

Background: Different regulatory strategies are utilized by bacteria to control central carbohydrate metabolism.
Results: Transcriptional factor HexR is a global regulator of the central carbohydrate metabolism genes in various groups of proteobacterial genomes.
Conclusion: HexR in Shewanella is a repressor/activator of the glycolytic/gluconeogenic genes.
Significance: Integration of the comparative genomics and experimental approaches is efficient for reconstruction of transcriptional regulons in bacteria.
Keywords: Bacteria, Carbohydrate Metabolism, Computational Biology, Glycolysis, Repressor Protein, Transcription Regulation, HexR, Proteobacteria
Abstract
Bacteria exploit multiple mechanisms for controlling central carbon metabolism (CCM). Thus, a bioinformatic analysis together with some experimental data implicated the HexR transcriptional factor as a global CCM regulator in some lineages of Gammaproteobacteria operating as a functional replacement of the Cra regulator characteristic of Enterobacteriales. In this study, we combined a large scale comparative genomic reconstruction of HexR-controlled regulons in 87 species of Proteobacteria with the detailed experimental analysis of the HexR regulatory network in the Shewanella oneidensis model system. Although nearly all of the HexR-controlled genes are associated with CCM, remarkable variations were revealed in the scale (from 1 to 2 target operons in Enterobacteriales up to 20 operons in Aeromonadales) and gene content of HexR regulons between 11 compared lineages. A predicted 17-bp pseudo-palindrome with a consensus tTGTAATwwwATTACa was confirmed as a HexR-binding motif for 15 target operons (comprising 30 genes) by in vitro binding assays. The negative effect of the key CCM intermediate, 2-keto-3-deoxy-6-phosphogluconate, on the DNA-regulator complex formation was verified. A dual mode of HexR action on various target promoters, repression of genes involved in catabolic pathways and activation of gluconeogenic genes, was for the first time predicted by the bioinformatic analysis and experimentally verified by changed gene expression pattern in S. oneidensis ΔhexR mutant. Phenotypic profiling revealed the inability of this mutant to grow on lactate or pyruvate as a single carbon source. A comparative metabolic flux analysis of wild-type and mutant strains of S. oneidensis using [13C]lactate labeling and GC-MS analysis confirmed the hypothesized HexR role as a master regulator of gluconeogenic flux from pyruvate via the transcriptional activation of phosphoenolpyruvate synthase (PpsA).
Introduction
Fine-tuning of the carbohydrate catabolic pathway expression is key to successful adaptation of microorganisms that occupy niches with variable carbon availability. Escherichia coli and related Enterobacteria utilize two global transcription factors (TFs),3 Crp and Cra (FruR), to control central and peripheral carbohydrate metabolism (1). The cAMP receptor protein, Crp, mediates catabolic repression of target genes in the presence of high levels of glucose, a preferable carbon source for Enterobacteria. In the presence of cAMP, Crp positively regulates its target genes by binding the cognate DNA sites. cAMP is generated by adenylate cyclase that is activated by components of the glucose-specific phosphotransferase system in the absence of glucose (2). The LacI protein family catabolite repressor/activator, Cra, was initially characterized as the fructose repressor, FruR, because it negatively regulates transcription of the fructose utilization operon (3). Later, Cra was characterized as a pleiotropic transcriptional regulator that represses multiple genes from the central glycolytic pathways, namely the EMP and ED pathways, and activates genes involved in gluconeogenesis and oxidative phosphorylation (4). DNA binding activity of Cra is triggered by two central intermediates of the EMP pathway, fructose 1-phosphate and fructose 1,6-biphosphate. Comparative genomic analysis of the Cra (FruR) regulon revealed substantial variability in regulon gene content among Enterobacteriales and that it acted as a local regulator of the fructose utilization operon in other γ-proteobacteria such as Vibrio and Pseudomonas species (5).
A different regulatory strategy is utilized to control central carbohydrate metabolism in the genus Pseudomonas, which favors the utilization of organic acids and amino acids over various other carbon sources (6). In contrast to Enterobacteria, the Pseudomonas EM glycolytic pathway is nonfunctional due to the absence of 6-phosphofructokinase (7). Degradation of glucose proceeds ultimately via the ED pathway, where 6-phosphogluconate and KDPG are key intermediates. Expression of all genes encoding the ED pathway in Pseudomonas putida, including glucokinase (glk), glucose-6-phosphate dehydrogenase (zwf), 6-phosphogluconolactonase (pgl), 6-phosphogluconate dehydrogenase (edd), KDPG aldolase (eda), and glyceraldehyde-3-phosphate dehydrogenase (gap-1), is negatively regulated by HexR (8, 9). Two monomers of HexR bind to imperfect palindromic sites with consensus sequence of TTGTN7–8ACAA in the promoter regions of the zwf, edd, and gap-1 genes. Binding of the ED pathway intermediate KDPG to HexR releases the repressor from its target sites (8–11).
Previously, we have applied the integrated genomic and experimental approaches to predict and validate novel metabolic pathways and transcriptional regulons involved in carbohydrate utilization in the Shewanella genus of γ-proteobacteria (12, 13). The obtained genomic encyclopedia of sugar utilization included 17 distinct peripheral pathways with committed TFs (e.g. the N-acetylglucosamine utilization pathway controlled by NagR regulon) and a core set of central carbohydrate metabolism genes, of which 12 genes were predicted to be controlled by the HexR regulator (Fig. 1B). In this study, we expand this analysis toward comparative genomic reconstruction of the HexR regulon in all sequenced Proteobacteria. We report the identification of DNA-binding motifs for HexR regulators and provide a detailed description of corresponding regulons in the genomes of 62 γ-proteobacteria and 25 β-proteobacteria. The comparative analysis of reconstructed regulons revealed considerable variability in the regulon content between the analyzed Proteobacteria. By correlating the binding site position within promoter regions with expression patterns of downstream genes, it was possible to predict the activation mode for HexR regulation of several target genes in Shewanella spp. A combination of in vivo and in vitro experimental techniques was used to validate the predicted HexR-dependent regulatory network in Shewanella oneidensis MR-1.
FIGURE 1.

Metabolic context of the reconstructed HexR regulons in Proteobacteria (A) and S. oneidensis MR-1 (B). Numbers in blue circles show the number of genomes where the target gene is preceded by a candidate HexR-binding site.
EXPERIMENTAL PROCEDURES
Bioinformatics Techniques and Resources
Genomic sequences were obtained from GenBankTM (14). Identification of orthologs and gene neighborhood analysis were performed in Microbes OnLine (15). Functional annotations of genes involved in central carbohydrate metabolism and related pathways were derived from the SEED comparative genomic database (16). Phylogenetic trees were built using the maximum likelihood method in the PHYLIP Package and visualized with Dendroscope (17). Sequence alignments were made with MUSCLE (18). Distant homologs were identified using pBLAST (19).
We used a well established comparative genomics method of regulon reconstruction (reviewed in Ref. 5) implemented in the RegPredict webserver (regpredict.lbl.gov) (20). We started from the genomic identification of a reference set of genomes that encodes HexR orthologs (according to the phylogenetic analysis of HexR proteins on supplemental Fig. S1). To find the conserved DNA-binding motif for HexR in each group of phylogenetically related genomes, we used initial training sets of known HexR targets from Pseudomonas spp., and then we updated each set by the most likely HexR-regulated genes confirmed by the comparative genomics tests as well as the functional and genome context considerations.
In each of 13 studied groups of Proteobacteria, an iterative motif detection procedure implemented in the RegPredict web tool was used to identify common regulatory DNA motifs in a set of upstream gene fragments and to construct the motif recognition profiles as described previously (21). For each clade of HexR proteins on the phylogenetic tree, we used a separate training gene set. The initial recognition profile was used to scan the genomes in this clade and to predict novel genes in the regulon. Scores of candidate sites were calculated as the sum of positional nucleotide weights. The score threshold was defined as the lowest score observed in the training set. The conserved regulatory interactions with high scored binding sites that involve target genes involved in central carbohydrate metabolism were included in the reconstructed HexR regulons. Candidate sites associated with new members of regulon were added to the training set, and the respective group profile was rebuilt to improve search accuracy. Sequence logos for the derived group-specific DNA-binding motifs were drawn using WebLogo package (22). The details of the reconstructed HexR regulons are captured and displayed in RegPrecise, a specialized database of bacterial regulons (regprecise.lbl.gov) (23).
Cloning of hexR and Overproduction in E. coli
The hexR gene (SO2490) from S. oneidensis MR-1 was PCR-amplified using as a template a pET-derived vector encoding SO2490 fused to an N-terminal His6 tag (24) and the following primers: 5′-CGATCATGGATCCatgaataccctagaaaagg (forward) and 5′-CTGCAGTCGAAGCTTttacagtatcggatc (reverse). The PCR products were digested with BamHI/HindIII, and the resulting 861-bp fragment encoding hexR was then cloned into the pSMT3 expression vector (25). The obtained pSMT3 vector encodes a fusion between the HexR protein and N-terminal hexahistidine Smt3 polypeptide (a yeast SUMO ortholog), allowing us to enhance protein solubility. The resulting construct was transformed in E. coli BL21/DE3 cells (Invitrogen). The cells were grown in 50 ml of LB medium to an A600 = 1.2 at 20 °C containing 0.3 mm isopropyl 1-thio-β-d-galactopyranoside and harvested after 12 h. The recombinant Smt3-His6-HexR protein was purified using a rapid nickel-nitrilotriacetic acid-agarose column as described previously (26). Briefly, harvested cells were resuspended in 20 mm HEPES buffer (pH 7) containing 100 mm NaCl, 0.03% Brij 35, and 2 mm β-mercaptoethanol supplemented with 2 mm phenylmethylsulfonyl fluoride and a protease inhibitor mixture (Sigma). Lysozyme was added to 1 mg/ml, and the cells were lysed by freeze-thaw cycle followed by sonication. The resulting crude extract was heated for 30 min at 70 °C. After centrifugation, the supernatant was supplemented with Tris-HCl buffer (pH 8) to achieve a final concentration of 50 mm and then was loaded onto a nickel-nitrilotriacetic acid-agarose minicolumn (0.3 ml). After washing the column with the starting buffer containing 1 m NaCl and 0.3% Brij-35, bound proteins were eluted with 0.3 ml of the starting buffer supplemented with 250 mm imidazole. The eluted protein was suspended in a solution of 20 mm HEPES (pH 7.0), 1 mm DTT, and 0.5 mm EDTA. Protein size, expression level, distribution between soluble and insoluble forms, and extent of purification were assessed with 12% SDS-polyacrylamide gels.
DNA Binding Assays
Interaction of purified recombinant-tagged HexR protein with its cognate DNA-binding sites in S. oneidensis MR-1 was assessed by two techniques, electrophoretic mobility shift assay and fluorescence polarization assay, using two sets of synthetic double-stranded oligonucleotides 3′-labeled with either biotin or 6-carboxyfluorescein, respectively. The dsDNA fragments containing the predicted HexR-binding sites were obtained by annealing custom-synthesized complementary oligonucleotides at a 1:10 ratio of labeled to unlabeled complementary oligonucleotides (supplemental Table S1).
For EMSAs, the 49-bp biotin-labeled dsDNA fragments (0.1 nm) were incubated with 500 nm purified HexR in a total volume of 20 μl as described previously (27). The binding buffer contained 20 mm Tris-HCl (pH 8.0), 150 mm KCl, 5 mm MgCl2, 1 mm DTT, 1 mm EDTA, 0.05% Nonidet P-40, and 2.5% glycerol. Poly(dI-dC) was added as a nonspecific competitor DNA at ∼104-fold molar excess over labeled target DNA to reduce nonspecific binding. After 25 min of incubation at 37 °C, the reaction mixtures were separated by electrophoresis on a 5% native polyacrylamide gel in 0.5× Tris borate/EDTA (100 min, 90 V, room temperature). The DNA was transferred by electrophoresis (30 min at 380 mA) onto a Hybond-N+ membrane and fixed by UV cross-linking. Biotin-labeled DNA was detected with the LightShift chemiluminescent EMSA kit. The effect of phosphorylated intermediates of the ED pathway on HexR-DNA binding was tested by addition of 2-keto-3-deoxy-6-phosphogluconate, glucose 6-phosphate, 6-phosphogluconate, and phosphoenolpyruvate to the incubation mixture (2 mm).
For FPA assays, the 27-bp fluorescence-labeled dsDNA fragments (1 nm) were incubated with the increasing concentrations of purified HexR (10–1000 nm) in 100 μl of reaction mixture in 96-well black plates (VWR, Radnor, PA). Binding buffer contained 20 mm Tris-HCl (pH 7.5), 100 mm NaCl, 0.3 mg/ml BSA, and 1 μg of poly(dI-dC). Fluorescence measurements were taken on Beckman DTX 880 multimode plate reader with excitation and emission filters at 495 and 520 nm, respectively. Background fluorescence from buffer was subtracted, and the fluorescence polarization value was defined as follows: FP = (I1 − G· I2)/(I1 + G·I2), where I1 and I2 are the fluorescence intensities measured in the parallel and perpendicular directions respective to the orientation of the excitation polarizer, and G is a correction factor (28).
Phenotypic Analysis and Metabolite Measurements
Chromosomal in-frame deletion mutants of hexR (SO2490) or ppsA (SO2644) genes were constructed using previously published methods (13). S. oneidensis MR-1 wild type (WT) and ΔhexR and ΔppsA mutant strains were precultured on LB medium to late exponential growth phase, and washed with M1 minimal medium without any carbon sources. Duplicate 50-ml cultures were inoculated to the same absorbance (A600 = 0.01) in M1 minimal medium supplemented with 10 mm N-acetyl-d-glucosamine or 20 mm of d-glycerate, dl-lactate, or pyruvate as the sole carbon source and incubated at 30 °C. Cell growth was monitored spectrophotometrically at 600 nm. Lactate, glycerate, acetate, and pyruvate were detected in culture supernatants by high pressure liquid chromatography with the Agilent model 1100 instrument equipped with Shodex RSpak KC-811. A mobile phase consisting of 6 mm HClO4 at a flow rate of 1.0 ml/min was used, and the column was operated at 50 °C.
RT-PCR Analysis
Genomic RNA was isolated from S. oneidensis MR-1 WT and ΔhexR cells grown in minimal medium supplied with either d-glycerate or inosine and collected at the same absorbance (A600 = 0.5) using the RNA purification kit from Promega. Reverse transcription of total RNA was performed with random primers using the iScript cDNA synthesis kit from Bio-Rad. RT-PCR was performed using the SYBR® GreenERTM qPCR SuperMix Universal kit from Invitrogen. Transcript levels were measured for 13 candidate regulated genes, and the results were normalized to the expression level of 16 S mRNA, and the gene expression fold change was calculated using the 2−ΔCT method (29) as a ratio of normalized mRNA levels in ΔhexR mutant and WT MR-1 strains.
GC-MS and Metabolic Flux Analysis
Cells were grown on a mixture containing 4 mm 13C-labeled lactate, 16 mm unlabeled lactate, and 20 mm glycerate or on lactate only containing 4 mm [U-13C]lactate and 16 mm unlabeled lactate. Aliquots of cultures were harvested at mid-exponential growth phase (A600 of 0.3). After centrifugation, the cell pellet was washed with 1 ml of 0.9% (w/v) NaCl and then hydrolyzed in 200 μl of 6 m HCl at 105 °C for 24 h. The filtrate was dried in a vacuum centrifuge at room temperature and derivatized at 85 °C for 1 h in 120 μl of pyridine and 30 μl of N-methyl-N-(tert-butyldimethylsilyl) trifluoroacetamide. After filtration, 5 μl of derivatized sample was injected into Agilent 6890-5973 GC-MS system with a HP-5MS column (30 m × 0.25 mm × 0.25 μm). GC oven temperature was programmed from 60 to 180 °C at 5 °C per min and from 180 to 260 °C at 10 °C per min. The flow rate of carrier gas (helium) was set at 1 ml min−1. The mass spectrometer was operated in the electron impact mode at 70 eV.
The GC-MS data were analyzed as described previously (30). Briefly, prior to analysis of cellular metabolism, mass spectra of the derivatized amino acids were corrected for the natural abundance of all stable isotopes. From the mass isotopomer distribution vectors of the phenylalanine, tyrosine, alanine, valine, aspartate, and threonine, the isotopomer distribution vectors of their respective precursor intermediates (i.e. phosphoenolpyruvate, pyruvate, and oxaloacetate) could be easily derived. The fractions of phosphoenolpyruvate originating from pyruvate (fPEP←PYR) and from oxaloacetate (fPEP←OAA) can then be determined by using Equation 1 implemented in the MATLAB program.
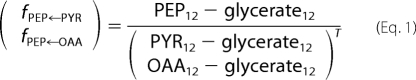 |
In Equation 1, the division is a left-hand matrix division.
For quantification of intracellular carbon fluxes in S. oneidensis grown on lactate, a bioreaction network was constructed based on the Internet-accessible genome database. This network includes the reactions of the gluconeogenic pathway and the tricarboxylic acid cycle, as well as the reactions catalyzed by phosphoenolpyruvate carboxylase, phosphoenolpyruvate carboxykinase, and malic enzyme. The glyoxylate shunt and the pyruvate kinase were identified to be inactive in this study; thus they are omitted from the bioreaction network for net flux analysis. From the bioreaction network, a stoichiometric matrix containing 21 unknown fluxes and 19 metabolite balances was constructed. Net fluxes were then calculated based on three different data sets: (i) substrate uptake and product formation rates; (ii) macromolecular biomass composition; and (iii) the calculated fPEP←PYR and fPEP←OAA. The macromolecular biomass composition of S. oneidensis MR-1 was taken from Ref. 31. The carbon flux distribution in the bioreaction network was determined with a MATLAB-based program by minimizing the sum of the weighted square residuals of the constraints from both metabolite balances and flux ratios as described previously (30).
RESULTS
Genomic Reconstruction of HexR Regulons in Proteobacteria
Phylogenetic Distribution of hexR Orthologs
Orthologs to the P. putida hexR were identified by BLAST searches against a nonredundant set of sequenced bacterial genomes and used to analyze the phylogenetic relationships between these proteins (supplemental Fig. S1). A single copy of hexR gene was detected in most lineages of γ- and β-proteobacteria but not in other taxonomic groups (supplemental Table S2). Among the γ-proteobacteria, hexR is missing in all of the Pasteurellales, Xanthomonadales, and Moraxellaceae lineages and in some Alteromonadales and Oceanospirillales genomes. Two highly diverged hexR paralogs were detected in Pseudomonadales. The experimentally studied P. putida paralog PP1021 is similar to hexR genes from other γ-proteobacteria, whereas the second paralog (termed hexR1) is more related to hexR from β-proteobacteria. The Burkholderia genomes also possess a second copy of hexR gene, which is translationally fused to the glucokinase gene, glk. Because of the absence of other glk homologs in Burkholderia, the chimeric hexR2-glk gene is the only candidate to fulfill the essential glucokinase function, and the role of its HexR2 domain is to be elucidated.
Chromosomal Co-localization of hexR Genes
Functionally related genes (e.g. genes involved in the same pathway or a transcriptional regulator and its target genes) often occur close on the bacterial chromosome (32). Indeed, the previously characterized in P. putida hexR regulator is divergently transcribed with the zwf-pgl-eda operon (9). Similar divergent chromosomal arrangement is retained in the Pseudomonas and other lineages of γ-proteobacteria, although the content of zwf-containing operons is different in some genomes (Fig. 2). A monocystronic zwf gene previously detected in E. coli (33) is conserved in other Enterobacteriales with the exception of Edwardsiella tarda, which encodes a zwf-pgl-edd-eda operon. Similar four-gene operons encoding the complete ED pathway were found in the Alteromonadales lineage (e.g. in all Shewanella spp.). We also noted that in many γ-proteobacteria, hexR is co-localized on the chromosome with glucokinase, glk, and pyruvate kinase, pykA. Among β-proteobacteria, hexR is divergently transcribed with edd-eda in Burkholderia or it belongs to either zwf-hexR operon in Comamonadaceae or the zwf-pgl-glk-hexR-pgi operon in Neisseriales. This analysis further confirmed functional coupling between hexR and the central carbohydrate metabolism genes and allowed us to define a training set of upstream gene sequences for identification of HexR binding motifs.
FIGURE 2.

Genomic organization of the hexR-containing loci and HexR-regulated genes in representative bacterial genomes. Genes and candidate HexR-binding sites are shown by arrows and circles, respectively.
Identification of HexR-binding Sites and Regulons
To reconstruct the HexR regulons in Proteobacteria, we applied the integrative comparative genomics approach (as implemented in RegPredict webserver (20)) that combines identification of TFs and candidate transcription factor-binding sites with cross-genomic comparison of regulons and with the genomic and functional context analysis of candidate target genes. First, we collected training sets of prospective target genes for each out of 11 defined taxonomic groups possessing HexR orthologs. These training sets were initially defined by the analysis of chromosomal co-localization of hexR genes and a previous knowledge of the HexR regulon in P. putida and then were further expanded by identifying orthologous target genes in the analyzed genomes. For each taxonomic group, we extracted DNA upstream regions of putative operons in this training set and applied a transcription factor-binding site motif recognition program to derive conserved HexR-binding motifs. After construction of a positional weight matrix for each identified motif, we searched for additional HexR-binding sites in the analyzed genomes, and we finally performed a consistency check or cross-species comparison of the predicted regulons (reviewed in Ref. 5). For those groups of genomes where the HexR regulon was expanded by multiple novel candidate target genes, the above procedure was repeated to include these targets into the transcription factor-binding site motif model and to revise the final gene content of the regulon.
A highly conserved palindromic signal with consensus TGTARnnnnnYTACA (where R and Y denote purines and pyrimidines, respectively) was identified as a candidate HexR-binding motif in nine groups of analyzed genomes (supplemental Fig. S1). Two different motifs were detected for two groups of HexR paralogs in the Pseudomonas lineage. Three previously characterized HexR-binding sites in P. putida coincide with the binding motif for the HexR group inferred in this study (consensus sequence aTGTTGT-4–8 nucleotides-ACaAcAt). The candidate binding motif for the second group of regulators in Pseudomonas (HexR1) is similar to HexR-binding motifs in other Proteobacteria. Significant difference between the predicted binding motifs of HexR paralogs in Pseudomonas suggests that the respective regulons should not cross-talk with each other. A different conserved motif, which does not have a palindromic symmetry but which has some resemblance to a right part of the classical HexR-binding motif, was predicted for HexR regulons in Hahella and Marinobacter spp.
The content of the reconstructed HexR regulons in 11 taxonomic groups of γ- and β-proteobacteria (totally 87 species) is summarized in supplemental Table S3. Detailed information about the predicted DNA-binding sites and downstream regulated genes is provided in the RegPrecise database (regprecise.lbl.gov) (23). The reconstructed HexR regulons control the central carbohydrate metabolism in all analyzed proteobacteria (Fig. 1A). However, as we describe it below, the specific content of HexR regulons is highly variable between different lineages.
Conserved Members of HexR Regulons
Based on overall appearance and taxonomic distribution of HexR-regulated genes, we classified them into several groups (Table 1). The first group of genes includes the most conserved regulon members that are regulated by HexR in at least 30 species from at least six different lineages. This group includes zwf, pgl, edd, eda, gapA, pykA, glk, and pgi genes that encode key glycolytic enzymes from the ED and EMP pathways. In addition, the hexR gene was predicted to be autoregulated in most genomes. The second group includes genes that belong to the HexR regulons in at least 10 species from at least two lineages. Genes in the second group are involved in glycolysis (gpmM and tpiA), gluconeogenesis (ppsA, gapB, and pckA), the pentose phosphate pathway (tal), pyruvate metabolism (aceEF and ppc), fermentation (adhE, pflBA, and grcA), glyoxylate metabolism (aceBA), amino acid biosynthesis (gltBD), and NADPH re-oxidation (pntAB). The third group includes genes that were found within the HexR regulons in two or more lineages but appeared in less than 10 species. In this group, there are genes involved in glycolysis (aldE, pgk, and eno), formate fermentation (focA), and glucose and mannitol utilization (ptsG, ptsHI-crr, and mtlADR).
TABLE 1.
Conserved core of HexR regulons in 11 groups of proteobacteria
This table shows the number of genomes in each taxonomic group where the target gene is preceded by a conserved HexR-binding site. Genes without a HexR binding site are indicated by -. The absence of orthologous gene in analyzed genomes is indicated as NA.

1The number of genomes in the analyzed taxonomic group is indicated in parentheses.
Lineage-specific HexR-regulated Genes
The rest of the predicted target genes belong to a group of lineage-specific regulon members (Table 2). At most, this group includes genes with candidate HexR-binding sites that are conserved in at least two species within the same lineage but that are not conserved outside of this lineage. This group also includes the Colwellia psychrerythraea fructose-biphosphate aldolase gene, fba, which is preceded by a strong HexR-binding site. The largest set of lineage-specific HexR targets was detected in the Shewanella species, and it includes genes from the central carbohydrate metabolism (gnd, phk, and adhB), nucleoside/deoxynucleoside utilization (deoABD, nupC, and cdd), respiratory chain (nqr operon), and glycine utilization (gcvTHP). The second largest pool of predicted lineage-specific HexR-regulated genes was detected in Vibrio species; it includes glycogen metabolism genes, a nitrite reductase, a peptidase, and a lactate permease. Extended HexR regulons in other lineages contain genes involved in utilization of galactosides and glycerol (mgl and glpT), glycolysis (glpN), the tricarboxylic acid cycle (gltA), lactate, and acetate fermentation (ldhA and ackA-pta).
TABLE 2.
Lineage-specific predicted members of HexR regulons
| Taxonomic group(s)a | Target gene(s) | No.b | Functional role |
|---|---|---|---|
| Shewanellaceae (16) | phk | 16 | Phosphoketolase |
| gnd | 14 | 6-Phosphogluconate dehydrogenase | |
| nqrABCDEF | 15 | NADH:ubiquinone oxidoreductase | |
| deoABD | 14 | Deoxynucleoside utilization | |
| nupC | 15 | Nucleoside transporter | |
| cdd | 10 | Cytidine deaminase | |
| gcvTHP | 12 | Glycine cleavage | |
| adhB | 8 | Alcohol dehydrogenase II | |
| mcp | 9 | Chemotaxis protein | |
| SO1118 | 10 | Hypothetical protein | |
| Psychromonadaceae/Aeromonadales (6) | mglBAC | 3 | Galactoside utilization |
| glpT | 2 | Glycerol utilization | |
| ldhA | 3 | Lactate dehydrogenase | |
| ackA-pta | 4 | Acetate kinase | |
| Oceanospirillales/Alteromonadales (6) | gltA | 2 | Citrate synthase |
| mgsA | 2 | Methylglyoxal synthase | |
| PF00248 | 2 | Putative aldo/keto reductase | |
| Alteromonadales (5) | fba | 1 | Fructose-bisphosphate aldolase |
| glgP | 2 | Glycogen phosphorylase | |
| cpsA | 2 | Capsular polysaccharide synthesis enzyme | |
| Enterobacteriales (11) | ybfA | 10 | Putative exported protein |
| Vibrionales (10) | pepD | 9 | Aminoacyl-histidine dipeptidase |
| nirBD | 7 | Nitrite reductase (NAD(P)H) | |
| glgX | 6 | Glycogen debranching enzyme | |
| glgCA | 5 | Glycogen synthase, glucose-1-phosphate adenylyltransferase | |
| lctP | 2 | l-Lactate permease | |
| ygaW | 8 | Putative inner membrane protein | |
| Pseudomonadales (8) | gapN | 2 | Putative glyceraldehyde-3P dehydrogenase |
| gltRS | 7 | Glucose uptake regulatory system |
a The number of genomes in the analyzed taxonomic group is indicated in parentheses.
b Number of genomes in taxonomic group where target gene has a conserved HexR-binding site.
Experimental Validation of HexR Regulon in S. oneidensis MR-1
Dual Mode of HexR Regulation
The reconstructed HexR regulon in S. oneidensis MR-1 contains 30 genes (organized in 15 operons) that are involved in various metabolic pathways (Fig. 1B). These include two opposite central carbon metabolism pathways, the glycolytic (ED) pathway for hexose catabolism (zwf-pgl-edd-eda, gapA, and pykA) and the gluconeogenic pathway for glucose biosynthesis (ppsA and gapB). This observation suggests that HexR may have an opposite regulatory effect on the target gene expression, similar to the Cra dual repressor/activator control of carbon metabolism in E. coli (4). To reveal a negative or positive mode of HexR regulation in Shewanella we used several complementary approaches as follows: (i) the comparative analysis of upstream promoter sequences in multiple Shewanella genomes; (ii) the analysis of correlations of gene expression profiles in the compendium of microarray data available for S. oneidensis MR-1; and (iii) RT-PCR analysis of regulon gene expression in hexR knock-out mutant (see below).
For each predicted HexR-regulated gene in S. oneidensis MR-1, DNA upstream regions of orthologous genes from other Shewanella genomes were aligned, and candidate HexR-binding sites and potential promoter elements (the −35 and −10 promoter boxes) were located (supplemental Fig. S2). For 9 genes, namely zwf, pykA, gapA, gnd, nqrA, deoA, cdd, phk, and mcp, HexR operators either overlap with or are located downstream of the putative conserved promoter elements, suggesting that HexR is a repressor of these genes. In contrast, five other genes (ppsA, gapB, gcvT, tal, and nupC) have HexR operators located upstream of predicted promoters (distance between the center of HexR site and the 5′ position of the −35 promoter box equal to either 29, 21, or 14 nucleotides), suggesting that HexR may be acting as an activator.
To evaluate pairwise correlations in expression of all HexR-regulated genes, we have computed the Pearson correlation between each pair of genes in the S. oneidensis MR-1 genome based on the expression of each gene in ∼200 microarray experiments available in the MicrobesOnline database (supplemental Fig. S3). This analysis allowed us to identify subregulons that have different expression patterns. There is a strong correlation within the first group of zwf, gapA, pykA, and tal genes and the second group of phk, deoA, cdd, and nqrA genes, although correlation between these two groups is also significant. The third group of highly correlated genes gapB, ppsA, and gcvT shows no correlation with the genes from the first two groups. Indeed, the expression data previously obtained for S. oneidensis MR-1 grown on various carbon sources (34) confirm up-regulation of the deoA, zwf, phk, nupC, gapA genes during growth on inosine, whereas ppsA, gapB, and gcvT genes were down-regulated in these conditions compared with the growth on lactate. The observed different patterns of gene expression in the HexR regulon and the sequence analysis of promoter gene regions in S. oneidensis MR-1 suggest that HexR is a dual mode regulator that can be either repressor or activator of genes expression depending on the relative position of its operators and promoters.
HexR Binds Its Cognate DNA Sites in Vitro
To test the ability of HexR to specifically bind to the predicted DNA sites and to assess effectors, hexR (SO2490) from S. oneidensis, MR-1 was cloned and overexpressed in E. coli. The Smt3-His6-tagged recombinant protein HexR was purified by Ni2+-chelating chromatography, followed by gel filtration as described under “Experimental Procedures.” Electrophoretic mobility shift assay (EMSA) and FPA were used to test specific binding of the purified HexR protein to its predicted operator sites in upstream regions of 15 genes in S. oneidensis MR-1 (Table 3).
TABLE 3.
In vitro and in vivo assessment of S. oneidensis HexR regulon
EMSA and FPA testing of the purified HexR protein was for its ability to bind its target sites in the S. oneidensis genome. For FPA, the apparent Kd values for HexR protein interacting with the tested DNA fragments are indicated. Differential expressions of HexR target genes in S. oneidensis MR-1 WT and ΔhexR mutant strains grown on various carbon sources are shown.
| Gene | Operon | HexR-binding site | EMSA shifta | FPA, Kd | ΔhexR/WT glycerate | ΔhexR/WT inosine |
|---|---|---|---|---|---|---|
| nm | ||||||
| SO0779 | gvcTHP | ATGTAAAAAAATTACAT | No shift | 52 | 0.32 | 1.87 |
| SO0902 | nqrABCDEF | TTGTTATAAAACTACAT | Shift | NA | 4.12 | 6.20 |
| SO1214 | nupC | TTGAAATTAAACTCCAA | Shift | NA | 0.48 | 0.45 |
| SO1218 | deoABD | TTGTAATATTGCTACAG | Shift | NA | 5.08 | 1.85 |
| SO1483 | aceBA | TGGTAATTTAATTACGT | NA | 62 | NA | NA |
| SO1902 | gnd | TGGTAATTAAGTTACAA | Weak shift | 49 | 0.90 | 1.91 |
| SO2345 | gapA | TGGTAATTTTACTACCA | Shift | 71 | 1.87 | 1.64 |
| SO2347 | gapB | CTCTAATAAAATTACAG | Shift | NA | 0.14 | 0.36 |
| SO2489 | zwf-pgl-edd-eda | CTGTTGTTATATTACAA | Shift | 39 | 7.33 | 4.93 |
| SO2491 | pykA | TTGTAACTTTACTACAT | Shift | NA | 9.81 | 2.33 |
| SO2644 | ppsA | TAGTAATAAAATTACAA | Shift | 37 | 0.06 | 0.11 |
| SO2791 | cdd | CTGTAACAAAATTACTA | Shift | NA | 3.18 | 1.38 |
| SO3542 | phk | TTGTAAGAAAATTACAA | Shift | 38 | 12.79 | 4.71 |
| SO3546 | tal-pgi | TTGTAATTTTTTTACAT | Shift | 28 | 1.31 | 1.00 |
| SO4557 | mcp | CTGTAAATTTTTTACAG | Shift | NA | 2.07 | 0.82 |
a Genes that were not tested in any experiment are indicated as NA.
Binding of purified HexR to the SO2489 dsDNA fragment containing the predicted HexR-binding site resulted in the reduced electrophoretic mobility of the DNA fragment in a concentration-dependent manner (supplemental Fig. S4A). Of the tested intermediary metabolites associated with the ED pathway (2 mm KDPG, 6-phosphoglucose, phosphoenolpyruvate, or 6-phosphogluconate), only KDPG demonstrated significant suppression of the HexR-DNA binding-mediated shift. These findings are in agreement with the previous results obtained for the P. putida HexR regulator, which binds DNA in the absence of an effector, whereas KDPG induces the HexR-DNA complex dissociation (8). Specific binding at 0.5 μm HexR was also confirmed for all other tested DNA fragments (supplemental Table S1) with the single exception of the SO0779 dsDNA fragment (supplemental Fig. S4B). The latter fragment did not show a clear shift in the EMSA experiments but was confirmed to bind HexR using the FPA assay (see below).
The FPA binding assay was used to assess the specific binding of eight predicted HexR-binding sites in S. oneidensis with increasing concentrations of the HexR protein (Fig. 3). All tested DNA fragments demonstrated the HexR concentration-dependent increase of fluorescence polarization, confirming specific interactions between HexR and DNA. The apparent Kd values for HexR protein interacting with the tested DNA fragments were in the range of 28–71 nm (Table 3). As a negative control, we assessed interaction between a different S. oneidensis transcription factor (NagR or SO3516) with the SO2489 DNA fragment, and the HexR regulator with the SO3507 DNA fragment containing the previously confirmed NagR-binding site (12), and in both experiments no significant change of fluorescence polarization was detected. These results confirm that HexR is a KDPG-responsive regulator that binds specifically to the predicted operator sites in S. oneidensis MR-1.
FIGURE 3.

Fluorescence polarization binding assay of HexR with target DNA operators in S. oneidensis MR-1. Increasing HexR protein concentrations (0.01–1 μm) were mixed with 27-bp fluorescence-labeled DNA fragments (1 nm) of gene regions containing HexR-binding sites. A, SO1483, SO2489, SO2644, SO3546, and SO0779; B, SO1902, SO3542, and SO2345. DNA-protein complex formation was monitored by an increase in the fluorescence polarization value. As a negative control, the S. oneidensis NagR regulator and its target DNA fragment (SO3507) were used.
HexR Affects Expression of Its Target Genes in Vivo
To validate the predicted mode of HexR regulation on gene expression in vivo, a ΔhexR targeted deletion mutant strain of S. oneidensis MR-1 was constructed, and the relative transcript levels of the predicted HexR target genes analyzed by quantitative RT-PCR (Table 3). Relative mRNA levels of nine genes were elevated more than 1.5-fold in the ΔhexR mutant compared with the WT strain when grown in the minimal medium supplied with either glycerate or inosine. The most prominent effect of hexR mutation was observed for the zwf, pykA, nqrA, phk, deoA, and gapA genes, suggesting that HexR represses expression of these genes. The cdd and mcp genes were up-regulated 2–3-fold in ΔhexR strain in the media supplied with glycerate, whereas gnd was up-regulated near 2-fold in the cells grown on inosine. Expression of the tal gene was not significantly affected by hexR mutation in both conditions. In contrast, the ppsA, gapB, and nupC genes were significantly down-regulated in the ΔhexR strain. For instance, expression of the ppsA gene was decreased 9- and 17-fold in the cells grown on inosine and glycerate, respectively. Finally, expression of gcvT was down-regulated 3-fold in the ΔhexR strain when the cells were grown on glycerate but was up-regulated 2-fold in the inosine-supplied cells. In our previous study of metabolic regulons in Shewanella spp. (12), we have identified several regulatory motifs in the upstream region of the gcvTHP operon (supplemental Fig. S2), which is presumably regulated by several factors, including the HexR activator, the glycine-responsive activator GcvA, and the previously unknown regulator of purine metabolism operated by binding to PUR-boxes. These overlapping regulatory interactions could possibly explain the observed differences in the expression patterns of the gcvH genes in ΔhexR strain grown on either glycerate or inosine, as the purine nucleoside could potentially repress the gcv operon via a PUR-box operator. The observed differences in the regulation of gnd and mcp promoters in strains grown on different carbon sources could be explained by possible involvement in their regulation of other still unknown regulatory mechanisms that can differentiate the tested carbon sources. These observations confirm the predicted negative mode of HexR regulation for the zwf, pykA, nqrA, phk, deoA, gapA, gnd, cdd, and mcp genes and its positive mode of action on the ppsA, gapB, nupC, and gcvT genes.
Phenotype Characterization of S. oneidensis ΔhexR Mutant
The previous phenotypic and genomic analyses suggest that S. oneidensis can use GlcNAc, glycerate, inosine, lactate, and pyruvate as the sole carbon source (13, 34, 35). Because of the absence of phosphofructokinase in Shewanella spp., utilization of GlcNAc proceeds through glucose 6-phosphate, which is catabolized via the HexR-regulated ED pathway (Fig. 1B). Utilization of inosine requires the HexR-regulated nupC and deoABD genes that mediate nucleoside uptake, release of ribose base, and its further utilization via the pentose phosphate pathway. The catabolic pathways produce pyruvate, which is further utilized via the fermentation/respiration routes to gain energy and the gluconeogenesis for biosynthetic needs. To determine whether the loss of HexR function impairs the utilization of carbon sources, the S. oneidensis MR-1 WT and ΔhexR mutant strains were tested for the ability to grow on the above five carbon sources. The hexR mutant was unable to grow on lactate or pyruvate but grew similar to the WT on GlcNAc (Fig. 4) or inosine (data not shown). An enhanced growth on glycerate was observed for the ΔhexR mutant compared with the WT strain (Fig. 4). The inability of the S. oneidensis ΔhexR strain to grow on lactate as a sole carbon source confirms that HexR is an important transcriptional activator of the gluconeogenic gene ppsA, whose activity is known to be essential for the growth of E. coli on lactate (36). Therefore, based on a combination of regulon analysis, metabolic reconstruction (31), and physiological data, we hypothesized that HexR controls the main gluconeogenic flux from lactate via activation of PpsA. Disruption of this flux in ΔhexR mutant might be the main cause of its inability to grow on lactate as a single carbon source. However, the flux from lactate through the tricarboxylic acid cycle appears to be HexR-independent, and thus, it might continue in its role in energy production even in the ΔhexR mutant (in the presence of an additional carbon source, see below).
FIGURE 4.

Growth phenotypes of S. oneidensis MR-1 ΔhexR mutant on various carbon sources. Cells were grown in the minimal medium containing 10 mm N-acetyl-d-glucosamine (GlcNAc) or 20 mm glycerate, lactate, and pyruvate as the sole carbon source. The cell growth was monitored by measuring the absorbance at 600 nm.
Metabolic Flux Analysis of S. oneidensis ΔhexR Mutant
To test this hypothesis, the intracellular carbon fluxes in the WT and the ΔhexR mutant strains were quantified by using the 13C-based metabolic flux analysis that relies on the [U-13C]lactate labeling and GC-MS analysis of the mass isotopomer pattern in proteinogenic amino acids (supplemental Table S4). From the labeling patterns of the respective amino acids, the origins of phosphoenolpyruvate were quantitatively determined (Fig. 5). Phosphoenolpyruvate can be derived from pyruvate via phosphoenolpyruvate synthase encoded by ppsA, from oxaloacetate via phosphoenolpyruvate carboxykinase, or from glycerate through the glycolytic conversion of 3-phosphoglycerate to phosphoenolpyruvate. In the S. oneidensis MR-1 WT strain grown on the mixture of lactate and glycerate, more than 40% of phosphoenolpyruvate originated from the phosphoenolpyruvate synthase reaction (Fig. 5). In contrast, the phosphoenolpyruvate synthase flux was absent, and the majority of phosphoenolpyruvate was derived from glycerate in the ΔhexR mutant strain. A similar result was also found for the ΔppsA mutant strain (supplemental Table S4). These results indicate that the ppsA-encoding phosphoenolpyruvate synthase is largely inactive in vivo in the ΔhexR mutant. Moreover, the HexR deficiency also resulted in significant changes in central metabolic fluxes. Compared with the WT, a significant increase in the uptake fluxes of lactate and glycerate was observed for the ΔhexR mutant. This mutant also exhibited much higher fluxes of secretion of acetate and pyruvate.
FIGURE 5.

Origins of phosphoenolpyruvate (A) and in vivo phosphoenolpyruvate-synthesizing fluxes (B) in S. oneidensis MR-1 WT and ΔhexR mutant. Results were obtained from the labeling experiments with 4 mm [U-13C]lactate and 16 mm unlabeled lactate. Unlabeled glycerate (20 mm) was added to the medium as indicated. The numbers in B are the molar net fluxes (mmol g−1 h−1) determined. Double-headed arrows indicate reactions assumed to be reversible, and the arrows with bigger heads represent the direction of the determined net fluxes. Gray arrows indicate precursor withdrawal for the amino acid analyzed by GC-MS. Metabolite abbreviations: 3PG, 3-phosphoglycerate; PYR, pyruvate; OAA, oxaloacetate. Net fluxes are in supplemental Table S4.
DISCUSSION
Transcriptional control of metabolic pathways for carbohydrate utilization in bacteria is mediated by a variety of TFs from different protein families (e.g. AraC, LacI, GntR, DeoR, and ROK). Peripheral sugar catabolic pathways are usually controlled by committed transcription factors with presumably local regulons (e.g. the GlcNAc utilization pathway is regulated by NagR repressor in Shewanella) (13), whereas central glycolytic pathways are regulated by global TFs (e.g. the EMP pathway is under control of Crp and FruR in E. coli) (1). Previous studies show that P. putida HexR is a KDPG-responsive transcriptional regulator that negatively controls the edd-glk-gltRS, zwf-pgl-eda, and gap genes involved in the ED and EMP pathways of glucose utilization (8–11). Here, we performed the comparative genomics reconstruction of HexR regulons in 87 species of γ- and β-proteobacteria by combining the identification candidate HexR-binding sites with the analysis of functional genomic context. We report a high variability in gene content of the reconstructed HexR regulons in different taxa, although the cognate DNA motifs of HexR orthologs share a common palindromic consensus sequence TGTA-N7-TACA. The combined bioinformatics, in vitro and in vivo characterization of the extended HexR regulon in Shewanella spp., revealed that HexR functions as both a repressor and activator on different promoters involved in the catabolic and gluconeogenic pathways, respectively. The discovered dual mode of action and the functional context of target genes indicate that HexR is a novel pleiotropic regulator of the central carbon metabolism in Proteobacteria. In Shewanella, HexR usually controls various genes from the central carbon metabolism, as well as the deoxynucleoside and glycine utilization.
HexR belongs to the RpiR family of TFs that are characterized by an N-terminal DNA binding helix-turn-helix domain (HTH_6 or PF01418 in Pfam database) and a C-terminal sugar phosphate binding domain (SIS or PF01380) (37). The presence of the SIS effector binding domain in regulators from the RpiR family correlates with their known ability to bind various sugar phosphates, including KDPG for HexR in P. putida (8, 10), allose 6-phosphate for RpiR/AlsR in E. coli (38), N-acetylmuramic acid 6-phosphate for MurR in E. coli (39), and glucosamine 6-phosphate for SiaR in Haemophilus influenzae (40). Multiple alignment of HexR proteins revealed that among the residues that were previously predicted to be more important for the recognition of DNA in P. putida HexR (according to a homology-based three-dimensional model of HexR (8)), only Arg-54 is absolutely conserved in orthologs; Glu-49 and Arg-57 are present in more that 90% of orthologs, whereas Gln-43 and Lys-46 are present only in HexR orthologs in Pseudomonas spp. These observations correlate with the relative conservation of the identified HexR-binding DNA motifs. Interestingly, the same residues are retained in a HexR paralog translationally fused to a glucokinase domain in Burkholderia spp., suggesting that this chimeric protein could function both as a kinase and a transcriptional regulator. Two residues likely involved in effector recognition in P. putida HexR, Ser-140 and Ser-184 (8), were found absolutely conserved in all HexR orthologs, suggesting that its effector molecule KDPG validated in P. putida (8) and S. oneidensis (this study) is conserved in all HexR orthologs.
Because of the absence of 6-phosphofructokinase, the ED pathway is an essential sugar catabolic route, and KDPG is its key intermediate in Shewanella, as well as in Pseudomonas, Burkholderia, Ralstonia and some other Proteobacteria. In this work, we demonstrated that the HexR protein in Shewanella is a KDPG-responsive repressor/activator of the central carbon metabolism (Fig. 1). The proposed model of HexR-dependent control in Shewanella is presented below. In the presence of carbohydrates (hexoses or pentoses), KDPG levels in the cell increases due to the consecutive action of peripheral and central catabolic pathways. Binding of KDPG to the HexR SIS domain promotes dissociation of HexR-DNA complexes, leading to de-repression of transcription of the central glycolytic genes (zwf-pgl-edd-eda, gapA, pykA, gnd, and phk) and de-activation of the ppsA and gapB genes involved in the gluconeogenesis. In contrast, when carbohydrates are limited in the medium (e.g. when the cell is growing on lactate or pyruvate as the sole carbon source), the KDPG level is decreased; and HexR binding to its operator sites promotes repression of the glycolytic genes and activation of the gluconeogenic genes (Fig. 6).
FIGURE 6.

Model of HexR-dependent regulation in Shewanella. In the unliganded form, HexR binds to the operator (O) regions in the target operons. When HexR operator is upstream of the RNA polymerase (RNAP)-binding site, activation of transcription is observed. When HexR operator overlaps or is downstream of the RNA polymerase-binding site, inhibition of transcription is observed. In the presence of exogenous sugars, their utilization results in accumulation of glycolytic intermediates. One of these catabolites, KDPG, bind to HexR, causing it to dissociate from the DNA. Dissociation reverses the activating effect of HexR, as in the case of ppsA gene of S. oneidensis, which encodes phosphoenolpyruvate synthase (catabolite repression, left), and reverses the inhibiting effect of HexR, as in the case of the pykA gene, which encodes pyruvate kinase (catabolite activation, right).
Lactate is a major source of carbon and energy for Shewanellae, a diverse group of dissimilatory metal-reducing bacteria commonly found in aquatic and sedimentary environments (35). Based on the combination of regulatory and phenotype analysis of S. oneidensis wild-type and ΔhexR mutant (Fig. 4), we concluded that HexR is a master regulator of the gluconeogenic flux from pyruvate (and lactate). Thus, when lactate is present as a single carbon source, it is used for both energy production (via the tricarboxylic acid cycle) and biosynthesis (via gluconeogenesis). This is achieved via a low level of KDPG leading to transcriptional activation of PpsA and down-regulation of PykA (to avoid futile cycling). When lactate is present simultaneously with other carbon sources such as GlcNAc, most of the lactate would route to the tricarboxylic acid cycle for energy, whereas biosynthetic needs are supported by GlcNAc. This is achieved via accumulation of KDPG, which interacts with HexR, resulting in de-activation of PpsA and de-repression of PykA. According to this model, the pyruvate kinase PykA up-regulation would promote disposal of excess glycolytic substrate via an elevated flux from phosphoenolpyruvate to pyruvate. This situation is modeled by deletion of HexR or by deletion of PpsA (Fig. 5).
The HexR regulon in Shewanella can be considered as a partial functional replacement of the Enterobacterial Cra (FruR) regulon that plays a pleiotropic role, modulating the direction of carbon flow through the different metabolic pathways of energy metabolism. The catabolite repressor/activator Cra in E. coli binds DNA operators in the absence of fructose 1-phosphate or fructose 1,6-biphosphate and is inactivated upon the interaction with the effectors (3). When bound to its targets, it acts as an activator of gluconeogenic (ppsA and pckA) and glyoxylate shunt (aceBA) genes and a repressor of glycolytic genes (pykF, gapA, edd-eda, eno, pgk, and pfkA) (41). Thus, both HexR and Cra are dual transcriptional regulators that mediate cAMP-independent catabolite control in response to key catabolite intermediates. Shewanella spp. does not have Cra and are not able to grow on fructose (13). The Cra (FruR) regulons reconstructed in other γ-proteobacteria (Aeromonadales, Pseudomonadales, Psychromonadaceae, are Vibrionales) are implicated in the local control of the fructose utilization operon (5). The HexR regulons are local (regulate 1–2 operons) in some lineages (e.g. in Enterobacteriales, Ralstonia, and Burkholderia) and global (control 15–20 operons) in other lineages (e.g. in Shewanella, Aeromonadales, Psychromonadaceae, and Vibrionales). For instance, in E. coli HexR has only two predicted target genes (zwf and ybfA), whereas in Photobacterium profundum it controls near 20 operons implicated in several central metabolic pathways. These observations suggest that regulatory networks for the central carbon metabolism are very flexible in Proteobacteria. Global rewiring of these regulatory networks includes multiple regulon expansion and reduction events.
Interestingly, the KDG-responsive KdgR regulon negatively controlling the pectin catabolism in plant pathogenic Erwinia is largely extended in comparison with other enterobacteria and include the positively regulated gene ppsA (42). Therefore, the ppsA gene is activated by at least three dual-mode catabolic regulators (HexR, FruR, and KdgR) in three lineages of γ-proteobacteria (Shewanella, Escherichia, and Erwinia, respectively).
Concluding Remarks
This study provided a comprehensive bioinformatic analysis of the HexR regulon that constitutes a pleiotropic system of transcriptional regulation of central carbohydrate metabolism in several groups of Proteobacteria. The key conjectures delivered by this analysis, the global regulatory effect and dual mode of HexR-dependent regulation, were experimentally confirmed in the S. oneidensis model providing additional support for the reconstruction of the entire HexR regulon in Proteobacteria. This study demonstrates the value of comparative genomics supported by focused validation experiments for the ab initio reconstruction regulatory networks in large sets of previously unexplored biological species.
This work was supported in part by the United States Department of Energy, Office of Science, as part of the Genomic Science Program under Contract DE-AC05-76RLO with Pacific Northwest National Laboratory (Genomic Science Program Foundational Scientific Focus Area), Sanford-Burnham Medical Research Institute, Lawrence Berkeley National Laboratory Grant DE-SC0004999, National Science Foundation Grant DBI-0850546, Russian Foundation for Basic Research Grant 10-04-01768, and the Russian Academy of Sciences Program Molecular and Cellular Biology.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1–S4 and Figs. S1–S4.
- TF
- transcription factor
- EMP pathway
- Embden Meyerhof-Parnas pathway
- ED pathway
- Entner-Doudoroff pathway
- KDPG
- 2-keto-3-deoxy-6-phosphogluconate
- FPA
- fluorescence polarization assay.
REFERENCES
- 1. Saier M. H., Jr. (1998) Biotechnol. Bioeng. 58, 170–174 [DOI] [PubMed] [Google Scholar]
- 2. Gosset G., Zhang Z., Nayyar S., Cuevas W. A., Saier M. H., Jr. (2004) J. Bacteriol. 186, 3516–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Saier M. H., Jr., Ramseier T. M. (1996) J. Bacteriol. 178, 3411–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ramseier T. M., Bledig S., Michotey V., Feghali R., Saier M. H., Jr. (1995) Mol. Microbiol. 16, 1157–1169 [DOI] [PubMed] [Google Scholar]
- 5. Rodionov D. A. (2007) Chem. Rev. 107, 3467–3497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Collier D. N., Hager P. W., Phibbs P. V., Jr. (1996) Res. Microbiol. 147, 551–561 [DOI] [PubMed] [Google Scholar]
- 7. Lessie T. G., Phibbs P. V., Jr. (1984) Annu. Rev. Microbiol. 38, 359–388 [DOI] [PubMed] [Google Scholar]
- 8. Daddaoua A., Krell T., Ramos J. L. (2009) J. Biol. Chem. 284, 21360–21368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. del Castillo T., Duque E., Ramos J. L. (2008) J. Bacteriol. 190, 2331–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim J., Jeon C. O., Park W. (2008) Microbiology 154, 3905–3916 [DOI] [PubMed] [Google Scholar]
- 11. Petruschka L., Adolf K., Burchhardt G., Dernedde J., Jürgensen J., Herrmann H. (2002) FEMS Microbiol. Lett. 215, 89–95 [DOI] [PubMed] [Google Scholar]
- 12. Rodionov D. A., Novichkov P. S., Stavrovskaya E. D., Rodionova I. A., Li X., Kazanov M. D., Ravcheev D. A., Gerasimova A. V., Kazakov A. E., Kovaleva G. Y., Permina E. A., Laikova O. N., Overbeek R., Romine M. F., Fredrickson J. K., Arkin A. P., Dubchak I., Osterman A. L., Gelfand M. S. (2011) BMC Genomics 12, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rodionov D. A., Yang C., Li X., Rodionova I. A., Wang Y., Obraztsova A. Y., Zagnitko O. P., Overbeek R., Romine M. F., Reed S., Fredrickson J. K., Nealson K. H., Osterman A. L. (2010) BMC Genomics 11, 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benson D. A., Karsch-Mizrachi I., Lipman D. J., Ostell J., Sayers E. W. (2010) Nucleic Acids Res. 38, D46–D51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dehal P. S., Joachimiak M. P., Price M. N., Bates J. T., Baumohl J. K., Chivian D., Friedland G. D., Huang K. H., Keller K., Novichkov P. S., Dubchak I. L., Alm E. J., Arkin A. P. (2010) Nucleic Acids Res. 38, D396–D400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Overbeek R., Begley T., Butler R. M., Choudhuri J. V., Chuang H. Y., Cohoon M., de Crécy-Lagard V., Diaz N., Disz T., Edwards R., Fonstein M., Frank E. D., Gerdes S., Glass E. M., Goesmann A., Hanson A., Iwata-Reuyl D., Jensen R., Jamshidi N., Krause L., Kubal M., Larsen N., Linke B., McHardy A. C., Meyer F., Neuweger H., Olsen G., Olson R., Osterman A., Portnoy V., Pusch G. D., Rodionov D. A., Rückert C., Steiner J., Stevens R., Thiele I., Vassieva O., Ye Y., Zagnitko O., Vonstein V. (2005) Nucleic Acids Res. 33, 5691–5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huson D. H., Richter D. C., Rausch C., Dezulian T., Franz M., Rupp R. (2007) BMC Bioinformatics 8, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Edgar R. C. (2004) Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Novichkov P. S., Rodionov D. A., Stavrovskaya E. D., Novichkova E. S., Kazakov A. E., Gelfand M. S., Arkin A. P., Mironov A. A., Dubchak I. (2010) Nucleic Acids Res. 38, W299–W307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mironov A. A., Koonin E. V., Roytberg M. A., Gelfand M. S. (1999) Nucleic Acids Res. 27, 2981–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crooks G. E., Hon G., Chandonia J. M., Brenner S. E. (2004) Genome Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Novichkov P. S., Laikova O. N., Novichkova E. S., Gelfand M. S., Arkin A. P., Dubchak I., Rodionov D. A. (2010) Nucleic Acids Res. 38, D111–D118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin C. T., Moore P. A., Auberry D. L., Landorf E. V., Peppler T., Victry K. D., Collart F. R., Kery V. (2006) Protein Expr. Purif. 47, 16–24 [DOI] [PubMed] [Google Scholar]
- 25. Mossessova E., Lima C. D. (2000) Mol. Cell 5, 865–876 [DOI] [PubMed] [Google Scholar]
- 26. Osterman A. L., Lueder D. V., Quick M., Myers D., Canagarajah B. J., Phillips M. A. (1995) Biochemistry 34, 13431–13436 [DOI] [PubMed] [Google Scholar]
- 27. Rodionov D. A., De Ingeniis J., Mancini C., Cimadamore F., Zhang H., Osterman A. L., Raffaelli N. (2008) Nucleic Acids Res. 36, 2047–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Titolo S., Brault K., Majewski J., White P. W., Archambault J. (2003) J. Virol. 77, 5178–5191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Livak K. J., Schmittgen T. D. (2001) Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 30. Zamboni N., Fendt S. M., Rühl M., Sauer U. (2009) Nat. Protoc. 4, 878–892 [DOI] [PubMed] [Google Scholar]
- 31. Pinchuk G. E., Hill E. A., Geydebrekht O. V., De Ingeniis J., Zhang X., Osterman A., Scott J. H., Reed S. B., Romine M. F., Konopka A. E., Beliaev A. S., Fredrickson J. K., Reed J. L. (2010) PLoS Comput. Biol. 6, e1000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Osterman A., Overbeek R. (2003) Curr. Opin. Chem. Biol. 7, 238–251 [DOI] [PubMed] [Google Scholar]
- 33. Rowley D. L., Wolf R. E., Jr. (1991) J. Bacteriol. 173, 968–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Driscoll M. E., Romine M. F., Juhn F. S., Serres M. H., McCue L. A., Beliaev A. S., Fredrickson J. K., Gardner T. S. (2007) Genome Inform. 18, 287–298 [PubMed] [Google Scholar]
- 35. Pinchuk G. E., Rodionov D. A., Yang C., Li X., Osterman A. L., Dervyn E., Geydebrekht O. V., Reed S. B., Romine M. F., Collart F. R., Scott J. H., Fredrickson J. K., Beliaev A. S. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 2874–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Niersbach M., Kreuzaler F., Geerse R. H., Postma P. W., Hirsch H. J. (1992) Mol. Gen. Genet. 231, 332–336 [DOI] [PubMed] [Google Scholar]
- 37. Bateman A. (1999) Trends Biochem. Sci. 24, 94–95 [DOI] [PubMed] [Google Scholar]
- 38. Sørensen K. I., Hove-Jensen B. (1996) J. Bacteriol. 178, 1003–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jaeger T., Mayer C. (2008) J. Bacteriol. 190, 6598–6608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnston J. W., Shamsulddin H., Miller A. F., Apicella M. A. (2010) BMC Microbiol. 10, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shimada T., Yamamoto K., Ishihama A. (2011) J. Bacteriol. 193, 649–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rodionov D. A., Gelfand M. S., Hugouvieux-Cotte-Pattat N. (2004) Microbiology 150, 3571–3590 [DOI] [PubMed] [Google Scholar]