprisingly, both these Npa3 point mutations are lethal and unable to support RNAPII transport in vivo. The Npa3-RNAPII binding data are consistent with the idea that Npa3 works in a manner analogous to Ran, which binds partner proteins in its GTP-associated form, whereas binding ceases upon GTP hydrolysis. Although other possibilities cannot be ruled out, our data on the GTP-regulated Npa3-RNAPII binding cycle would thus be in agreement with a model in which GTP-bound Npa3 associates with RNAPII in the cytoplasm, assists in/triggers its transport across the nuclear membrane, and then releases it upon GTP hydrolysis in the nucleus. Npa3 would then be recycled to the cytoplasm, in all likelihood via a mechanism that involves Xpo1 (Crm1).
Somewhat surprisingly, it has recently been suggested that an important pathway for the import of S. cerevisiae RNAPII into the nucleus is through association with the Iwr1 protein, which possesses an NLS and appears to be imported into the nucleus via the classical, importin α-dependent pathway (10). The connection between Iwr1 and Npa3, if any, remains unclear, as does the importance of the human Iwr1 homologue in the transport of RNAPII in higher cells.
Together, the data accumulated to date in several laboratories are best explained by the existence of distinct pathways for RNAPII nuclear import in both yeast and humans. Thus, although several players in the nuclear import of RNAPII have now been identified, the precise mechanism of transport and the relationship between the possibly several mechanisms still need to be resolved.
Acknowledgments
We thank members of the Svejstrup laboratory for helpful discussions. Drs. Patrick Cramer (Munich, Germany) and Karim Labib (Manchester, United Kingdom) are thanked for kind gifts of plasmid and strain, respectively.
This work was supported by a grant from the European Community (Integrated Project DNA Repair (LSHG-CT-2005-512113)) and by an in-house grant from Cancer Research UK (to J. Q. S.).
- RNAPII
- RNA polymerase II
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- NLS
- nuclear localization signal
- EGFP
- enhanced green fluorescent protein
- TEV
- tobacco etch virus
- WCE
- whole-cell extract.
REFERENCES
- 1. Kornberg R. D. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 12955–12961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cramer P., Armache K. J., Baumli S., Benkert S., Brueckner F., Buchen C., Damsma G. E., Dengl S., Geiger S. R., Jasiak A. J., Jawhari A., Jennebach S., Kamenski T., Kettenberger H., Kuhn C. D., Lehmann E., Leike K., Sydow J. F., Vannini A. (2008) Annu. Rev. Biophys. 37, 337–352 [DOI] [PubMed] [Google Scholar]
- 3. Fuda N. J., Ardehali M. B., Lis J. T. (2009) Nature 461, 186–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Selth L. A., Sigurdsson S., Svejstrup J. Q. (2010) Annu. Rev. Biochem. 79, 271–293 [DOI] [PubMed] [Google Scholar]
- 5. Ho J. H., Kallstrom G., Johnson A. W. (2000) J. Cell Biol. 151, 1057–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gadal O., Strauss D., Kessl J., Trumpower B., Tollervey D., Hurt E. (2001) Mol. Cell. Biol. 21, 3405–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boulon S., Pradet-Balade B., Verheggen C., Molle D., Boireau S., Georgieva M., Azzag K., Robert M. C., Ahmad Y., Neel H., Lamond A. I., Bertrand E. (2010) Mol. Cell 39, 912–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Forget D., Lacombe A. A., Cloutier P., Al-Khoury R., Bouchard A., Lavallée-Adam M., Faubert D., Jeronimo C., Blanchette M., Coulombe B. (2010) Mol. Cell. Proteomics 9, 2827–2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carre C., Shiekhattar R. (2011) Mol. Cell. Biol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Czeko E., Seizl M., Augsberger C., Mielke T., Cramer P. (2011) Mol. Cell 42, 261–266 [DOI] [PubMed] [Google Scholar]
- 11. Dez C., Froment C., Noaillac-Depeyre J., Monsarrat B., Caizergues-Ferrer M., Henry Y. (2004) Mol. Cell. Biol. 24, 6324–6337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huh W. K., Falvo J. V., Gerke L. C., Carroll A. S., Howson R. W., Weissman J. S., O'Shea E. K. (2003) Nature 425, 686–691 [DOI] [PubMed] [Google Scholar]
- 13. Giaever G., Chu A. M., Ni L., Connelly C., Riles L., Véronneau S., Dow S., Lucau-Danila A., Anderson K., André B., Arkin A. P., Astromoff A., El-Bakkoury M., Bangham R., Benito R., Brachat S., Campanaro S., Curtiss M., Davis K., Deutschbauer A., Entian K. D., Flaherty P., Foury F., Garfinkel D. J., Gerstein M., Gotte D., Güldener U., Hegemann J. H., Hempel S., Herman Z., Jaramillo D. F., Kelly D. E., Kelly S. L., Kötter P., LaBonte D., Lamb D. C., Lan N., Liang H., Liao H., Liu L., Luo C., Lussier M., Mao R., Menard P., Ooi S. L., Revuelta J. L., Roberts C. J., Rose M., Ross-Macdonald P., Scherens B., Schimmack G., Shafer B., Shoemaker D. D., Sookhai-Mahadeo S., Storms R. K., Strathern J. N., Valle G., Voet M., Volckaert G., Wang C. Y., Ward T. R., Wilhelmy J., Winzeler E. A., Yang Y., Yen G., Youngman E., Yu K., Bussey H., Boeke J. D., Snyder M., Philippsen P., Davis R. W., Johnston M. (2002) Nature 418, 387–391 [DOI] [PubMed] [Google Scholar]
- 14. Nitta M., Saijo M., Kodo N., Matsuda T., Nakatsu Y., Tamai H., Tanaka K. (2000) Nucleic Acids Res. 28, 4212–4218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lembo F., Pero R., Angrisano T., Vitiello C., Iuliano R., Bruni C. B., Chiariotti L. (2003) Mol. Cell. Biol. 23, 1656–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kanemaki M., Sanchez-Diaz A., Gambus A., Labib K. (2003) Nature 423, 720–724 [DOI] [PubMed] [Google Scholar]
- 17. Otero G., Fellows J., Li Y., de Bizemont T., Dirac A. M., Gustafsson C. M., Erdjument-Bromage H., Tempst P., Svejstrup J. Q. (1999) Mol. Cell 3, 109–118 [DOI] [PubMed] [Google Scholar]
- 18. Kong S. E., Svejstrup J. Q. (2002) DNA Repair 1, 731–741 [DOI] [PubMed] [Google Scholar]
- 19. Rondón A. G., Jimeno S., García-Rubio M., Aguilera A. (2003) J. Biol. Chem. 278, 39037–39043 [DOI] [PubMed] [Google Scholar]
- 20. Sayre M. H., Tschochner H., Kornberg R. D. (1992) J. Biol. Chem. 267, 23376–23382 [PubMed] [Google Scholar]
- 21. Selth L., Svejstrup J. Q. (2007) J. Biol. Chem. 282, 12358–12362 [DOI] [PubMed] [Google Scholar]
- 22. Langmead B., Trapnell C., Pop M., Salzberg S. L. (2009) Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robinson M. D., McCarthy D. J., Smyth G. K. (2010) Bioinformatics 26, 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ayscough K. R., Drubin D. G. (1998) Immunofluorescence Microscopy of Yeast Cells, Academic Press, New York [Google Scholar]
- 25. Aygün O., Svejstrup J., Liu Y. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 8580–8584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fousteri M., Vermeulen W., van Zeeland A. A., Mullenders L. H. (2006) Mol. Cell 23, 471–482 [DOI] [PubMed] [Google Scholar]
- 27. Greenwood C., Selth L. A., Dirac-Svejstrup A. B., Svejstrup J. Q. (2009) J. Biol. Chem. 284, 141–149 [DOI] [PubMed] [Google Scholar]
- 28. Gras S., Chaumont V., Fernandez B., Carpentier P., Charrier-Savournin F., Schmitt S., Pineau C., Flament D., Hecker A., Forterre P., Armengaud J., Housset D. (2007) EMBO Rep. 8, 569–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Graziano M. P., Gilman A. G. (1989) J. Biol. Chem. 264, 15475–15482 [PubMed] [Google Scholar]
- 30. Lange A., Mills R. E., Lange C. J., Stewart M., Devine S. E., Corbett A. H. (2007) J. Biol. Chem. 282, 5101–5105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yen Y. M., Roberts P. M., Johnson R. C. (2001) Traffic 2, 449–464 [DOI] [PubMed] [Google Scholar]
- 32. Hanover J. A., Love D. C., DeAngelis N., O'Kane M. E., Lima-Miranda R., Schulz T., Yen Y. M., Johnson R. C., Prinz W. A. (2007) J. Biol. Chem. 282, 33743–33751 [DOI] [PubMed] [Google Scholar]
- 33. Fagotto F., Glück U., Gumbiner B. M. (1998) Curr. Biol. 8, 181–190 [DOI] [PubMed] [Google Scholar]